Summary
Clinical characteristics.
Mitochondrial DNA (mtDNA)-associated Leigh syndrome and NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa) are part of a continuum of progressive neurodegenerative disorders caused by abnormalities of mitochondrial energy generation.
- Leigh syndrome (or subacute necrotizing encephalomyelopathy) is characterized by onset of symptoms typically between ages three and 12 months, often following a viral infection. Decompensation (often with elevated lactate levels in blood and/or CSF) during an intercurrent illness is typically associated with psychomotor retardation or regression. Neurologic features include hypotonia, spasticity, movement disorders (including chorea), cerebellar ataxia, and peripheral neuropathy. Extraneurologic manifestations may include hypertrophic cardiomyopathy. About 50% of affected individuals die by age three years, most often as a result of respiratory or cardiac failure.
- NARP is characterized by proximal neurogenic muscle weakness with sensory neuropathy, ataxia, and pigmentary retinopathy. Onset of symptoms, particularly ataxia and learning difficulties, is often in early childhood. Individuals with NARP can be relatively stable for many years, but may suffer episodic deterioration, often in association with viral illnesses.
Diagnosis/testing.
The diagnosis of mtDNA-associated Leigh syndrome is established clinically in a proband with progressive neurologic disease with motor and intellectual developmental delay, signs and symptoms of brain stem and/or basal ganglia disease, raised lactate concentration in blood and/or cerebrospinal fluid, and any one of the following:
- Characteristic features on brain imaging
- Typical neuropathologic changes
- Typical neuropathology in a similarly affected sib
Identification of a pathogenic variant in one of the 14 mitochondrial genes known to be involved in mtDNA-associated Leigh syndrome confirms the diagnosis.
The diagnosis of NARP is established in a proband with suggestive clinical features and a heteroplasmic pathogenic variant in one of the three mitochondrial genes known to be involved in NARP identified by molecular genetic testing.
Management.
Treatment of manifestations: Supportive treatment includes use of sodium bicarbonate or sodium citrate for acute exacerbations of acidosis and anti-seizure medication for seizures. Dystonia is treated with benzhexol, baclofen, tetrabenazine, and gabapentin alone or in combination, or by injections of botulinum toxin. Anticongestive therapy may be required for cardiomyopathy. Regular nutritional assessment of daily caloric intake and adequacy of diet and psychological support for the affected individual and family are essential.
Surveillance: Neurologic, ophthalmologic, and cardiologic evaluations at regular intervals to monitor progression and appearance of new symptoms. Care is frequently coordinated by a biochemical geneticist in North America, and by a metabolic physician/pediatrician elsewhere in the world.
Agents/circumstances to avoid: Sodium valproate and barbiturates, anesthesia, and dichloroacetate.
Genetic counseling.
Mitochondrial DNA-associated Leigh syndrome and NARP are transmitted by maternal inheritance. The father of a proband is not at risk of having the mtDNA pathogenic variant. The mother of a proband usually has the mtDNA pathogenic variant and may or may not have symptoms. In most cases, the mother has a much lower proportion of abnormal mtDNA than the proband and usually remains asymptomatic or develops only mild symptoms. Occasionally the mother has a substantial proportion of abnormal mtDNA and develops severe symptoms in adulthood. Offspring of males with a mtDNA pathogenic variant are not at risk; all offspring of females with a mtDNA pathogenic variant are at risk of inheriting the pathogenic variant. The risk to offspring of a female proband of developing symptoms depends on the tissue distribution and proportion of abnormal mtDNA. Prenatal testing and preimplantation genetic testing for couples at increased risk of having children with mtDNA-associated Leigh syndrome or NARP are possible by analysis of mtDNA extracted from non-cultured fetal cells or from single blastomeres, respectively. However, long-term outcome cannot be reliably predicted on the basis of molecular genetic test results.
GeneReview Scope
Table
Leigh syndrome (mtDNA pathogenic variant) Leigh-like syndrome
Diagnosis
Suggestive Findings
Mitochondrial DNA-Associated Leigh Syndrome
Mitochondrial DNA-associated Leigh syndrome should be suspected in individuals with the following findings.
Clinical features
- Motor and intellectual developmental delay, usually with neurodevelopmental regression
- Signs and symptoms of brain stem and/or basal ganglia disease (e.g., respiratory abnormalities, nystagmus, ophthalmoparesis, optic atrophy, ataxia, dystonia)
- Seizures
- Progressive neurologic disease
Laboratory findings
- Lactate concentration in blood is increased. Elevation tends to be more marked in postprandial samples.
- Lactate concentration in cerebrospinal fluid (CSF) is increased. Increased lactate is more consistent in CSF than blood samples, but is not an invariant finding.
- Plasma amino acids may show increased alanine concentration, reflecting persistent hyperlactatemia.
- Decreased plasma citrulline concentration was reported in individuals with the m.8993T>G pathogenic variant [Rabier et al 1998].
- Urine organic acid analysis often detects lactic aciduria and Krebs cycle intermediates.Note: Identification of increased methylmalonic acid or proprionic acid is suggestive of other specific types of Leigh syndrome or organic acidemias (e.g., 3-methylglutaconic aciduria with sensorineural deafness, encephalopathy and Leigh-like (MEGDEL) syndrome, succinylCoA-ligase deficiency, methylmalonic aciduria, propionic aciduria) (see Differential Diagnosis).
Radiographic findings on brain imaging
- Characteristic bilateral symmetric hypodensities in the basal ganglia on computed tomography or bilateral symmetric hyperintense signal abnormality in the brain stem and/or basal ganglia on T2-weighted magnetic resonance imaging (MRI) [Bonfante et al 2016]
- Proton magnetic resonance spectroscopy can also be useful in detecting regional elevations in brain lactate levels.
- In individuals with NARP, cerebral and cerebellar atrophy may be noted on brain MRI.Note: Specific brain lesions affecting the mammillothalamic tracts, substantia nigra, medial lemniscus, medial longitudinal fasciculus, spinothalamic tracts, and cerebellum appear to be characteristic of Leigh syndrome caused by pathogenic variants in the nuclear gene NDUFAF2 [Barghuti et al 2008, Hoefs et al 2008, Herzer et al 2010]. MEGDEL syndrome is associated with a distinctive brain MRI pattern affecting the basal ganglia, especially the putamen. Initially there are T2-weighted signal changes of the pallidum, and later swelling of the putamen and caudate nucleus with an "eye" representing early sparing of the dorsal putamen, followed by progressive involvement of the putamina [Wortmann et al 2015] (see Differential Diagnosis).
Histopathology of muscle tissue shows only minimal if any changes, such as accumulation of intracytoplasmic neutral lipid droplets. Ragged red fibers are rarely (if ever) seen. Cytochrome c oxidase-negative fibers are occasionally found in individuals with Leigh syndrome caused by certain mtDNA and nuclear gene variants.
Note: (1) Although muscle biopsy is only occasionally abnormal, when it is abnormal it can be as much of a contributor to diagnostic certainty as respiratory chain enzymes or molecular testing. (2) If an affected individual is having a muscle biopsy for enzyme testing, histologic examination should also be performed.
Respiratory chain enzyme studies. Biochemical analysis of tissue biopsies or cultured cells often detects deficient activity of one or more of the respiratory chain enzyme complexes. Isolated defects of complex I or complex IV are the most common enzyme abnormalities observed and can help guide subsequent molecular genetic testing of mtDNA or nuclear genes. Biochemical results can also be normal, usually in individuals with mtDNA pathogenic variants affecting complex V subunits such as the pathogenic variants at mitochondrial nucleotides 8993 and 9176 (see Table 5).
- Skeletal muscle is usually the tissue of choice for enzyme studies.
- Skin fibroblasts can be used, but only about 50% of respiratory chain enzyme defects identified in skeletal muscle are also identified in skin fibroblasts.
- Approximately 10%-20% of individuals with normal skeletal muscle respiratory chain enzymes may have an enzyme defect detected in liver or cardiac muscle, particularly if those tissues are involved clinically [Thorburn et al 2004].
NARP
NARP should be suspected in individuals with the clinical, electrophysiologic, and radiographic features listed below. However, not all features may be present, at least initially, and the diagnosis should be suspected in individuals with several of the following features:
- Muscle weakness
- Neuropathy
- Ataxia
- Seizures
- Retinitis pigmentosa or optic atrophy
- Learning difficulties
- Other:
- Electromyography and nerve conduction studies may demonstrate peripheral neuropathy (which may be a sensory or sensorimotor axonal polyneuropathy).
- Cerebral and cerebellar atrophy may be noted on brain MRI.
- Electroretinogram may reveal abnormalities (including small-amplitude waveform) or may be normal.
Establishing the Diagnosis
Leigh syndrome. The diagnosis of mtDNA-associated Leigh syndrome is established in a proband fulfilling the criteria for Leigh syndrome (see following) in whom a heteroplasmic or homoplasmic pathogenic variant in one of the genes listed in Table 1a or Table 1b has been identified.
Stringent diagnostic criteria for Leigh syndrome were defined by Rahman et al [1996]:*
- Progressive neurologic disease with motor and intellectual developmental delay
- Signs and symptoms of brain stem and/or basal ganglia disease
- Raised lactate concentration in blood and/or cerebrospinal fluid (CSF)
- One or more of the following:
- Characteristic features of Leigh syndrome on neuroradioimaging (See Suggestive Findings.)
- Typical neuropathologic changes: multiple focal symmetric necrotic lesions in the basal ganglia, thalamus, brain stem, dentate nuclei, and optic nerves. Histologically, lesions have a spongiform appearance and are characterized by demyelination, gliosis, and vascular proliferation. Neuronal loss can occur, but typically the neurons are relatively spared.
- Typical neuropathology in a similarly affected sib
* Note: Prior to the development of modern imaging techniques, definitive diagnosis of Leigh syndrome was based on characteristic neuropathologic features and thus could only be made postmortem.
Baertling et al [2014] described similar diagnostic criteria that allow for the diagnosis of Leigh syndrome in the absence of raised lactate levels. Their criteria include the following:
- Neurodegenerative disease with variable symptoms resulting from mitochondrial dysfunction
- Mitochondrial dysfunction caused by a hereditary genetic defect
- Bilateral CNS lesions that can be associated with further abnormalities in diagnostic imaging
Lake et al [2016] revised the diagnostic criteria to include "abnormal energy metabolism indicated by a severe defect in oxidative phosphorylation (OXPHOS) or pyruvate dehydrogenase complex (PDHc) activity, a molecular diagnosis in a gene related to mitochondrial energy generation, or elevated serum or CSF lactate."
NARP. Strict diagnostic criteria for NARP have not yet been established. The diagnosis of NARP is established in a proband with the above suggestive clinical features and a mtDNA pathogenic variant identified by molecular genetic testing.
Molecular genetic testing approaches for Leigh syndrome and NARP can include targeted single-gene testing, mitochondrial genome sequencing, and more comprehensive genomic testing.
Option 1 (preferred in children)*
- 1.
Targeted analysis for the two common MT-ATP6 pathogenic variants (see Table 1) is performed concurrently with deletion/duplication analysis on leukocyte DNA.
- 2.
Mitochondrial genome sequencing is performed next if an MT-ATP6 pathogenic variant or deletion/duplication is not detected.
Option 2 (preferred in adults)*
- 1.
Targeted sequence analysis of leukocyte DNA for the two common MT-ATP6 pathogenic variants can be performed first (see Table 1).
- 2.
Mitochondrial genome sequencing is performed next if an MT-ATP6 pathogenic variant is not detected by targeted analysis.
Option 3. Mitochondrial genome sequencing is performed first (see Table 1).
* Note: (1) Most mtDNA pathogenic variants are "heteroplasmic" (i.e., mutated mtDNA coexists with wild type mtDNA) and for some pathogenic variants, the mutation load may vary among different tissues and may increase or decrease with age. (2) Mitochondrial DNA pathogenic variants may be lost from the leukocyte population with increasing age [Rahman et al 2001]; therefore, in adults with milder symptoms and for asymptomatic older maternal relatives, the pathogenic variant may only be detected in tissues such as hair follicles, urine sediment cells, or skeletal muscle, which is the most reliable source of mtDNA for analysis [McDonnell et al 2004, Shanske et al 2004]. (3) Leukocyte (vs skeletal muscle) testing is acceptable in children, particularly when using next-generation sequencing methods, which allow detection of very low heteroplasmy levels. (4) Deletions are not usually detectable in leukocyte DNA from adults; in this age group, muscle (or urinary epithelial cells) is the tissue of choice for analysis. (5) Deletions/duplications of mtDNA are an extremely rare cause of Leigh syndrome in adults. The authors are not aware of any published reports of children with Leigh syndrome, Leigh-like syndrome, or NARP where a pathogenic mtDNA variant was not detected in blood.
More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation). For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
See Table 1a for the most common genetic causes (i.e., pathogenic variants of any one of the mtDNA-encoded genes included in this table account for >1% of mtDNA-associated Leigh syndrome and NARP) and Table 1b for less common genetic causes (i.e., pathogenic variants of any one of the mitochondrial genes included in this table are reported in only a few families).
Table 1a.
Molecular Genetics of Mitochondrial DNA-Associated Leigh Syndrome and NARP: Most Common Genetic Causes
Table 1b.
Molecular Genetics of mtDNA-Associated Leigh Syndrome and NARP: Less Common Genetic Causes
Clinical Characteristics
Clinical Description
Mitochondrial DNA-associated Leigh syndrome (subacute necrotizing encephalomyelopathy). Onset of symptoms can be from the neonatal period through adulthood but is typically between age three and 12 months, often following a viral infection. Later onset (i.e., age >1 year, including presentation in adulthood) and slower progression occur in up to 25% of individuals [Sofou et al 2014].
Leigh syndrome is a progressive neurodegenerative disorder. Initial features may be nonspecific, such as failure to thrive and persistent vomiting. Decompensation (often with raised blood and/or CSF lactate concentrations) during an intercurrent illness is typically associated with psychomotor retardation or regression. A period of recovery may follow the initial decompensation, but the individual rarely returns to the developmental status achieved prior to the presenting illness.
Neurologic features include hypotonia, spasticity, dystonia, muscle weakness, hypo- or hyperreflexia, seizures (myoclonic or generalized tonic-clonic), infantile spasms, movement disorders (including chorea), cerebellar ataxia, and peripheral neuropathy. Brain stem lesions may cause respiratory difficulty (apnea, hyperventilation, or irregular respiration), bulbar problems such as abnormal swallowing and speech, persistent vomiting, and abnormalities of thermoregulation (hypo- and hyperthermia).
Ophthalmologic findings include optic atrophy, retinitis pigmentosa, and eye movement disorders. Pigmentary retinopathy occurs in up to 40% of individuals with a mtDNA 8993 pathogenic variant [Santorelli et al 1993].
Other. Individuals with Leigh syndrome may present with extraneurologic multisystem manifestations. These can include cardiac (hypertrophic or dilated cardiomyopathy [Wang et al 2008, Hadzsiev et al 2010]), hepatic (hepatomegaly or liver failure [Van Hove et al 2010, Duff et al 2015]), or renal (renal tubulopathy or diffuse glomerulocystic kidney damage [López et al 2006, Naess et al 2009] manifestations. Leigh syndrome as a whole is the most phenotypically heterogeneous mitochondrial disease, with more than 200 associated phenotypes [Rahman et al 2017].
Most affected individuals have episodic deterioration interspersed with "plateaus" during which development may be quite stable or even show some progress. The duration of these plateaus is variable and in rare cases may be ten years or more. More typically, death occurs by age two to three years, most often from respiratory or cardiac failure. In undiagnosed individuals, death may appear to be sudden and unexpected.
Leigh-like syndrome. The term "Leigh-like syndrome" is often used for individuals with clinical and other features that are strongly suggestive of Leigh syndrome but who do not fulfill the stringent diagnostic criteria because of atypical neuropathology (variation in the distribution or character of lesions or with the additional presence of unusual features such as extensive cortical destruction), atypical or normal neuroimaging, normal blood and CSF lactate levels, or incomplete evaluation. The heterogeneous clinical presentation that occurs in Leigh syndrome is also present in Leigh-like syndromes.
NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa). Onset of symptoms, particularly ataxia and learning difficulties, is often in early childhood.
NARP is characterized by proximal neurogenic muscle weakness with sensory neuropathy, ataxia, pigmentary retinopathy, seizures, learning difficulties, and dementia. Other clinical features include short stature, sensorineural hearing loss, progressive external ophthalmoplegia, cardiac conduction defects (heart block) and a mild anxiety disorder [Santorelli et al 1997, Sembrano et al 1997, Rawle & Larner 2013]. Visual symptoms may be the only clinical feature. One individual had obstructive sleep apnea requiring tracheostomy and nocturnal mechanical ventilation [Sembrano et al 1997].
Individuals with NARP can be relatively stable for many years, but may experience episodic deterioration, often in association with viral illnesses.
Intermediate phenotypes in the continuum. Maternal relatives of individuals with Leigh syndrome or NARP can have any one or a combination of the individual symptoms associated with Leigh syndrome, NARP, or other mitochondrial disorders. These include mild learning difficulties, muscle weakness, night blindness, deafness, diabetes mellitus, migraine, or sudden unexpected death.
Genotype-Phenotype Correlations
For most mtDNA pathogenic variants, it is difficult to distinguish a correlation between genotype and phenotype because clinical expression of a mtDNA pathogenic variant is influenced not only by the pathogenicity of the variant itself but also by the relative amount of mutated and wild type mtDNA (the heteroplasmic mutant load), the variation in the proportion of abnormal mtDNA among different tissues, and the energy requirements of brain and other tissues, which may vary with age.
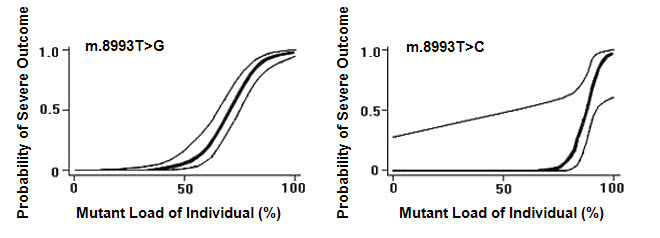
The m.8993T>G and m.8993T>C pathogenic variants probably show the strongest genotype-phenotype correlation of any mtDNA pathogenic variants. Notably, they show very little tissue-dependent or age-dependent variation in the proportion of abnormal mtDNA [White et al 1999c] as well as a strong correlation between the proportion of abnormal mtDNA and disease severity, allowing White et al [1999a] to generate logistic regression models that predict the probability of a severe outcome in an individual based on the measured proportion of abnormal mtDNA of m.8993T>G and m.8993T>C (Figure 1). Note, however, that in such retrospective studies it is not possible to completely avoid ascertainment bias, and the data should be regarded as broadly indicative rather than precise.
- m.8993T>G. Individuals in whom the proportion of abnormal mtDNA is below 60% are usually asymptomatic, or have only mild pigmentary retinopathy or migraine headaches; however, asymptomatic adults with levels of abnormal mtDNA as high as 75% have been reported [Tatuch et al 1992, Ciafaloni et al 1993]. As a generalization, individuals with moderate levels (~70%-90%) of the m.8993T>G pathogenic variant present with the NARP phenotype, while those with more than 90% abnormal mtDNA have maternally inherited Leigh syndrome [Claeys et al 2016].Note: Overlap in the proportion of abnormal mtDNA is observed between some asymptomatic individuals and others with NARP, and between some individuals with NARP and others with Leigh syndrome.
- m.8993T>C is a less severe variant than m.8993T>G, and virtually all symptomatic individuals with m.8993T>C ve more than 90% abnormal mtDNA.
Genotype-phenotype correlations are much weaker for other mtDNA pathogenic variants detected in multiple unrelated individuals with Leigh syndrome (e.g., m.3243A>G in MT-TL1, m.8344A>G in MT-TK, m.9176T>C in MT-ATP6, m.14459G>A and m.14487T>C in MT-ND6, m.10158T>C and m.10191T>C in MT-ND3, and m.13513G>A in MT-ND5). The presence of any of these variants in individuals with symptoms of Leigh syndrome identifies the genetic cause of the disorder. However, unlike the m.8993T>G and m.8993T>C variants, it is usually not possible to interpret the heteroplasmic mutant load to predict outcome (e.g., in asymptomatic family members or in prenatal diagnosis) unless the value is near 0% or near 100%.
Penetrance
Nomenclature
Leigh syndrome was originally described by Leigh [1951] as "subacute necrotizing encephalomyelopathy" in an infant age seven months.
Individuals with Leigh syndrome caused by a mtDNA pathogenic variant are often referred to as having "maternally inherited Leigh syndrome" (MILS) [Ciafaloni et al 1993].
Prevalence
The following prevalence data are for all Leigh syndrome. In southeastern Australia, Leigh syndrome occurs in 1:77,000 infants, and the combined birth prevalence of Leigh syndrome plus Leigh-like syndrome was 1:40,000 [Rahman et al 1996]. In western Sweden, the prevalence of Leigh syndrome in preschool children was 1:34,000 [Darin et al 2001]. Thus, the prevalence of Leigh syndrome is likely to be 1:30,000 to 1:40,000.
Analyses of a large series of 67 individuals with Leigh or Leigh-like syndrome reported by Rahman et al [1996] have identified mtDNA pathogenic variants in 34% [Author, personal communication]. The prevalence of mtDNA-associated Leigh syndrome is likely to be 1:100,000 to 1:140,000.
No data on the prevalence of NARP exist. NARP is substantially less common than Leigh syndrome.
Genetically Related Disorders
Mitochondrial DNA pathogenic variants can also be associated with a variety of disorders including MELAS, MERRF, Leber hereditary optic neuropathy (LHON), infantile bilateral striatal necrosis, progressive external ophthalmoplegia, diabetes mellitus, cardiomyopathy, deafness, or sudden (unexplained) death in infancy, childhood, or adulthood (see Mitochondrial Disorders Overview).
Differential Diagnosis
Leigh syndrome and Leigh-like syndrome. In most individuals with Leigh syndrome, the disease is not caused by a mtDNA pathogenic variant but by an autosomal recessive or X-linked disorder of mitochondrial energy generation. Pathogenic variants in nuclear genes that result in respiratory chain complex deficiencies and Leigh and Leigh-like syndromes are summarized in Tables 2-4. See also Nuclear Gene-Encoded Leigh Syndrome Spectrum Overview.
Table 2.
Autosomal Recessive Leigh Syndrome
Table 3.
Autosomal Recessive Leigh-Like Syndromes
Table 4.
X-Linked Leigh Syndrome and Leigh-Like Syndrome
Other disorders that cause or resemble Leigh syndrome:
- Bilateral striatal necrosis [De Meirleir et al 1995, Thyagarajan et al 1995]. Autosomal recessive infantile bilateral striatal necrosis may be caused by mutation of:
- NUP62 (OMIM 605815), encoding a component of the nuclear pore [Basel-Vanagaite et al 2006]; and
- ADAR (OMIM 146920), encoding the editing protein adenosine deaminase acting on RNA [Livingston et al 2014].
- Acute necrotizing encephalopathy, which may be triggered by viral infections. Mutation of RANBP2, encoding another nuclear pore component, is associated with susceptibility to infection-induced acute encephalopathy 3 (OMIM 608033).
- Viral encephalopathies [Suwa et al 1999]
- Other neurodegenerative disorders with similar changes on neuroimaging. These include pantothenate kinase-associated neurodegeneration, neuroferritinopathy, and methylmalonic acidemia and propionic acidemia.
NARP. Disorders in the differential diagnosis:
- Neurogenic weakness and neuropathy (See Charcot-Marie-Tooth Hereditary Neuropathy Overview.)
- Ataxia (See Hereditary Ataxia Overview.)
- Retinitis pigmentosa (See Retinitis Pigmentosa Overview.)
Management
Evaluation Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with mtDNA-associated Leigh syndrome or NARP, the following evaluations are recommended:
- Developmental assessment
- Neurologic evaluation, MRI, MRS, EEG (if seizures are suspected), and nerve conduction studies (if neuropathy is suspected)
- Metabolic evaluation, including plasma and cerebrospinal fluid lactate and pyruvate concentrations, urine organic acids
- Ophthalmologic evaluation
- Cardiac evaluation
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
As for most mitochondrial diseases, no specific curative treatment for mtDNA-associated Leigh syndrome and NARP exists [Kanabus et al 2014]. Supportive management includes treatment of the following:
- Acidosis. Sodium bicarbonate or sodium citrate for acute exacerbations of acidosis
- Seizures. Appropriate anti-seizure medication tailored to the type of seizure under the supervision of a neurologist. Sodium valproate and barbiturates should be avoided because of their inhibitory effects on the mitochondrial respiratory chain [Anderson et al 2002].
- Dystonia
- Benzhexol, baclofen, tetrabenazine, and gabapentin may be useful, alone or in various combinations; an initial low dose should be started and gradually increased until symptom control is achieved or intolerable side effects occur.
- Botulinum toxin injection has also been used in individuals with Leigh syndrome and severe intractable dystonia.
- Cardiomyopathy. Anticongestive therapy may be required and should be supervised by a cardiologist.
Regular assessment of daily caloric intake and adequacy of dietary structure including micronutrients and feeding management is indicated.
Psychological support for the affected individual and family is essential.
Surveillance
Affected individuals should be followed at regular intervals (typically every 6-12 months) to monitor progression and the appearance of new symptoms. Neurologic, ophthalmologic, and cardiologic evaluations are recommended.
Agents/Circumstances to Avoid
Sodium valproate and barbiturates should be avoided because of their inhibitory effect on the mitochondrial respiratory chain [Anderson et al 2002].
Anesthesia can potentially aggravate respiratory symptoms and precipitate respiratory failure, so careful consideration should be given to its use and to monitoring of the individual prior to, during, and after anesthetic procedures [Parikh et al 2015].
Dichloroacetate (DCA) reduces blood lactate by activating the pyruvate dehydrogenase complex.
- Anecdotal reports have suggested that DCA may cause some short-term clinical improvement in mtDNA-associated Leigh syndrome [Fujii et al 2002].
- A double-blind, placebo-controlled trial of DCA in a different mitochondrial disease, MELAS, found no benefit and in fact documented a toxic effect of DCA on peripheral nerves [Kaufmann et al 2006].
- A subsequent report described the results of long-term administration of DCA to 36 children with congenital lactic acidosis (randomized control trial followed by an open label extension) [Stacpoole et al 2008]. This study concluded that oral DCA is well tolerated in young children with congenital lactic acidosis and that it was not possible to determine whether the peripheral neuropathy associated with long-term DCA administration is attributable to the drug or to the underlying disease process. It therefore appears prudent for individuals with mtDNA-associated Leigh syndrome or NARP to avoid DCA, in view of the underlying risk of peripheral neuropathy caused by the disease itself in these conditions.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Antioxidants, including coenzyme Q10 and analogs such as idebenone, can enhance the function and viability of cultured cells from individuals with the m.8993T>G pathogenic variant [Geromel et al 2001, Mattiazzi et al 2004] but have no proven efficacy in the treatment of Leigh syndrome. Newer mitochondrial-targeted antioxidants (e.g., mitoQ) that show much greater protection against oxidative stress in cultured cell and animal models [Jauslin et al 2003, Adlam et al 2005] have been proposed as potential therapies for a range of oxidative stress-related disorders. However, no clinical trials relevant to Leigh syndrome have been reported.
EPI-743 is a structurally modified variant of CoQ10 (bis-methyl instead of bis-methoxy groups on the quinone ring, and chain length of 3 instead of 10 prenyl units) and was identified in a drug screen to have 1000-fold increased antioxidant properties compared to native CoQ10 [Enns et al 2012]. Open-label trials in end-of-life settings appeared to slow disease progression compared to historical natural history data [Enns et al 2012, Martinelli et al 2012], but the extremely unpredictable natural history of Leigh syndrome causes difficulty in interpretation of open-label studies. A Phase 2B randomized placebo-controlled double-blind crossover clinical trial of EPI-743 in children with Leigh syndrome completed in May 2015; findings have not yet been reported (ClinicalTrials.gov).
Gene therapy provides a potential approach to decreasing the proportion of abnormal mtDNA in the cells of an individual. However, all of the approaches discussed below are still a long way from clinical applicability.
In allotopic gene expression, mtDNA genes are recoded so that they can be inserted into and expressed from the nucleus. This technique was used successfully to transfer recoded mitochondrial MT-ATP6 and thereby rescue the ATP synthesis defect in cybrids containing the m.8993T>G pathogenic variant associated with maternally inherited Leigh syndrome and NARP [Manfredi et al 2002].
Studies in cultured cells have shown that a mitochondrially targeted restriction endonuclease can recognize and degrade mtDNA containing the m.8993T>G pathogenic variant found in NARP and mtDNA-associated Leigh syndrome, while leaving wild-type mtDNA intact [Tanaka et al 2002].
Another study used an adenoviral vector to deliver the restriction endonuclease to the mitochondrion and showed that there was no evidence of nuclear DNA damage in treated cells [Alexeyev et al 2008].
Transcription activator-like effector nucleases (TALENs) engineered to localize to mitochondria ("mito-TALENs") were used to eliminate mutated mtDNA from cybrids containing the m.14459G>A pathogenic variant, a maternally inherited variant that can cause Leigh syndrome [Bacman et al 2013]. More recently mito-TALENs were demonstrated to be efficacious in eliminating NARP-associated m.9176T>C mutated mtDNA from artificial mammalian oocytes [Reddy et al 2015]. Selective elimination of mutated mtDNA using mitochondrially targeted zinc-finger nucleases (mtZFNs) has also been achieved for the m.8993T>G NARP-causing variant [Gammage et al 2016].
Promising results have been obtained using a similar proof-of-principle approach in a mouse model of mtDNA heteroplasmy to shift the mtDNA heteroplasmy in muscle and brain transduced with recombinant viruses [Bayona-Bafaluy et al 2005]. This strategy could potentially prevent disease onset or reverse clinical symptoms in individuals harboring certain heteroplasmic pathogenic variants in mtDNA.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Other
A range of vitamins and other compounds are often used in the hope of improving mitochondrial function. Most commonly these include riboflavin, thiamine, and coenzyme Q10 (each at 50-100 mg/3x/day) [Kanabus et al 2014].
The ketogenic diet (KD) has been proposed as a therapy for mitochondrial disease, in particular complex I deficiency. Although this high-fat, low-carbohydrate diet has proven efficacy for refractory epilepsies, evidence for its use in the treatment of primary mitochondrial disorders is currently lacking. Preliminary reports of reducing mtDNA deletion load in cybrid models using ketones [Santra et al 2004] were not replicated in a mouse model of mitochondrial disease, although there did appear to be some improvement of myopathy in these mice [Ahola-Erkkilä et al 2010]. More recently KD was reported to attenuate liver disease in a mouse model of complex III deficiency [Purhonen et al 2017]. However, a modified Atkins diet (also high fat, low carbohydrate) led to muscle pain and elevation of creatine kinase in a small series of individuals with mitochondrial myopathy, suggesting that KD may not be well tolerated in individuals with mitochondrial disease [Ahola et al 2016]. Further clinical trials are required to determine the efficacy of KD in mitochondrial disorders. Another promising option is supplementation with the fatty acid decanoic acid, which is thought to be the active component of the KD and appears to stimulate mitochondrial biogenesis in cell models [Hughes et al 2014], including fibroblasts from individuals with complex I-deficient Leigh syndrome [Kanabus et al 2016]; clinical trials have yet to be performed.
Biotin, creatine, succinate, and idebenone have also been used. Some of these agents may show partial efficacy in some individuals with milder mitochondrial disorders, but sustained therapeutic response in individuals with NARP or Leigh syndrome has not been described.
Several studies have investigated whether upregulation of mitochondrial biogenesis may provide an effective therapeutic approach for mitochondrial respiratory chain diseases. This approach involves using agonists such as bezafibrate or resveratrol to stimulate the peroxisome proliferator-activated receptor gamma (PPARgamma) coactivator alpha (PGC-1alpha) pathway. Alternatively, nicotinamide analogs such as nicotinamide riboside or nicotinamide mononucleotide have been used to boost nicotinamide adenine dinucleotide (NAD) levels and induce mitochondrial biogenesis via the same PGC1-alpha pathway.
- A study by Bastin et al [2008] showed promising results in fibroblasts from individuals with a range of respiratory chain enzyme defects; nine of 14 patient cell lines tested exhibited a significant increase in the activity of the deficient respiratory chain enzyme after bezafibrate treatment. These findings are likely to prompt clinical trials; however, no data showing that such approaches will be effective in individuals with mitochondrial disorders have been reported to date.
- Oral administration of nicotinamide riboside to two mouse models with predominantly myopathic phenotypes showed improvements in NAD levels in mouse tissues and induced mitochondrial biogenesis, delaying disease progression [Cerutti et al 2014, Khan et al 2014]. Nicotinamide riboside also showed promising results in fibroblasts from an individual with pathogenic variants in NDUFS1, a Leigh syndrome-associated nuclear gene [Felici et al 2015], with increased NAD levels and restoration of mitochondrial membrane potential. It remains unclear whether nicotinamide riboside can effectively boost NAD levels in brain, and nicotinamide mononucleotide may show more promise for neurologic disorders such as Leigh syndrome, since it has been shown to boost mitochondrial respiratory function in a mouse model of Alzheimer disease [Long et al 2015]. Clinical trials of nicotinamide analogs in individuals with Leigh syndrome have yet to be reported.
Another study explored the use of alpha-ketoglutarate and aspartate in transmitochondrial cybrids heteroplasmic for the m.8993T>G pathogenic variant [Sgarbi et al 2009]. The rationale was that these substrates would increase flux through the citric acid cycle, thereby increasing ATP production independently of oxidative phosphorylation (so-called "substrate level phosphorylation"). Initial results were promising, but further studies are needed before clinical applications can be considered.
Finally, two promising approaches have been suggested by recent studies in the Ndufs4-/- mouse model of Leigh syndrome.
- Rapamycin markedly delayed the onset and progression of symptoms in Ndufs4-/- mice [Johnson et al 2013]. The mechanism of action was unclear, as it did not appear to be acting via known mechanisms such as immune suppression, stimulating macroautophagy or induction of the mitochondrial unfolded protein response. However, the Ndufs4-/- mouse brains showed activation of the rapamycin target mTOR, which is a central coordinator of nutrient sensing and growth. Rapamycin suppressed mTOR activation, indicating that restoration of cellular signaling pathways may be a key to the beneficial effect. Rapamycin has a number of side effects (e.g., immunosuppression, hyperlipidemia, decreased wound healing) that may limit its clinical utility; however, this report identifies a potential new pathway to target for treatment of Leigh syndrome and other mitochondrial disorders.
- Chronic exposure of Ndufs4-/- mice to hypoxia (11% O2 instead of 21% O2, equivalent to an elevation of ~4500 m) extended life span and alleviated physiologic abnormalities such as defects in locomotor activity, body temperature instability, and poor weight gain [Jain et al 2016]. While humans can acclimatize to comparable oxygen tensions, the authors emphasized that caution was needed in subjecting affected individuals to altered O2 levels. Further studies are needed to elucidate whether partial or intermittent hypoxia or pharmacologic agents influencing the hypoxic response may be suitable therapeutic approaches.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Mitochondrial DNA-associated Leigh syndrome and NARP are transmitted by mitochondrial (maternal) inheritance.
Risk to Family Members
Parents of a proband
- The father of a proband is not at risk of having the mtDNA pathogenic variant.
- The mother of a proband usually has the mtDNA pathogenic variant and may have symptoms.
- In most cases, the mother has a much lower proportion of abnormal mtDNA than the proband and usually remains asymptomatic or develops only mild symptoms.
- Occasionally the mother has a substantial proportion of abnormal mtDNA and develops severe symptoms in adulthood, as described in de Vries et al [1993].
- With the exception of the m.8993T>G and m.8993T>C variants, a lower proportion of abnormal mtDNA in maternal blood does not exclude a higher proportion in tissues such as brain or muscle.
- Some affected individuals have no known family history of Leigh syndrome, NARP, or other mitochondrial disorder. The explanation for apparently simplex cases may be absence of a comprehensive and/or reliable family history or a de novo mtDNA pathogenic variant in the proband.
- Apparently de novo mtDNA pathogenic variants have been reported quite frequently in small studies, with one report describing apparently de novo m.8993T>G pathogenic variants being present in five of ten pedigrees [White et al 1999c].
- Recently, a study of 105 index cases with a range of different mtDNA pathogenic variants suggested that approximately 25% of cases resulted from de novo pathogenic variants [Sallevelt et al 2017].
Sibs of a proband
- The risk to the sibs depends on the genetic status of the mother.
- If the mother of the proband has the mtDNA pathogenic variant, all sibs are at risk of inheriting the variant.
- For the m.8993T>G and m.8993T>C pathogenic variants, if the mother of the proband has undetectable mutated mtDNA in blood, sibs of the proband are at very low risk (substantially <10%) of having inherited sufficient mutated mtDNA to cause symptoms.Note: White et al [1999a] generated logistic regression models that gave predictive curves for m.8993T>G and m.8993T>C predicting the recurrence risks for sibs of a proband based on the mother's proportion of abnormal mtDNA in blood (Figure 2). A strong positive relationship exists between the mother's proportion of abnormal mtDNA and the predicted recurrence risk. However, the 95% confidence interval of the risk estimate was wide and these data are of limited use for genetic counseling.
- For pathogenic variants other than m.8993T>G and m.8993T>C, the abnormal mtDNA may be undetectable in maternal blood but detectable in other tissues including oocytes. Thus, sibs of a proband are at risk of developing symptoms, depending on the tissue distribution and proportion of abnormal mtDNA.
- If the proband is presumed to have a de novo mtDNA pathogenic variant (i.e., the pathogenic variant is absent from the blood or other samples from the mother and any maternal relatives who were tested), sibs are at low risk [Sallevelt et al 2017]. The Sallevelt et al [2017] study implies that the potential for de novo pathogenic variants extends to variants other than m.8993T>G/C.
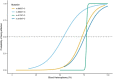
Figure 2.
Predicted recurrence risks (95% CI) for NARP or Leigh syndrome caused by the mtDNA m.8993T>G or m.8993T>C variant based on the mother's measured proportion of abnormal mtDNA (mutant load) in blood [White et al 1999a]
Offspring of a proband
- Offspring of a male proband with a mtDNA pathogenic variant are not at risk.
- All offspring of a female proband with a mtDNA pathogenic variant are at risk of inheriting the variant. The risk to offspring of a female proband of developing symptoms depends on the tissue distribution and proportion of abnormal mtDNA. Retrospective studies for some of the most common mtDNA pathogenic variants can be used to indicate an approximate (empiric) recurrence risk for women who have or are at risk of having these variants. See White et al [1999a] for m.8993T>G and m.8993T>C; see Chinnery et al [1998] for m.3243A>G and m.8344A>G; see Sallevelt et al [2017] for other mtDNA pathogenic variants.
Other family members. The risk to other family members depends on the genetic status of the proband's mother: if the proband's mother has a mtDNA pathogenic variant, her sibs and mother are also at risk.
Related Genetic Counseling Issues
Phenotypic variability. The phenotype of an individual with a mtDNA pathogenic variant results from a combination of factors including the severity of the pathogenic variant, the percentage of mutated mitochondria (mtDNA heteroplasmy), and the organs and tissues in which they are found (tissue distribution). Different family members often inherit different percentages of mutated mtDNA and therefore can have a wide range of clinical symptoms.
Interpretation of test results of asymptomatic at-risk family members is extremely difficult. Prediction of phenotype based on test results is not possible. Furthermore, absence of the mtDNA pathogenic variant in one tissue (e.g., blood) does not guarantee that the variant is absent in other tissues.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy. Similarly, decisions regarding testing to determine the genetic status of at-risk asymptomatic family members are best made before pregnancy.
- It is appropriate to offer genetic counseling (including general discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk; however, it is not possible to make specific predictions about the potential severity of disease in individuals or their offspring.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Genetic counseling and prenatal diagnosis of disorders caused by mutation of mitochondrial DNA present considerable challenges. A Consensus Workshop on this topic was held in 1999, sponsored by the European Neuromuscular Disease Centre and involving representatives from 14 major international centers specializing in mtDNA diseases. The major findings of the workshop [Poulton & Turnbull 2000] (full text) relating to Leigh syndrome and NARP are summarized here. The important issues for both counseling and prenatal diagnosis depend on the following:
- Is there a close relationship between the proportion of abnormal mtDNA and disease severity?
- Is mutated mtDNA uniformly distributed in all tissues?
- Does the proportion of abnormal mtDNA change with time?
Four conclusions were reached:
- Genetic counseling and prenatal diagnosis for women known to have or suspected of having a mtDNA pathogenic variant require the involvement of professionals with up-to-date experience in this area to ensure that prospective parents are counseled regarding all potential outcomes of prenatal diagnosis or assisted reproduction technologies and that possible limitations of interpretation are explained.
- Practice is limited by lack of available information. Collection and analysis of more information on the outcome of pregnancies is warranted.
- No general rules allow for precise prediction of the inheritance risks for heteroplasmic mtDNA variants. Each variant must therefore be assessed separately.
- Despite the difficulties currently associated with counseling for mtDNA pathogenic variants, affected families are seeking advice and help. Furthermore, extensive investigation has shown that the transmission of a heteroplasmic mtDNA pathogenic variant can be predicted within some broad range of possibilities. Thus, a consensus was reached on recommendations for prenatal testing of some mtDNA pathogenic variants. See Prenatal Testing.
Prenatal Testing and Preimplantation Genetic Testing
Once the mtDNA pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk is possible.
- Available evidence suggests that the proportion of abnormal mtDNA in all extra-embryonic and embryonic tissues is similar and does not change substantially during pregnancy [Thorburn & Dahl 2001].
- Analysis should be done on the biopsy, not on cultured cells.
- The major limitation of this approach is potential difficulty in interpreting the results to predict outcome:
- An intermediate proportion of abnormal mtDNA would represent a "gray zone" in which interpretation is difficult or impossible.
- For the m.8993T>G and m.8993T>C pathogenic variants, Poulton & Turnbull [2000] state that it is reasonable to offer this form of prenatal testing to asymptomatic women with <50% levels of mutated mtDNA.
- CVS and amniocentesis can also potentially be offered to women with a low proportion of abnormal mtDNA in blood in other pathogenic variants including m.3243A>G, m.8344A>G, and rare mtDNA single-nucleotide variants, but the weaker correlation between proportion of abnormal mtDNA and disease severity means that couples would require careful counseling before embarking on these procedures.
- Prenatal testing for m.8993T>G [Harding et al 1992, Bartley et al 1996, Ferlin et al 1997, White et al 1999b], m.8993T>C [Leshinsky-Silver et al 2003], m.9176T>C [Jacobs et al 2005], and m.3243A>G [Monnot et al 2011] has been reported.
- A recent multicenter study reported prenatal diagnosis for 17 pregnancies at risk for maternally inherited mitochondrial disease, including several mtDNA pathogenic variants associated with Leigh syndrome [Nesbitt et al 2014].
- A recent review concluded that prenatal diagnosis is "the best option for female carriers with low-level mutations demonstrating skewing to 0% or 100%" [Smeets et al 2015].
Other Reproductive Options
Oocyte donation accompanied by IVF using the partner's sperm. Use of a maternal relative as the oocyte donor should be avoided since the relative may have oocytes with a high proportion of abnormal mtDNA even though her leukocytes may lack detectable abnormal mtDNA.
The two major limitations of oocyte donation are:
- Limited availability of donor oocytes;
- Personal or cultural views regarding the use of donor gametes or the desire for a child who is genetically related to both parents.
Preimplantation genetic testing (PGT) may be an option for some families with mtDNA pathogenic variants [Thorburn & Dahl 2001, Dean et al 2003, Jacobs et al 2006]. Successful use of PGT has been reported for m.8993T>G [Steffann et al 2006, Sallevelt et al 2013]. Embryos should only be regarded as suitable for implantation if they have a very low proportion of abnormal mtDNA, preferably 0%. It has been suggested that PGT can be used to transfer embryos with 18% abnormal mtDNA with low recurrence risk, and PGT is currently considered the preferred reproductive option for familial heteroplasmic mtDNA single-nucleotide variants [Smeets et al 2015].
The high copy number of mtDNA (>104 copies per cell in an 8-cell embryo) means that mtDNA analysis for pathogenic variants should be less prone to artifacts (e.g., amplification failure, allele dropout) that can complicate analysis for nuclear gene defects in single cells.
In some women, particularly those with a high proportion of mutated mtDNA in blood or urine, a large proportion of oocytes may have substantial abnormal mtDNA, in which case even multiple cycles of ovarian stimulation may not result in an embryo suitable for implantation.
PGT for mtDNA pathogenic variants may provide valuable information even if a successful unaffected conception is not achieved.
- If most of the embryos tested have a substantial proportion of abnormal mtDNA, oocyte donation is likely to be the only current option for ensuring an unaffected embryo.
- In contrast, if most of the embryos tested have undetectable mutated mtDNA, the parents may opt for prenatal testing in subsequent unassisted (natural) pregnancies.
A workshop on PGT for mtDNA pathogenic variants was held in 2010, sponsored by the European Neuromuscular Centre (ENMC) and involving representatives from 15 international centers specializing in mtDNA diseases. Attendees described data on PGT studies in a total of nine families; a summary of the workshop discussions has been published [Poulton & Bredenoord 2010].
Mitochondrial donation. Transfer of nuclear material (the pronucleus or the maternal spindle) from an unfertilized oocyte or single-cell embryo into an enucleated donor cell could potentially block transmission of mutated mtDNA into the developing embryo. This approach may be suitable even for women with a high proportion of mutated mtDNA, in whom PGT is unlikely to be an effective reproductive option. Studies in mice and macaque have shown that nuclear transfer approaches can prevent transmission of a substantial proportion of abnormal mtDNA to offspring [Sato et al 2005, Tachibana et al 2009]. Proof-of-principle studies with abnormally fertilized human zygotes also demonstrated minimal carryover of donor zygote mtDNA and allowed onward development to the blastocyst stage in vitro [Craven et al 2010]. A recent study reported efficient replacement of oocyte mutated mtDNA after spindle transfer, resulting in embryos containing >99% donor mtDNA [Kang et al 2016]. However, some embryonic stem cell lines derived from these embryos gradually lost donor mtDNA, with reversal to the maternal haplotype, a phenomenon that needs to be investigated further [Kang et al 2016]. The first preclinical study on pronunclear transfer using normally fertilized human embryos has also now been reported and shown to be effective [Hyslop et al 2016]. After extensive scientific, ethical, and public consultation, the UK government has given permission for mitochondrial donation therapies to prevent the transmission of severe mitochondrial disease caused by specific mtDNA pathogenic variants; the first clinics are currently being licensed [Herbert & Turnbull 2017]. Very recently, the first child born after spindle transfer has been reported, born to a woman carrying the m.8993T>G Leigh syndrome-causing pathogenic variant [Zhang et al 2017]. The boy is said to be well at seven months but long-term neurodevelopmental follow up is clearly needed.
Thorburn & Dahl [2001], Jacobs et al [2006], and Poulton & Bredenoord [2010] provide more detailed discussions of reproductive options for women with mtDNA pathogenic variants.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- MedlinePlus
- United Mitochondrial Disease FoundationPhone: 888-317-UMDF (8633)Email: info@umdf.org
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Mito FoundationAustraliaPhone: 61-1-300-977-180Email: info@mito.org.au
- Muscular Dystrophy Association (MDA) - USAPhone: 833-275-6321
- People Against Leigh Syndrome (PALS)Phone: 713-248-8782
- Retina InternationalIrelandPhone: 353 1 961 9259Email: info@retina-International.org
- The Lily FoundationUnited KingdomEmail: liz@thelilyfoundation.org.uk
- eyeGENE – National Ophthalmic Disease Genotyping Network RegistryPhone: 301-435-3032Email: eyeGENEinfo@nei.nih.gov
- RDCRN Patient Contact Registry: North American Mitochondrial Disease Consortium
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Mitochondrial DNA-Associated Leigh Syndrome and NARP: Genes and Databases
Table B.
OMIM Entries for Mitochondrial DNA-Associated Leigh Syndrome and NARP (View All in OMIM)
Molecular Pathogenesis
All mtDNA genes lack introns and are transcribed as large polycistronic transcripts that are processed into monocistronic mRNAs. Protein-coding genes are then translated by the mitochondrial-specific translational machinery.
Benign variants. Mitochondrial DNA is highly polymorphic; information on known polymorphisms can be obtained at MITOMAP: A Human Mitochondrial Genome Database, which provides a compendium of benign variants and pathogenic variants of the human mtDNA.
The highly polymorphic nature of mtDNA adds complexity to variant interpretation, particularly when using indirect genotyping assays such as PCR-RFLP. For example, several benign variants introduce or abolish a restriction site such that fragments produced by restriction digest may suggest a false positive or false negative result [Johns & Neufeld 1993, Kirby et al 1998, White et al 1998].
Pathogenic variants. Mitochondrial DNA pathogenic variants that have been shown to cause Leigh syndrome, Leigh-like syndrome, or NARP are listed in Table 5.
Table 5.
Selected Pathogenic Variants in Mitochondrial DNA-Associated Leigh Syndrome, Leigh-Like Syndrome, and NARP
Normal gene product. Human mtDNA encodes 37 genes, including 13 genes encoding protein subunits of the mitochondrial respiratory chain and oxidative phosphorylation, 22 tRNA genes, and two rRNA genes. The mitochondrial-specific translational machinery is required because translation of mtDNA-encoded genes is physically separated from the cytosolic translational machinery and because the mtDNA genetic code differs from the universal genetic code in several codons.
Abnormal gene product. For some mtDNA pathogenic variants associated with NARP and Leigh syndrome, strong correlation exists between the proportion of abnormal to wild type mtDNA and severity of the biochemical phenotype in cultured cells. For some variants, such as m.8993T>G and m.8993T>C, a strong correlation also exists between the proportion of abnormal to wild type mtDNA and clinical severity. However, the mechanism by which some mtDNA pathogenic variants cause a phenotype such as Leigh syndrome, while others cause myopathy, deafness, or diabetes mellitus, is not known.
Pathogenic mtDNA variants causing NARP and Leigh fall into two major classes, namely those in tRNA genes and those in protein-coding genes.
- Transfer RNA pathogenic variants cause decreased mitochondrial protein synthesis by mechanisms that involve abnormalities in both base modification and aminoacylation of the mutated tRNA and in some cases processing of the polycistronic mtRNA transcript, as discussed elsewhere (see MELAS and MERRF).
- Pathogenic variants in protein-coding mtDNA genes typically cause decreased activity of the respiratory chain complex of which that subunit is a part.
The most common mtDNA variant in the NARP and Leigh syndrome (mtDNA pathogenic variants) continuum, m.8993T>G pathogenic variant is the best understood. The m.8993T>G variant changes a conserved leucine to an arginine (p.Leu156Arg) in subunit 6 of the mitochondrial F1F0 ATP synthase. ATP synthase (or complex V) uses the proton gradient generated by respiratory chain complexes I to IV to drive ATP synthesis. Subunit 6 forms part of the F0 proton channel of the ATP synthase and the p.Leu156Arg amino acid substitution appears to block proton translocation and inhibit ATP synthesis [Tatuch & Robinson 1993]. The pathogenic variant may also interfere with assembly or stability of the ATP synthase [García et al 2000, Nijtmans et al 2001]. Inhibition of ATP synthesis by the m.8993T>G variant is expected to increase mitochondrial membrane potential and lead to increased production of superoxide, perhaps triggering increased cell death [Geromel et al 2001, Mattiazzi et al 2004]. These pathogenic mechanisms must contribute to the specific pattern of tissue involvement and cell loss seen in the NARP and Leigh syndrome (mtDNA pathogenic variants) continuum.
The m.8993T>C pathogenic variant changes p.Leu156Pro, and presumably results in less severe interference with proton translocation and a milder clinical phenotype than the m.8993T>G pathogenic variant [Santorelli et al 1996].
The MT-ND6 m.14459G>A and m.14487T>C pathogenic variants result in a marked decrease in the steady-state amounts of fully assembled complex I [Kirby et al 2003, Ugalde et al 2003].
There are limited data on the molecular pathogenesis of other mtDNA subunit pathogenic variants associated with the NARP and Leigh syndrome (mtDNA pathogenic variants) continuum, but most presumably cause either (1) a catalytic defect or (2) instability of the subunit and complex in which it is incorporated, or both.
Chapter Notes
Revision History
- 4 May 2023 (aa) Revision: in Table 3, hypertrichosis removed as distinguishing clinical feature in manganese-dependent β-galactosyltransferase deficiency
- 28 September 2017 (sw) Comprehensive update posted live
- 17 April 2014 (me) Comprehensive update posted live
- 3 May 2011 (cd) Revision: MT-ND2 added as gene in which mutation is causative
- 8 February 2011 (me) Comprehensive update posted live
- 3 February 2006 (me) Comprehensive update posted live
- 30 October 2003 (me) Review posted live
- 3 July 2003 (dt) Original submission
References
Published Guidelines / Consensus Statements
- Poulton J, Turnbull DM. 74th European Neuromuscular Centre International Consensus Workshop on genetic counseling and prenatal diagnosis of mitochondrial DNA disorders. 19-20 November 1999, Naarden, The Netherlands. Available online. 2000. Accessed 4-12-23.
Literature Cited
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–95. [PubMed: 15985532]
- Ahola S, Auranen M, Isohanni P, Niemisalo S, Urho N, Buzkova J, Velagapudi V, Lundbom N, Hakkarainen A, Muurinen T, Piirila P, Pietilainen KH, Suomalainen A. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol Med. 2016;8:1234–47. [PMC free article: PMC5090657] [PubMed: 27647878]
- Ahola-Erkkilä S, Carroll CJ, Peltola-Mjösund K, Tulkki V, Mattila I, Seppänen-Laakso T, Oresic M, Tyynismaa H, Suomalainen A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet. 2010;19:1974–84. [PubMed: 20167576]
- Ahola S, Isohanni P, Euro L, Brilhante V, Palotie A, Pihko H, Lönnqvist T, Lehtonen T, Laine J, Tyynismaa H, Suomalainen A. Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy. Neurology. 2014;83:743–51. [PMC free article: PMC4150129] [PubMed: 25037205]
- Alexeyev MF, Venediktova N, Pastukh V, Shokolenko I, Bonilla G, Wilson GL. Selective elimination of mutant mitochondrial genomes as therapeutic strategy for the treatment of NARP and MILS syndromes. Gene Ther. 2008;15:516–23. [PMC free article: PMC10416612] [PubMed: 18256697]
- Anderson CM, Norquist BA, Vesce S, Nicholls DG, Soine WH, Duan S, Swanson RA. Barbiturates induce mitochondrial depolarization and potentiate excitotoxic neuronal death. J Neurosci. 2002;22:9203–9. [PMC free article: PMC6758030] [PubMed: 12417645]
- Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, Kennaway NG, Harding CO, Jaksch M, Shoubridge EA. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet. 2003;12:2693–702. [PubMed: 12928484]
- Antonicka H, Ostergaard E, Sasarman F, Weraarpachai W, Wibrand F, Pedersen AM, Rodenburg RJ, van der Knaap MS, Smeitink JA, Chrzanowska-Lightowlers ZM, Shoubridge EA. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet. 2010;87:115–22. [PMC free article: PMC2896764] [PubMed: 20598281]
- Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med. 2013;19:1111–3. [PMC free article: PMC4153471] [PubMed: 23913125]
- Baertling F, Rodenburg RJ, Schaper J, Smeitink JA, Koopman WJ, Mayatepek E, Morava E, Distelmaier F. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. 2014;85:257–65. [PubMed: 23772060]
- Baertling F, Sánchez-Caballero L, Timal S, van den Brand MA, Ngu LH, Distelmaier F, Rodenburg RJ, Nijtmans LG. Mutations in mitochondrial complex I assembly factor NDUFAF3 cause Leigh syndrome. Mol Genet Metab. 2017a;120:243–6. [PubMed: 27986404]
- Baertling F, Sánchez-Caballero L, van den Brand MAM, Wintjes LT, Brink M, van den Brandt FA, Wilson C, Rodenburg RJT, Nijtmans LGJ. NDUFAF4 variants are associated with Leigh syndrome and cause a specific mitochondrial complex I assembly defect. Eur J Hum Genet. 2017b;25:1273–7. [PMC free article: PMC5643967] [PubMed: 28853723]
- Baker PR 2nd, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, Aicher J, Creadon-Swindell G, Geiger E, MacLean KN, Lee WT, Deshpande C, Freckmann ML, Shih LY, Wasserstein M, Rasmussen MB, Lund AM, Procopis P, Cameron JM, Robinson BH, Brown GK, Brown RM, Compton AG, Dieckmann CL, Collard R, Coughlin CR 2nd, Spector E, Wempe MF, Van Hove JL. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain. 2014;137:366–79. [PMC free article: PMC3914472] [PubMed: 24334290]
- Barel O, Shorer Z, Flusser H, Ofir R, Narkis G, Finer G, Shalev H, Nasasra A, Saada A, Birk OS. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am J Hum Genet. 2008;82:1211–6. [PMC free article: PMC2427202] [PubMed: 18439546]
- Barghuti F, Elian K, Gomori JM, Shaag A, Edvardson S, Saada A, Elpeleg O. The unique neuroradiology of complex I deficiency due to NDUFA12L defect. Mol Genet Metab. 2008;94:78–82. [PubMed: 18180188]
- Bartley J, Senadheera D, Park P, Brar H, Abad D, Wong LJ. Prenatal diagnosis of T8993G mitochondrial DNA point mutation in amniocytes by heteroplasmy detection. Am J Hum Genet. 1996;59:A316.
- Basel-Vanagaite L, Muncher L, Straussberg R, Pasmanik-Chor M, Yahav M, Rainshtein L, Walsh CA, Magal N, Taub E, Drasinover V, Shalev H, Attia R, Rechavi G, Simon AJ, Shohat M. Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol. 2006;60:214–22. [PubMed: 16786527]
- Bastin J, Aubey F, Rötig A, Munnich A, Djouadi F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients' cells lacking its components. J Clin Endocrinol Metab. 2008;93:1433–41. [PubMed: 18211970]
- Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci U S A. 2005;102:14392–7. [PMC free article: PMC1242285] [PubMed: 16179392]
- Bénit P, Chretien D, Kadhom N, de Lonlay-Debeney P, Cormier-Daire V, Cabral A, Peudenier S, Rustin P, Munnich A, Rotig A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet. 2001;68:1344–52. [PMC free article: PMC1226121] [PubMed: 11349233]
- Bénit P, Slama A, Cartault F, Giurgea I, Chretien D, Lebon S, Marsac C, Munnich A, Rötig A, Rustin P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J Med Genet. 2004;41:14–7. [PMC free article: PMC1757256] [PubMed: 14729820]
- Berkovic SF, Carpenter S, Evans A, Karpati G, Shoubridge EA, Andermann F, Meyer E, Tyler JL, Diksic M, Arnold D, et al. Myoclonus epilepsy and ragged-red fibres (MERRF). 1. A clinical, pathological, biochemical, magnetic resonance spectrographic and positron emission tomographic study. Brain. 1989;112:1231–60. [PubMed: 2508988]
- Blok MJ, Spruijt L, de Coo IF, Schoonderwoerd K, Hendrickx A, Smeets HJ. Mutations in the ND5 subunit of complex I of the mitochondrial DNA are a frequent cause of oxidative phosphorylation disease. J Med Genet. 2007;44:e74. [PMC free article: PMC2598042] [PubMed: 17400793]
- Bonfante E, Koenig MK, Adejumo RB, Perinjelil V, Riascos RF. The neuroimaging of Leigh syndrome: case series and review of the literature. Pediatr Radiol. 2016;46:443–51. [PubMed: 26739140]
- Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Péquignot E, Munnich A, Rotig A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11:144–9. [PubMed: 7550341]
- Brautbar A, Wang J, Abdenur JE, Chang RC, Thomas JA, Grebe TA, Lim C, Weng SW, Graham BH, Wong LJ. The mitochondrial 13513G>A mutation is associated with Leigh disease phenotypes independent of complex I deficiency in muscle. Mol Genet Metab. 2008;94:485–90. [PubMed: 18495510]
- Buda P, Piekutowska-Abramczuk D, Karkucińska-Więckowska A, Jurkiewicz E, Chełstowska S, Pajdowska M, Migdał M, Książyk J, Kotulska K, Pronicka E. "Drop attacks" as first clinical symptoms in a child carrying MTTK m.8344A>G mutation. Folia Neuropathol. 2013;51:347–54. [PubMed: 24374964]
- Budde SM, van den Heuvel LP, Janssen AJ, Smeets RJ, Buskens CA, DeMeirleir L, Van Coster R, Baethmann M, Voit T, Trijbels JM, Smeitink JA. Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem Biophys Res Commun. 2000;275:63–8. [PubMed: 10944442]
- Bugiani M, Invernizzi F, Alberio S, Briem E, Lamantea E, Carrara F, Moroni I, Farina L, Spada M, Donati MA, Uziel G, Zeviani M. Clinical and molecular findings in children with complex I deficiency. Biochim Biophys Acta. 2004;1659:136–47. [PubMed: 15576045]
- Calvo SE, Tucker EJ, Compton AG, Kirby DM, Crawford G, Burtt NP, Rivas M, Guiducci C, Bruno DL, Goldberger OA, Redman MC, Wiltshire E, Wilson CJ, Altshuler D, Gabriel SB, Daly MJ, Thorburn DR, Mootha VK. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42:851–8. [PMC free article: PMC2977978] [PubMed: 20818383]
- Cameron JM, MacKay N, Feigenbaum A, Tarnopolsky M, Blaser S, Robinson BH, Schulze A. Exome sequencing identifies complex I NDUFV2 mutations as a novel cause of Leigh syndrome. Eur J Paediatr Neurol. 2015;19:525–32. [PubMed: 26008862]
- Campos Y, Martín MA, Rubio JC, Gutiérrez del Olmo MC, Cabello A, Arenas J. Bilateral striatal necrosis and MELAS associated with a new T3308C mutation in the mitochondrial ND1 gene. Biochem Biophys Res Commun. 1997;238:323–5. [PubMed: 9299504]
- Caporali L, Ghelli AM, Iommarini L, Maresca A, Valentino ML, La Morgia C, Liguori R, Zanna C, Barboni P, De Nardo V, Martinuzzi A, Rizzo G, Tonon C, Lodi R, Calvaruso MA, Cappelletti M, Porcelli AM, Achilli A, Pala M, Torroni A, Carelli V. Cybrid studies establish the causal link between the mtDNA m.3890G>A/MT-ND1 mutation and optic atrophy with bilateral brainstem lesions. Biochim Biophys Acta. 2013;1832:445–52. [PMC free article: PMC3778985] [PubMed: 23246842]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, Viscomi C, Zeviani M. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19:1042–9. [PMC free article: PMC4051987] [PubMed: 24814483]
- Chalmers RM, Lamont PJ, Nelson I, Ellison DW, Thomas NH, Harding AE, Hammans SR. A mitochondrial DNA tRNA(Val) point mutation associated with adult-onset Leigh syndrome. Neurology. 1997;49:589–92. [PubMed: 9270602]
- Chen Z, Zhao Z, Ye Q, Chen Y, Pan X, Sun B, Huang H, Zheng A. Mild clinical manifestation and unusual recovery upon coenzyme Q10 treatment in the first Chinese Leigh syndrome pedigree with mutation m.10197 G>A. Mol Med Rep. 2015;11:1956–62. [PubMed: 25384404]
- Ching CK, Mak CM, Au KM, Chan KY, Yuen YP, Yau EK, Ma LC, Chow HL, Chan AY. A patient with congenital hyperlactataemia and Leigh syndrome: an uncommon mitochondrial variant. Hong Kong Med J. 2013;19:357–61. [PubMed: 23918514]
- Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. MELAS and MERRF. The relationship between maternal mutation load and the frequency of clinically affected offspring. Brain. 1998;121:1889–94. [PubMed: 9798744]
- Chol M, Lebon S, Bénit P, Chretien D, de Lonlay P, Goldenberg A, Odent S, Hertz-Pannier L, Vincent-Delorme C, Cormier-Daire V, Rustin P, Rötig A, Munnich A. The mitochondrial DNA G13513A MELAS mutation in the NADH dehydrogenase 5 gene is a frequent cause of Leigh-like syndrome with isolated complex I deficiency. J Med Genet. 2003;40:188–91. [PMC free article: PMC1735406] [PubMed: 12624137]
- Ciafaloni E, Santorelli FM, Shanske S, Deonna T, Roulet E, Janzer C, Pescia G, DiMauro S. Maternally inherited Leigh syndrome. J Pediatr. 1993;122:419–22. [PubMed: 8095070]
- Claeys KG, Abicht A, Hausler M, Kleinle S, Wiesmann M, Schulz JB, Horvath R, Weis J. Novel genetic and neuropathological insights in neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP). Muscle Nerve. 2016;54:328–33. [PubMed: 27015314]
- Cox R, Platt J, Chen LC, Tang S, Wong LJ, Enns GM. Leigh syndrome caused by a novel m.4296G>A mutation in mitochondrial tRNA isoleucine. Mitochondrion. 2012;12:258–61. [PubMed: 21982779]
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, Herbert M, Turnbull DM. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–5. [PMC free article: PMC2875160] [PubMed: 20393463]
- Crimi M, Galbiati S, Moroni I, Bordoni A, Perini MP, Lamantea E, Sciacco M, Zeviani M, Biunno I, Moggio M, Scarlato G, Comi GP. A missense mutation in the mitochondrial ND5 gene associated with a Leigh-MELAS overlap syndrome. Neurology. 2003;60:1857–61. [PubMed: 12796552]
- Crimi M, Papadimitriou A, Galbiati S, Palamidou P, Fortunato F, Bordoni A, Papandreou U, Papadimitriou D, Hadjigeorgiou GM, Drogari E, Bresolin N, Comi GP. A new mitochondrial DNA mutation in ND3 gene causing severe Leigh syndrome with early lethality. Pediatr Res. 2004;55:842–6. [PubMed: 14764913]
- Danhauser K, Herebian D, Haack TB, Rodenburg RJ, Strom TM, Meitinger T, Klee D, Mayatepek E, Prokisch H, Distelmaier F. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur J Hum Genet. 2016;24:450–4. [PMC free article: PMC4755375] [PubMed: 26081641]
- Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA anbormalities. Ann Neurol. 2001;49:377–83. [PubMed: 11261513]
- de Lonlay P, Valnot I, Barrientos A, Gorbatyuk M, Tzagoloff A, Taanman JW, Benayoun E, Chrétien D, Kadhom N, Lombès A, de Baulny HO, Niaudet P, Munnich A, Rustin P, Rötig A. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat Genet. 2001;29:57–60. [PubMed: 11528392]
- De Meirleir L, Seneca S, Lissens W, Schoentjes E, Desprechins B. Bilateral striatal necrosis with a novel point mutation in the mitochondrial ATPase 6 gene. Pediatr Neurol. 1995;13:242–6. [PubMed: 8554662]
- de Vries DD, van Engelen BG, Gabreels FJ, Ruitenbeek W, van Oost BA. A second missense mutation in the mitochondrial ATPase 6 gene in Leigh's syndrome. Ann Neurol. 1993;34:410–2. [PubMed: 8395787]
- Dean NL, Battersby BJ, Ao A, Gosden RG, Tan SL, Shoubridge EA. Prospect of preimplantation genetic diagnosis for heritable mitochondrial DNA diseases. Mol Hum Reprod. 2003;9:631–8. [PubMed: 12970401]
- Debray FG, Morin C, Janvier A, Villeneuve J, Maranda B, Laframboise R, Lacroix J, Decarie JC, Robitaille Y, Lambert M, Robinson BH, Mitchell GA. LRPPRC mutations cause a phenotypically distinct form of Leigh syndrome with cytochrome c oxidase deficiency. J Med Genet. 2011;48:183–9. [PubMed: 21266382]
- Dermaut B, Seneca S, Dom L, Smets K, Ceulemans L, Smet J, De Paepe B, Tousseyn S, Weckhuysen S, Gewillig M, Pals P, Parizel P, De Bleecker JL, Boon P, De Meirleir L, De Jonghe P, Van Coster R, Van Paesschen W, Santens P. Progressive myoclonic epilepsy as an adult-onset manifestation of Leigh syndrome due to m.14487T>C. J Neurol Neurosurg Psychiatry. 2010;81:90–3. [PubMed: 20019223]
- Duff RM, Shearwood AM, Ermer J, Rossetti G, Gooding R, Richman TR, Balasubramaniam S, Thorburn DR, Rackham O, Lamont PJ, Filipovska A. A mutation in MT-TW causes a tRNA processing defect and reduced mitochondrial function in a family with Leigh syndrome. Mitochondrion. 2015;25:113–9. [PubMed: 26524491]
- Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, Eshhar S, Saada A. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76:1081–6. [PMC free article: PMC1196446] [PubMed: 15877282]
- Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Amagata A, Barnes A, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012;105:91–102. [PubMed: 22115768]
- Fassone E, Duncan AJ, Taanman JW, Pagnamenta AT, Sadowski MI, Holand T, Qasim W, Rutland P, Calvo SE, Mootha VK, Bitner-Glindzicz M, Rahman S. FOXRED1, encoding an FAD-dependent oxidoreductase complex-I-specific molecular chaperone, is mutated in infantile-onset mitochondrial encephalopathy. Hum Mol Genet. 2010;19:4837–47. [PMC free article: PMC4560042] [PubMed: 20858599]
- Fassone E, Wedatilake Y, DeVile CJ, Chong WK, Carr LJ, Rahman S. Treatable Leigh-like encephalopathy presenting in adolescence. BMJ Case Rep. 2013;2013:200838. [PMC free article: PMC3822156] [PubMed: 24099834]
- Felici R, Lapucci A, Cavone L, Pratesi S, Berlinguer-Palmini R, Chiarugi A. Pharmacological NAD-boosting strategies improve mitochondrial homeostasis in human complex I-mutant fibroblasts. Mol Pharmacol. 2015;87:965–71. [PubMed: 25788480]
- Ferdinandusse S, Waterham HR, Heales SJ, Brown GK, Hargreaves IP, Taanman JW, Gunny R, Abulhoul L, Wanders RJ, Clayton PT, Leonard JV, Rahman S. HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase. Orphanet J Rare Dis. 2013;8:188. [PMC free article: PMC4222069] [PubMed: 24299452]
- Ferlin T, Landrieu P, Rambaud C, Fernandez H, Dumoulin R, Rustin P, Mousson B. Segregation of the G8993 mutant mitochondrial DNA through generations and embryonic tissues in a family at risk of Leigh syndrome. J Pediatr. 1997;131:447–9. [PubMed: 9329425]
- Fernandez-Moreira D, Ugalde C, Smeets R, Rodenburg RJ, Lopez-Laso E, Ruiz-Falco ML, Briones P, Martin MA, Smeitink JA, Arenas J. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann Neurol. 2007;61:73–83. [PubMed: 17262856]
- Floyd BJ, Wilkerson EM, Veling MT, Minogue CE, Xia C, Beebe ET, Wrobel RL, Cho H, Kremer LS, Alston CL, Gromek KA, Dolan BK, Ulbrich A, Stefely JA, Bohl SL, Werner KM, Jochem A, Westphall MS, Rensvold JW, Taylor RW, Prokisch H, Kim JJ, Coon JJ, Pagliarini DJ. Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol Cell. 2016;63:621–32. [PMC free article: PMC4992456] [PubMed: 27499296]
- Fraidakis MJ, Jardel C, Allouche S, Nelson I, Auré K, Slama A, Lemière I, Thenint JP, Hamon JB, Zagnoli F, Heron D, Sedel F, Lombès A. Phenotypic diversity associated with the MT-TV gene m.1644G>A mutation, a matter of quantity. Mitochondrion. 2014;15:34–9. [PubMed: 24691472]
- Fujii T, Ito M, Miyajima T, Okuno T. Dichloroacetate therapy in Leigh syndrome with a mitochondrial T8993C mutation. Pediatr Neurol. 2002;27:58–61. [PubMed: 12160976]
- Fukumura S, Ohba C, Watanabe T, Minagawa K, Shimura M, Murayama K, Ohtake A, Saitsu H, Matsumoto N, Tsutsumi H. Compound heterozygous GFM2 mutations with Leigh syndrome complicated by arthrogryposis multiplex congenita. J Hum Genet. 2015;60:509–13. [PubMed: 26016410]
- Funalot B, Reynier P, Vighetto A, Ranoux D, Bonnefont JP, Godinot C, Malthièry Y, Mas JL. Leigh-like encephalopathy complicating Leber's hereditary optic neuropathy. Ann Neurol. 2002;52:374–7. [PubMed: 12205655]
- Gammage PA, Gaude E, Van Haute L, Rebelo-Guiomar P, Jackson CB, Rorbach J, Pekalski ML, Robinson AJ, Charpentier M, Concordet JP, Frezza C, Minczuk M. Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs. Nucleic Acids Res. 2016;44:7804–16. [PMC free article: PMC5027515] [PubMed: 27466392]
- García JJ, Ogilvie I, Robinson BH, Capaldi RA. Structure, functioning, and assembly of the ATP synthase in cells from patients with the T8993G mitochondrial DNA mutation. Comparison with the enzyme in Rho(0) cells completely lacking mtdna. J Biol Chem. 2000;275:11075–81. [PubMed: 10753912]
- Gerards M, Kamps R, van Oevelen J, Boesten I, Jongen E, de Koning B, Scholte HR, de Angst I, Schoonderwoerd K, Sefiani A, Ratbi I, Coppieters W, Karim L, de Coo R, van den Bosch B, Smeets H. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain. 2013;136:882–90. [PubMed: 23423671]
- Gerards M, Sluiter W, van den Bosch BJ, de Wit LE, Calis CM, Frentzen M, Akbari H, Schoonderwoerd K, Scholte HR, Jongbloed RJ, Hendrickx AT, de Coo IF, Smeets HJ. Defective complex I assembly due to C20orf7 mutations as a new cause of Leigh syndrome. J Med Genet. 2010;47:507–12. [PMC free article: PMC2921275] [PubMed: 19542079]
- Geromel V, Kadhom N, Cebalos-Picot I, Ouari O, Polidori A, Munnich A, Rotig A, Rustin P. Superoxide-induced massive apoptosis in cultured skin fibroblasts harboring the neurogenic ataxia retinitis pigmentosa (NARP) mutation in the ATPase-6 gene of the mitochondrial DNA. Hum Mol Genet. 2001;10:1221–8. [PubMed: 11371515]
- Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, Benna C, D'Adamo P, Diodato D, Costa R, Mariotti C, Uziel G, Smiderle C, Zeviani M. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet. 2011;43:259–63. [PubMed: 21278747]
- Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D'Adamo P, Novara F, Zuffardi O, Uziel G, Zeviani M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86:639–49. [PMC free article: PMC2850437] [PubMed: 20362274]
- Grafakou O, Oexle K, van den Heuvel L, Smeets R, Trijbels F, Goebel HH, Bosshard N, Superti-Furga A, Steinmann B, Smeitink J. Leigh syndrome due to compound heterozygosity of dihydrolipoamide dehydrogenase gene mutations. Description of the first E3 splice site mutation. Eur J Pediatr. 2003;162:714–8. [PubMed: 12925875]
- Haack TB, Gorza M, Danhauser K, Mayr JA, Haberberger B, Wieland T, Kremer L, Strecker V, Graf E, Memari Y, Ahting U, Kopajtich R, Wortmann SB, Rodenburg RJ, Kotzaeridou U, Hoffmann GF, Sperl W, Wittig I, Wilichowski E, Schottmann G, Schuelke M, Plecko B, Stephani U, Strom TM, Meitinger T, Prokisch H, Freisinger P. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab. 2014;111:342–52. [PubMed: 24461907]
- Hadzsiev K, Maasz A, Kisfali P, Kalman E, Gomori E, Pal E, Berenyi E, Komlosi K, Melegh B. Mitochondrial DNA 11777C>A mutation associated Leigh syndrome: case report with a review of the previously described pedigrees. Neuromolecular Med. 2010;12:277–84. [PubMed: 20502985]
- Hallmann K, Kudin AP, Zsurka G, Kornblum C, Reimann J, Stüve B, Waltz S, Hattingen E, Thiele H, Nürnberg P, Rüb C, Voos W, Kopatz J, Neumann H, Kunz WS. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain. 2016;139:338–45. [PubMed: 26685157]
- Han J, Lee YM, Kim SM, Han SY, Lee JB, Han SH. Ophthalmological manifestations in patients with Leigh syndrome. Br J Ophthalmol. 2015;99:528–35. [PubMed: 25351680]
- Harding AE, Holt IJ, Sweeney MG, Brockington M, Davis MB. Prenatal diagnosis of mitochondrial DNA8993 T----G disease. Am J Hum Genet. 1992;50:629–33. [PMC free article: PMC1684296] [PubMed: 1539598]
- Head RA, Brown RM, Zolkipli Z, Shahdadpuri R, King MD, Clayton PT, Brown GK. Clinical and genetic spectrum of pyruvate dehydrogenase deficiency: dihydrolipoamide acetyltransferase (E2) deficiency. Ann Neurol. 2005;58:234–41. [PubMed: 16049940]
- Herbert M, Turnbull D. Mitochondrial donation - clearing the final regulatory hurdle in the United Kingdom. N Engl J Med. 2017;376:171–3. [PubMed: 28030773]
- Herzer M, Koch J, Prokisch H, Rodenburg R, Rauscher C, Radauer W, Forstner R, Pilz P, Rolinski B, Freisinger P, Mayr JA, Sperl W. Leigh disease with brainstem involvement in complex I deficiency due to assembly factor NDUFAF2 defect. Neuropediatrics. 2010;41:30–4. [PubMed: 20571988]
- Hinttala R, Smeets R, Moilanen JS, Ugalde C, Uusimaa J, Smeitink JA, Majamaa K. Analysis of mitochondrial DNA sequences in patients with isolated or combined oxidative phosphorylation system deficiency. J Med Genet. 2006;43:881–6. [PMC free article: PMC2563189] [PubMed: 16738010]
- Hoefs SJ, Dieteren CE, Distelmaier F, Janssen RJ, Epplen A, Swarts HG, Forkink M, Rodenburg RJ, Nijtmans LG, Willems PH, Smeitink JA, van den Heuvel LP. NDUFA2 complex I mutation leads to Leigh disease. Am J Hum Genet. 2008;82:1306–15. [PMC free article: PMC2427319] [PubMed: 18513682]
- Hoefs SJ, van Spronsen FJ, Lenssen EW, Nijtmans LG, Rodenburg RJ, Smeitink JA, van den Heuvel LP. NDUFA10 mutations cause complex I deficiency in a patient with Leigh disease. Eur J Hum Genet. 2011;19:270–4. [PMC free article: PMC3061993] [PubMed: 21150889]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Hughes SD, Kanabus M, Anderson G, Hargreaves IP, Rutherford T, O'Donnell M, Cross JH, Rahman S, Eaton S, Heales SJ. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J.Neurochem. 2014;129:426–33. [PubMed: 24383952]
- Hyslop LA, Blakeley P, Craven L, Richardson J, Fogarty NM, Fragouli E, Lamb M, Wamaitha SE, Prathalingam N, Zhang Q, O'Keefe H, Takeda Y, Arizzi L, Alfarawati S, Tuppen HA, Irving L, Kalleas D, Choudhary M, Wells D, Murdoch AP, Turnbull DM, Niakan KK, Herbert M. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature. 2016;534:383–6. [PMC free article: PMC5131843] [PubMed: 27281217]
- Jacobs LJ, de Coo IF, Nijland JG, Galjaard RJ, Los FJ, Schoonderwoerd K, Niermeijer MF, Geraedts JP, Scholte HR, Smeets HJ. Transmission and prenatal diagnosis of the T9176C mitochondrial DNA mutation. Mol Hum Reprod. 2005;11:223–8. [PubMed: 15709156]
- Jacobs LJ, de Wert G, Geraedts JP, de Coo IF, Smeets HJ. The transmission of OXPHOS disease and methods to prevent this. Hum Reprod Update. 2006;12:119–36. [PubMed: 16199488]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, Zhang F, Goessling W, Zapol WM, Mootha VK. Hypoxia as a therapy for mitochondrial disease. Science. 2016;352:54–61. [PMC free article: PMC4860742] [PubMed: 26917594]
- Janer A, Prudent J, Paupe V, Fahiminiya S, Majewski J, Sgarioto N, Des Rosiers C, Forest A, Lin ZY, Gingras AC, Mitchell G, McBride HM, Shoubridge EA. SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol Med. 2016;8:1019–38. [PMC free article: PMC5009808] [PubMed: 27390132]
- Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–4. [PubMed: 12923074]
- Johns DR, Neufeld MJ. Pitfalls in the molecular genetic diagnosis of Leber hereditary optic neuropathy (LHON). Am J Hum Genet. 1993;53:916–20. [PMC free article: PMC1682383] [PubMed: 8213820]
- Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, Palmiter RD, Rabinovitch PS, Morgan PG, Sedensky MM, Kaeberlein M. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013;342:1524–8. [PMC free article: PMC4055856] [PubMed: 24231806]
- Joost K, Rodenburg R, Piirsoo A, van den Heuvel B, Zordania R, Ounap K. A novel mutation in the SCO2 gene in a neonate with early-onset cardioencephalomyopathy. Pediatr Neurol. 2010;42:227–30. [PubMed: 20159436]
- Kanabus M, Heales SJ, Rahman S. Development of pharmacological strategies for mitochondrial disorders. Br J Pharmacol. 2014;171:1798–817. [PMC free article: PMC3976606] [PubMed: 24116962]
- Kanabus M, Fassone E, Hughes SD, Bilooei SF, Rutherford T, Donnell MO, Heales SJ, Rahman S. The pleiotropic effects of decanoic acid treatment on mitochondrial function in fibroblasts from patients with complex I deficient Leigh syndrome. J Inherit Metab Dis. 2016;39:415–26. [PMC free article: PMC4851692] [PubMed: 27080638]
- Kang E, Wu J, Gutierrez NM, Koski A, Tippner-Hedges R, Agaronyan K, Platero-Luengo A, Martinez-Redondo P, Ma H, Lee Y, Hayama T, Van Dyken C, Wang X, Luo S, Ahmed R, Li Y, Ji D, Kayali R, Cinnioglu C, Olson S, Jensen J, Battaglia D, Lee D, Wu D, Huang T, Wolf DP, Temiakov D, Belmonte JC, Amato P, Mitalipov S. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature. 2016;540:270–5. [PubMed: 27919073]
- Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, De Vivo DC. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324–30. [PubMed: 16476929]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsström S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, Suomalainen A. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014;6:721–31. [PMC free article: PMC4203351] [PubMed: 24711540]
- Kirby DM, Boneh A, Chow CW, Ohtake A, Ryan MT, Thyagarajan D, Thorburn DR. Low mutant load of mitochondrial DNA G13513A mutation can cause Leigh's disease. Ann Neurol. 2003;54:473–8. [PubMed: 14520659]
- Kirby DM, Kahler SG, Freckmann ML, Reddihough D, Thorburn DR. Leigh disease caused by the mitochondrial DNA G14459A mutation in unrelated families. Ann Neurol. 2000;48:102–4. [PubMed: 10894222]
- Kirby DM, Milovac T, Thorburn DR. A false-positive diagnosis for the common MELAS (A3243G) mutation caused by a novel variant (A3426G) in the ND1 gene of mitochondria DNA. Mol Diagn. 1998;3:211–5. [PubMed: 10089279]
- Koch J, Feichtinger RG, Freisinger P, Pies M, Schrödl F, Iuso A, Sperl W, Mayr JA, Prokisch H, Haack TB. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J Med Genet. 2016;53:270–8. [PubMed: 26783368]
- Komaki H, Akanuma J, Iwata H, Takahashi T, Mashima Y, Nonaka I, Goto Y. A novel mtDNA C11777A mutation in Leigh syndrome. Mitochondrion. 2003;2:293–304. [PubMed: 16120329]
- Kopajtich R, Nicholls TJ, Rorbach J, Metodiev MD, Freisinger P, Mandel H, Vanlander A, Ghezzi D, Carrozzo R, Taylor RW, Marquard K, Murayama K, Wieland T, Schwarzmayr T, Mayr JA, Pearce SF, Powell CA, Saada A, Ohtake A, Invernizzi F, Lamantea E, Sommerville EW, Pyle A, Chinnery PF, Crushell E, Okazaki Y, Kohda M, Kishita Y, Tokuzawa Y, Assouline Z, Rio M, Feillet F, Mousson de Camaret B, Chretien D, Munnich A, Menten B, Sante T, Smet J, Régal L, Lorber A, Khoury A, Zeviani M, Strom TM, Meitinger T, Bertini ES, Van Coster R, Klopstock T, Rötig A, Haack TB, Minczuk M, Prokisch H. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am J Hum Genet. 2014;95:708–20. [PMC free article: PMC4259976] [PubMed: 25434004]
- Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2016;79:190–203. [PubMed: 26506407]
- Lake NJ, Webb BD, Stroud DA, Richman TR, Ruzzenente B, Compton AG, Mountford HS, Pulman J, Zangarelli C, Rio M, Bodaert N, Assouline Z, Sherpa MD, Schadt EE, Houten SM, Byrnes J, McCormick EM, Zolkipli-Cunningham Z, Haude K, Zhang Z, Retterer K, Bai R, Calvo SE, Mootha VEK, Christodoulou J, Rötig A, Filipovska A, Cristian I, Falk MJ, Metodiev MD, Thorburn DR. Biallelic mutations in MRPS34 lead to instability of the small mitoribosomal subunit and Leigh syndrome. Am J Hum Genet. 2017;101:239–54. [PMC free article: PMC5544391] [PubMed: 28777931]
- Lebon S, Chol M, Benit P, Mugnier C, Chretien D, Giurgea I, Kern I, Girardin E, Hertz-Pannier L, de Lonlay P, Rötig A, Rustin P, Munnich A. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J Med Genet. 2003;40:896–9. [PMC free article: PMC1735336] [PubMed: 14684687]
- Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14:216–21. [PMC free article: PMC499520] [PubMed: 14874135]
- Leshinsky-Silver E, Lev D, Malinger G, Shapira D, Cohen S, Lerman-Sagie T, Saada A. Leigh disease presenting in utero due to a novel missense mutation in the mitochondrial DNA-ND3. Mol Genet Metab. 2010;100:65–70. [PubMed: 20202874]
- Leshinsky-Silver E, Lev D, Tzofi-Berman Z, Cohen S, Saada A, Yanoov-Sharav M, Gilad E, Lerman-Sagie T. Fulminant neurological deterioration in a neonate with Leigh syndrome due to a maternally transmitted missense mutation in the mitochondrial ND3 gene. Biochem Biophys Res Commun. 2005;334:582–7. [PubMed: 16023078]
- Leshinsky-Silver E, Perach M, Basilevsky E, Hershkovitz E, Yanoov-Sharav M, Lerman-Sagie T, Lev D. Prenatal exclusion of Leigh syndrome due to T8993C mutation in the mitochondrial DNA. Prenat Diagn. 2003;23:31–3. [PubMed: 12533809]
- Leshinsky-Silver E, Shuvalov R, Inbar S, Cohen S, Lev D, Lerman-Sagie T. Juvenile Leigh syndrome, optic atrophy, ataxia, dystonia, and epilepsy due to T14487C mutation in the mtDNA-ND6 gene: a mitochondrial syndrome presenting from birth to adolescence. J Child Neurol. 2011;26:476–81. [PubMed: 21196529]
- Levy RJ, Ríos PG, Akman HO, Sciacco M, Vivo DC, DiMauro S. Long survival in patients with leigh syndrome and the m.10191T>C mutation in MT-ND3 : a case report and review of the literature. J Child Neurol. 2014;29:NP105–10. [PMC free article: PMC4035473] [PubMed: 24284231]
- Lim BC, Park JD, Hwang H, Kim KJ, Hwang YS, Chae JH, Cheon JE, Kim IO, Lee R, Moon HK. Mutations in ND subunits of complex I are an important genetic cause of childhood mitochondrial encephalopathies. J Child Neurol. 2009;24:828–32. [PubMed: 19617458]
- Lim SC, Smith KR, Stroud DA, Compton AG, Tucker EJ, Dasvarma A, Gandolfo LC, Marum JE, McKenzie M, Peters HL, Mowat D, Procopis PG, Wilcken B, Christodoulou J, Brown GK, Ryan MT, Bahlo M, Thorburn DR. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am J Hum Genet. 2014;2014;94:209–22. [PMC free article: PMC3928654] [PubMed: 24462369]
- Limongelli A, Schaefer J, Jackson S, Invernizzi F, Kirino Y, Suzuki T, Reichmann H, Zeviani M. Variable penetrance of a familial progressive necrotising encephalopathy due to a novel tRNA(Ile) homoplasmic mutation in the mitochondrial genome. J Med Genet. 2004;41:342–9. [PMC free article: PMC1735786] [PubMed: 15121771]
- Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A, Forte G, Jenkinson EM, Kasher PR, Szynkiewicz M, Rice GI, Crow YJ. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2014;51:76–82. [PubMed: 24262145]
- Loeffen J, Elpeleg O, Smeitink J, Smeets R, Stöckler-Ipsiroglu S, Mandel H, Sengers R, Trijbels F, van den Heuvel L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann Neurol. 2001;49:195–201. [PubMed: 11220739]
- Loeffen J, Smeitink J, Triepels R, Smeets R, Schuelke M, Sengers R, Trijbels F, Hamel B, Mullaart R, van den Heuvel L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet. 1998;63:1598–608. [PMC free article: PMC1377631] [PubMed: 9837812]
- Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS, Schuh RA. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer's disease-relevant murine model. BMC Neurol. 2015;15:19. [PMC free article: PMC4358858] [PubMed: 25884176]
- López LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125–9. [PMC free article: PMC1698707] [PubMed: 17186472]
- Malfatti E, Bugiani M, Invernizzi F, de Souza CF, Farina L, Carrara F, Lamantea E, Antozzi C, Confalonieri P, Sanseverino MT, Giugliani R, Uziel G, Zeviani M. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain. 2007;130:1894–904. [PubMed: 17535832]
- Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002;30:394–9. [PubMed: 11925565]
- Martikainen MH, Kytövuori L, Majamaa K. Juvenile parkinsonism, hypogonadism and Leigh-like MRI changes in a patient with m.4296G>A mutation in mitochondrial DNA. Mitochondrion. 2013;13:83–6. [PubMed: 23395828]
- Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi-Vici C, Pontrelli G, Corsetti T, Livadiotti S, Kheifets V, Hinman A, Shrader WD, Thoolen M, Klein MB, Bertini E, Miller G. EPI-743 reverses the progression of the pediatric mitochondrial disease--genetically defined Leigh Syndrome. Mol Genet Metab. 2012;107:383–8. [PubMed: 23010433]
- Mattiazzi M, Vijayvergiya C, Gajewski CD, DeVivo DC, Lenaz G, Wiedmann M, Manfredi G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum Mol Genet. 2004;13:869–79. [PubMed: 14998933]
- Mayr JA, Freisinger P, Schlachter K, Rolinski B, Zimmermann FA, Scheffner T, Haack TB, Koch J, Ahting U, Prokisch H, Sperl W. Thiamine pyrophosphokinase deficiency in encephalopathic children with defects in the pyruvate oxidation pathway. Am J Hum Genet. 2011;89:806–12. [PMC free article: PMC3234371] [PubMed: 22152682]
- McDonnell MT, Schaefer AM, Blakely EL, McFarland R, Chinnery PF, Turnbull DM, Taylor RW. Noninvasive diagnosis of the 3243A > G mitochondrial DNA mutation using urinary epithelial cells. Eur J Hum Genet. 2004;12:778–81. [PubMed: 15199381]
- McFarland R, Clark KM, Morris AA, Taylor RW, Macphail S, Lightowlers RN, Turnbull DM. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet. 2002;30:145–6. [PubMed: 11799391]
- McFarland R, Kirby DM, Fowler KJ, Ohtake A, Ryan MT, Amor DJ, Fletcher JM, Dixon JW, Collins FA, Turnbull DM, Taylor RW, Thorburn DR. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann Neurol. 2004;55:58–64. [PubMed: 14705112]
- Mineri R, Rimoldi M, Burlina AB, Koskull S, Perletti C, Heese B, von Döbeln U, Mereghetti P, Di Meo I, Invernizzi F, Zeviani M, Uziel G, Tiranti V. Identification of new mutations in the ETHE1 gene in a cohort of 14 patients presenting with ethylmalonic encephalopathy. J Med Genet. 2008;45:473–8. [PubMed: 18593870]
- Mitchell G, Ogier H, Munnich A, Saudubray JM, Shirrer J, Charpentier C, Rocchiccioli F. Neurological deterioration and lactic acidemia in biotinidase deficiency. A treatable condition mimicking Leigh's disease. Neuropediatrics. 1986;17:129–31. [PubMed: 3762868]
- Mkaouar-Rebai E, Chamkha I, Kammoun F, Kammoun T, Aloulou H, Hachicha M, Triki C, Fakhfakh F. Two new mutations in the MT-TW gene leading to the disruption of the secondary structure of the tRNA(Trp) in patients with Leigh syndrome. Mol Genet Metab. 2009;97:179–84. [PubMed: 19349200]
- Mkaouar-Rebai E, Ellouze E, Chamkha I, Kammoun F, Triki C, Fakhfakh F. Molecular-clinical correlation in a family with a novel heteroplasmic Leigh syndrome missense mutation in the mitochondrial cytochrome c oxidase III gene. J Child Neurol. 2011;26:12–20. [PubMed: 20525945]
- Monlleo-Neila L, Toro MD, Bornstein B, Garcia-Arumi E, Sarrias A, Roig-Quilis M, Munell F. Leigh syndrome and the mitochondrial m.13513G>A mutation: expanding the clinical spectrum. J Child Neurol. 2013;28:1531–4. [PubMed: 23034978]
- Monnot S, Gigarel N, Samuels DC, Burlet P, Hesters L, Frydman N, Frydman R, Kerbrat V, Funalot B, Martinovic J, Benachi A, Feingold J, Munnich A, Bonnefont JP, Steffann J. Segregation of mtDNA throughout human embryofetal development: m.3243A>G as a model system. Hum Mutat. 2011;32:116–25. [PMC free article: PMC3058134] [PubMed: 21120938]
- Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, Mitchell GA, Morin C, Mann M, Hudson TJ, Robinson B, Rioux JD, Lander ES. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100:605–10. [PMC free article: PMC141043] [PubMed: 12529507]
- Moslemi AR, Darin N, Tulinius M, Wiklund LM, Holme E, Oldfors A. Progressive encephalopathy and complex I deficiency associated with mutations in MTND1. Neuropediatrics. 2008;39:24–8. [PubMed: 18504678]
- Naess K, Freyer C, Bruhn H, Wibom R, Malm G, Nennesmo I, von Döbeln U, Larsson NG. MtDNA mutations are a common cause of severe disease phenotypes in children with Leigh syndrome. Biochim Biophys Acta. 2009;1787:484–90. [PubMed: 19103152]
- Negishi Y, Hattori A, Takeshita E, Sakai C, Ando N, Ito T, Goto Y, Saitoh S. Homoplasmy of a mitochondrial 3697G>A mutation causes Leigh syndrome. J Hum Genet. 2014;59:405–7. [PubMed: 24830958]
- Nesbitt V, Alston CL, Blakely EL, Fratter C, Feeney CL, Poulton J, Brown GK, Turnbull DM, Taylor RW, McFarland R. A national perspective on prenatal testing for mitochondrial disease. Eur J Hum Genet. 2014;22:1255–9. [PMC free article: PMC4200441] [PubMed: 24642831]
- Nijtmans LG, Henderson NS, Attardi G, Holt IJ. Impaired ATP synthase assembly associated with a mutation in the human ATP synthase subunit 6 gene. J Biol Chem. 2001;276:6755–62. [PubMed: 11076946]
- Ogawa E, Shimura M, Fushimi T, Tajika M, Ichimoto K, Matsunaga A, Tsuruoka T, Ishige M, Fuchigami T, Yamazaki T, Mori M, Kohda M, Kishita Y, Okazaki Y, Takahashi S, Ohtake A, Murayama K. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: a study of 106 Japanese patients. J Inherit Metab Dis. 2017;40:685–93. [PMC free article: PMC5579154] [PubMed: 28429146]
- Ohlenbusch A, Edvardson S, Skorpen J, Bjornstad A, Saada A, Elpeleg O, Gärtner J, Brockmann K. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J Rare Dis. 2012;7:69. [PMC free article: PMC3492161] [PubMed: 22995659]
- Oquendo CE, Antonicka H, Shoubridge EA, Reardon W, Brown GK. Functional and genetic studies demonstrate that mutation in the COX15 gene can cause Leigh syndrome. J Med Genet. 2004;41:540–4. [PMC free article: PMC1735852] [PubMed: 15235026]
- Ostergaard E, Hansen FJ, Sorensen N, Duno M, Vissing J, Larsen PL, Faeroe O, Thorgrimsson S, Wibrand F, Christensen E, Schwartz M. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–61. [PubMed: 17287286]
- Ostergaard E, Rodenburg RJ, van den Brand M, Thomsen LL, Duno M, Batbayli M, Wibrand F, Nijtmans L. Respiratory chain complex I deficiency due to NDUFA12 mutations as a new cause of Leigh syndrome. J Med Genet. 2011;48:737–40. [PubMed: 21617257]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–23. [PMC free article: PMC2778844] [PubMed: 18614015]
- Pagnamenta AT, Hargreaves IP, Duncan AJ, Taanman JW, Heales SJ, Land JM, Bitner-Glindzicz M, Leonard JV, Rahman S. Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol Genet Metab. 2006;89:214–21. [PubMed: 16798039]
- Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, Anselm I, Cohen BH, Falk MJ, Greene C, Gropman AL, Haas R, Hirano M, Morgan P, Sims K, Tarnopolsky M, Van Hove JL, Wolfe L, DiMauro S. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17:689–701. [PMC free article: PMC5000852] [PubMed: 25503498]
- Péquignot MO, Dey R, Zeviani M, Tiranti V, Godinot C, Poyau A, Sue C, Di Mauro S, Abitbol M, Marsac C. Mutations in the SURF1 gene associated with Leigh syndrome and cytochrome C oxidase deficiency. Hum Mutat. 2001;17:374–81. [PubMed: 11317352]
- Peters H, Buck N, Wanders R, Ruiter J, Waterham H, Koster J, Yaplito-Lee J, Ferdinandusse S, Pitt J. ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism. Brain. 2014;137:2903–8. [PubMed: 25125611]
- Petruzzella V, Di Giacinto G, Scacco S, Piemonte F, Torraco A, Carrozzo R, Vergari R, Dionisi-Vici C, Longo D, Tessa A, Papa S, Bertini E. Atypical Leigh syndrome associated with the D393N mutation in the mitochondrial ND5 subunit. Neurology. 2003;61:1017–8. [PubMed: 14557590]
- Pitceathly RD, Rahman S, Wedatilake Y, Polke JM, Cirak S, Foley AR, Sailer A, Hurles ME, Stalker J, Hargreaves I, Woodward CE, Sweeney MG, Muntoni F, Houlden H, Taanman JW, Hanna MG, et al. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013;3:1795–805. [PMC free article: PMC3701321] [PubMed: 23746447]
- Poulton J, Bredenoord AL. 174th ENMC international workshop: applying pre-implantation genetic diagnosis to mtDNA diseases: implications of scientific advances 19-21 March 2010, Naarden, The Netherlands. Neuromuscul Disord. 2010;20:559–63. [PubMed: 20627569]
- Poulton J, Turnbull DM. 74th ENMC international workshop: mitochondrial diseases 19-20 November 1999, Naarden, The Netherlands. Neuromuscul Disord. 2000;10:460–2. [PubMed: 10899455]
- Purhonen J, Rajendran J, Mörgelin M, Uusi-Rauva K, Katayama S, Krjutskov K, Einarsdottir E, Velagapudi V, Kere J, Jauhiainen M, Fellman V, Kallijärvi J. Ketogenic diet attenuates hepatopathy in mouse model of respiratory chain complex III deficiency caused by a Bcs1l mutation. Sci Rep. 2017;7:957. [PMC free article: PMC5430426] [PubMed: 28424480]
- Quinonez SC, Leber SM, Martin DM, Thoene JG, Bedoyan JK. Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr Neurol. 2013;48:67–72. [PMC free article: PMC4535688] [PubMed: 23290025]
- Quintana E, Mayr JA, García Silva MT, Font A, Tortoledo MA, Moliner S, Ozaez L, Lluch M, Cabello A, Ricoy JR, Koch J, Ribes A, Sperl W, Briones P. PDH E1β deficiency with novel mutations in two patients with Leigh syndrome. J Inherit Metab Dis. 2009;32 Suppl 1:S339–43. [PubMed: 19924563]
- Rabier D, Diry C, Rotig A, Rustin P, Heron B, Bardet J, Parvy P, Ponsot G, Marsac C, Saudubray JM, Munnich A, Kamoun P. Persistent hypocitrullinaemia as a marker for mtDNA NARP T 8993 G mutation? J Inherit Metab Dis. 1998;21:216–9. [PubMed: 9686360]
- Rahman J, Noronha A, Thiele I, Rahman S. Leigh map: a novel computational diagnostic resource for mitochondrial disease. Ann Neurol. 2017;81:9–16. [PMC free article: PMC5347854] [PubMed: 27977873]
- Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–51. [PubMed: 8602753]
- Rahman S, Poulton J, Marchington D, Suomalainen A. Decrease of 3243 A—>G mtDNA mutation from blood in MELAS syndrome: a longitudinal study. Am J Hum Genet. 2001;68:238–40. [PMC free article: PMC1234919] [PubMed: 11085913]
- Rantamäki MT, Soini HK, Finnilä SM, Majamaa K, Udd B. Adult-onset ataxia and polyneuropathy caused by mitochondrial 8993T-->C mutation. Ann Neurol. 2005;58:337–40. [PubMed: 16049925]
- Rawle MJ, Larner AJ. NARP syndrome: a 20-year follow-up. Case Rep Neurol. 2013;5:204–7. [PMC free article: PMC3919433] [PubMed: 24516410]
- Reddy P, Ocampo A, Suzuki K, Luo J, Bacman SR, Williams SL, Sugawara A, Okamura D, Tsunekawa Y, Wu J, Lam D, Xiong X, Montserrat N, Esteban CR, Liu GH, Sancho-Martinez I, Manau D, Civico S, Cardellach F, Del Mar O'Callaghan M, Campistol J, Zhao H, Campistol JM, Moraes CT, Izpisua Belmonte JC. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell. 2015;161:459–69. [PMC free article: PMC4505837] [PubMed: 25910206]
- Riley LG, Cowley MJ, Gayevskiy V, Roscioli T, Thorburn DR, Prelog K, Bahlo M, Sue CM, Balasubramaniam S, Christodoulou J. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. 2017;40:261–9. [PubMed: 27995398]
- Ronchi D, Cosi A, Tonduti D, Orcesi S, Bordoni A, Fortunato F, Rizzuti M, Sciacco M, Collotta M, Cagdas S, Capovilla G, Moggio M, Berardinelli A, Veggiotti P, Comi GP. Clinical and molecular features of an infant patient affected by Leigh Disease associated to m.14459G>A mitochondrial DNA mutation: a case report. BMC Neurol. 2011;11:85. [PMC free article: PMC3148968] [PubMed: 21749722]
- Rubegni A, Pisano T, Bacci G, Tessa A, Battini R, Procopio E, Giglio S, Pasquariello R, Santorelli FM, Guerrini R, Nesti C. Leigh-like neuroimaging features associated with new biallelic mutations in OPA1. Eur J Paediatr Neurol. 2017;21:671–7. [PubMed: 28442211]
- Ruiter EM, Siers MH, van den Elzen C, van Engelen BG, Smeitink JA, Rodenburg RJ, Hol FA. The mitochondrial 13513G > A mutation is most frequent in Leigh syndrome combined with reduced complex I activity, optic atrophy and/or Wolff-Parkinson-White. Eur J Hum Genet. 2007;15:155–61. [PubMed: 17106447]
- Sacconi S, Salviati L, Gooch C, Bonilla E, Shanske S, DiMauro S. Complex neurologic syndrome associated with the G1606A mutation of mitochondrial DNA. Arch Neurol. 2002;59:1013–5. [PubMed: 12056939]
- Sakai C, Yamaguchi S, Sasaki M, Miyamoto Y, Matsushima Y, Goto Y. ECHS1 mutations cause combined respiratory chain deficiency resulting in Leigh syndrome. Hum Mutat. 2015;36:232–9. [PubMed: 25393721]
- Sallevelt SC, de Die-Smulders CE, Hendrickx AT, Hellebrekers DM, de Coo IF, Alston CL, Knowles C, Taylor RW, McFarland R, Smeets HJ. De novo mtDNA point mutations are common and have a low recurrence risk. J Med Genet. 2017;54:73–83. [PMC free article: PMC5502310] [PubMed: 27450679]
- Sallevelt SC, Dreesen JC, Drüsedau M, Spierts S, Coonen E, van Tienen FH, van Golde RJ, de Coo IF, Geraedts JP, de Die-Smulders CE, Smeets HJ. Preimplantation genetic diagnosis in mitochondrial DNA disorders: challenge and success. J Med Genet. 2013;50:125–32. [PubMed: 23339111]
- Santorelli FM, Mak SC, Vazquez-Memije E, Shanske S, Kranz-Eble P, Jain KD, Bluestone DL, De Vivo DC, DiMauro S. Clinical heterogeneity associated with the mitochondrial DNA T8993C point mutation. Pediatr Res. 1996;39:914–7. [PubMed: 8726250]
- Santorelli FM, Shanske S, Macaya A, DeVivo DC, DiMauro S. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh's syndrome. Ann Neurol. 1993;34:827–34. [PubMed: 8250532]
- Santorelli FM, Tanji K, Sano M, Shanske S, El-Shahawi M, Kranz-Eble P, DiMauro S, De Vivo DC. Maternally inherited encephalopathy associated with a single-base insertion in the mitochondrial tRNATrp gene. Ann Neurol. 1997;42:256–60. [PubMed: 9266739]
- Santra S, Gilkerson RW, Davidson M, Schon EA. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann Neurol. 2004;56:662–9. [PubMed: 15389892]
- Sarzi E, Brown MD, Lebon S, Chretien D, Munnich A, Rotig A, Procaccio V. A novel recurrent mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am J Med Genet Part A. 2007;143A:33–41. [PubMed: 17152068]
- Sato A, Kono T, Nakada K, Ishikawa K, Inoue S, Yonekawa H, Hayashi J. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc Natl Acad Sci U S A. 2005;102:16765–70. [PMC free article: PMC1283814] [PubMed: 16275929]
- Saunders C, Smith L, Wibrand F, Ravn K, Bross P, Thiffault I, Christensen M, Atherton A, Farrow E, Miller N, Kingsmore SF, Ostergaard E. CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria. Am J Hum Genet. 2015;96:258–65. [PMC free article: PMC4320254] [PubMed: 25597511]
- Schiff M, Miné M, Brivet M, Marsac C, Elmaleh-Bergés M, Evrard P, Ogier de Baulny H. Leigh's disease due to a new mutation in the PDHX gene. Ann Neurol. 2006;59:709–14. [PubMed: 16566017]
- Schwartzentruber J, Buhas D, Majewski J, Sasarman F, Papillon-Cavanagh S, Thiffault I, Sheldon KM, Massicotte C, Patry L, Simon M, Zare AS, McKernan KJ, Michaud J, Boles RG, Deal CL, Desilets V, Shoubridge EA, Samuels ME, et al. Mutation in the nuclear-encoded mitochondrial isoleucyl-tRNA synthetase IARS2 in patients with cataracts, growth hormone deficiency with short stature, partial sensorineural deafness, and peripheral neuropathy or with Leigh syndrome. Hum Mutat. 2014;35:1285–9. [PubMed: 25130867]
- Sembrano E, Barthlen GM, Wallace S, Lamm C. Polysomnographic findings in a patient with the mitochondrial encephalomyopathy NARP. Neurology. 1997;49:1714–7. [PubMed: 9409376]
- Sgarbi G, Casalena GA, Baracca A, Lenaz G, DiMauro S, Solaini G. Human NARP mitochondrial mutation metabolism corrected with alpha-ketoglutarate/aspartate: a potential new therapy. Arch Neurol. 2009;66:951–7. [PubMed: 19667215]
- Shamseldin HE, Alshammari M, Al-Sheddi T, Salih MA, Alkhalidi H, Kentab A, Repetto GM, Hashem M, Alkuraya FS. FS. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J Med Genet. 2012;49:234–41. [PubMed: 22499341]
- Shanske S, Coku J, Lu J, Ganesh J, Krishna S, Tanji K, Bonilla E, Naini AB, Hirano M, DiMauro S. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol. 2008;65:368–72. [PubMed: 18332249]
- Shanske S, Pancrudo J, Kaufmann P, Engelstad K, Jhung S, Lu J, Naini A, DiMauro S, De Vivo DC. Varying loads of the mitochondrial DNA A3243G mutation in different tissues: implications for diagnosis. Am J Med Genet A. 2004;130A:134–7. [PubMed: 15372523]
- Silvestri G, Ciafaloni E, Santorelli FM, Shanske S, Servidei S, Graf WD, Sumi M, DiMauro S. Clinical features associated with the A-->G transition at nucleotide 8344 of mtDNA ("MERRF mutation"). Neurology. 1993;43:1200–6. [PubMed: 8170567]
- Simon M, Richard EM, Wang X, Shahzad M, Huang VH, Qaiser TA, Potluri P, Mahl SE, Davila A, Nazli S, Hancock S, Yu M, Gargus J, Chang R, Al-Sheqaih N, Newman WG, Abdenur J, Starr A, Hegde R, Dorn T, Busch A, Park E, Wu J, Schwenzer H, Flierl A, Florentz C, Sissler M, Khan SN, Li R, Guan MX, Friedman TB, Wu DK, Procaccio V, Riazuddin S, Wallace DC, Ahmed ZM, Huang T, Riazuddin S. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 2015;11:e1005097. [PMC free article: PMC4373692] [PubMed: 25807530]
- Smeets HJ, Sallevelt SC, Dreesen JC, de Die-Smulders CE, de Coo IF. Preventing the transmission of mitochondrial DNA disorders using prenatal or preimplantation genetic diagnosis. Ann N Y Acad Sci. 2015;1350:29–36. [PubMed: 26312584]
- Sofou K, De Coo IF, Isohanni P, Ostergaard E, Naess K, De Meirleir L, Tzoulis C, Uusimaa J, De Angst IB, Lönnqvist T, Pihko H, Mankinen K, Bindoff LA, Tulinius M, Darin N. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. 2014;9:52. [PMC free article: PMC4021638] [PubMed: 24731534]
- Soreze Y, Boutron A, Habarou F, Barnerias C, Nonnenmacher L, Delpech H, Mamoune A, Chrétien D, Hubert L, Bole-Feysot C, Nitschke P, Correia I, Sardet C, Boddaert N, Hamel Y, Delahodde A, Ottolenghi C, de Lonlay P. Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase. Orphanet J Rare Dis. 2013;8:192. [PMC free article: PMC3905285] [PubMed: 24341803]
- Spangenberg L, Graña M, Greif G, Suarez-Rivero JM, Krysztal K, Tapié A, Boidi M, Fraga V, Lemes A, Gueçaimburú R, Cerisola A, Sánchez-Alcázar JA, Robello C, Raggio V, Naya H. 3697G>A in MT-ND1 is a causative mutation in mitochondrial disease. Mitochondrion. 2016;28:54–9. [PubMed: 27017994]
- Spiegel R, Shaag A, Edvardson S, Mandel H, Stepensky P, Shalev SA, Horovitz Y, Pines O, Elpeleg O. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol. 2009;66:419–24. [PubMed: 19798730]
- Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 2008;121:e1223–8. [PMC free article: PMC3777225] [PubMed: 18411236]
- Steffann J, Frydman N, Gigarel N, Burlet P, Ray PF, Fanchin R, Feyereisen E, Kerbrat V, Tachdjian G, Bonnefont JP, Frydman R, Munnich A. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J Med Genet. 2006;43:244–7. [PMC free article: PMC2563237] [PubMed: 16155197]
- Sudo A, Honzawa S, Nonaka I, Goto Y. Leigh syndrome caused by mitochondrial DNA G13513A mutation: frequency and clinical features in Japan. J Hum Genet. 2004;49:92–6. [PubMed: 14730434]
- Sugiana C, Pagliarini DJ, McKenzie M, Kirby DM, Salemi R, Abu-Amero KK, Dahl HH, Hutchison WM, Vascotto KA, Smith SM, Newbold RF, Christodoulou J, Calvo S, Mootha VK, Ryan MT, Thorburn DR. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am J Hum Genet. 2008;83:468–78. [PMC free article: PMC2561934] [PubMed: 18940309]
- Suwa K, Yamagata T, Momoi MY, Kawakami A, Kikuchi Y, Miyao M, Hirokawa H, Oikawa T. Acute relapsing encephalopathy mimicking acute necrotizing encephalopathy in a 4-year-old boy. Brain Dev. 1999;21:554–8. [PubMed: 10598058]
- Taanman JW, Rahman S, Pagnamenta AT, Morris AA, Bitner-Glindzicz M, Wolf NI, Leonard JV, Clayton PT, Schapira AH. Analysis of mutant DNA polymerase gamma in patients with mitochondrial DNA depletion. Hum Mutat. 2009;30:248–54. [PubMed: 18828154]
- Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, Li Y, Ramsey C, Kolotushkina O, Mitalipov S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–72. [PMC free article: PMC2774772] [PubMed: 19710649]
- Tanaka M, Borgeld HJ, Zhang J, Muramatsu S, Gong JS, Yoneda M, Maruyama W, Naoi M, Ibi T, Sahashi K, Shamoto M, Fuku N, Kurata M, Yamada Y, Nishizawa K, Akao Y, Ohishi N, Miyabayashi S, Umemoto H, Muramatsu T, Furukawa K, Kikuchi A, Nakano I, Ozawa K, Yagi K. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci. 2002;9:534–41. [PubMed: 12372991]
- Tarnopolsky M, Meaney B, Robinson B, Sheldon K, Boles RG. Severe infantile leigh syndrome associated with a rare mitochondrial ND6 mutation, m.14487T>C. Am J Med Genet A. 2013;161A:2020–3. [PubMed: 23813926]
- Tatuch Y, Christodoulou J, Feigenbaum A, Clarke JT, Wherret J, Smith C, Rudd N, Petrova-Benedict R, Robinson BH. Heteroplasmic mtDNA mutation (T----G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am J Hum Genet. 1992;50:852–8. [PMC free article: PMC1682643] [PubMed: 1550128]
- Tatuch Y, Robinson BH. The mitochondrial DNA mutation at 8993 associated with NARP slows the rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem Biophys Res Commun. 1993;192:124–8. [PubMed: 8476414]
- Taylor RW, Morris AA, Hutchinson M, Turnbull DM. Leigh disease associated with a novel mitochondrial DNA ND5 mutation. Eur J Hum Genet. 2002;10:141–4. [PubMed: 11938446]
- Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, Smertenko T, Alston CL, Neeve VC, Best A, Yarham JW, Kirschner J, Schara U, Talim B, Topaloglu H, Baric I, Holinski-Feder E, Abicht A, Czermin B, Kleinle S, Morris AA, Vassallo G, Gorman GS, Ramesh V, Turnbull DM, Santibanez-Koref M, McFarland R, Horvath R, Chinnery PF. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2014;312:68–77. [PMC free article: PMC6558267] [PubMed: 25058219]
- Thorburn DR, Chow CW, Kirby DM. Respiratory chain enzyme analysis in muscle and liver. Mitochondrion. 2004;4:363–75. [PubMed: 16120398]
- Thorburn DR, Dahl HH. Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. Am J Med Genet. 2001;106:102–14. (Semin Med Genet) [PubMed: 11579429]
- Thyagarajan D, Shanske S, Vazquez-Memije M, De Vivo D, DiMauro S. A novel mitochondrial ATPase 6 point mutation in familial bilateral striatal necrosis. Ann Neurol. 1995;38:468–72. [PubMed: 7668837]
- Tiranti V, Corona P, Greco M, Taanman J-W, Carrara F, Lamantea E, Nijtmans L, Uziel G, Zeviani M. A novel frameshift mutation of the mtDNA COIII gene leads to impaired assembly of cytochrome c oxidase in a patient affected by Leigh-like syndrome. Hum Mol Genet. 2000;9:2733–42. [PubMed: 11063732]
- Tort F, Ferrer-Cortès X, Thió M, Navarro-Sastre A, Matalonga L, Quintana E, Bujan N, Arias A, García-Villoria J, Acquaviva C, Vianey-Saban C, Artuch R, García-Cazorla À, Briones P, Ribes A. Mutations in the lipoyltransferase LIPT1 gene cause a fatal disease associated with a specific lipoylation defect of the 2-ketoacid dehydrogenase complexes. Hum Mol Genet. 2014;23:1907–15. [PubMed: 24256811]
- Triepels RH, van den Heuvel LP, Loeffen JL, Buskens CA, Smeets RJ, Rubio Gozalbo ME, Budde SM, Mariman EC, Wijburg FA, Barth PG, Trijbels JM, Smeitink JA. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann Neurol. 1999;45:787–90. [PubMed: 10360771]
- Tucker EJ, Hershman SG, Köhrer C, Belcher-Timme CA, Patel J, Goldberger OA, Christodoulou J, Silberstein JM, McKenzie M, Ryan MT, Compton AG, Jaffe JD, Carr SA, Calvo SE, Rajbhandary UL, Thorburn DR, Mootha VK. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011;14:428–34. [PMC free article: PMC3486727] [PubMed: 21907147]
- Tulinius M, Moslemi AR, Darin N, Westerberg B, Wiklund LM, Holme E, Oldfors A. Leigh syndrome with cytochrome-c oxidase deficiency and a single T insertion nt 5537 in the mitochondrial tRNATrp gene. Neuropediatrics. 2003;34:87–91. [PubMed: 12776230]
- Uehara N, Mori M, Tokuzawa Y, Mizuno Y, Tamaru S, Kohda M, Moriyama Y, Nakachi Y, Matoba N, Sakai T, Yamazaki T, Harashima H, Murayama K, Hattori K, Hayashi J, Yamagata T, Fujita Y, Ito M, Tanaka M, Nibu K, Ohtake A, Okazaki Y. New MT-ND6 and NDUFA1 mutations in mitochondrial respiratory chain disorders. Ann Clin Transl Neurol. 2014;1:361–9. [PMC free article: PMC4184687] [PubMed: 25356405]
- Ugalde C, Hinttala R, Timal S, Smeets R, Rodenburg RJ, Uusimaa J, van Heuvel LP, Nijtmans LG, Majamaa K, Smeitink JA. Mutated ND2 impairs mitochondrial complex I assembly and leads to Leigh syndrome. Mol Genet Metab. 2007;90:10–4. [PubMed: 16996290]
- Ugalde C, Triepels RH, Coenen MJ, van den Heuvel LP, Smeets R, Uusimaa J, Briones P, Campistol J, Majamaa K, Smeitink JA, Nijtmans LG. Impaired complex I assembly in a Leigh syndrome patient with a novel missense mutation in the ND6 gene. Ann Neurol. 2003;54:665–9. [PubMed: 14595656]
- Valente L, Piga D, Lamantea E, Carrara F, Uziel G, Cudia P, Zani A, Farina L, Morandi L, Mora M, Spinazzola A, Zeviani M, Tiranti V. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim Biophys Acta. 2009;1787:491–501. [PubMed: 18977334]
- Valente L, Tiranti V, Marsano RM, Malfatti E, Fernandez-Vizarra E, Donnini C, Mereghetti P, De Gioia L, Burlina A, Castellan C, Comi GP, Savasta S, Ferrero I, Zeviani M. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet. 2007;80:44–58. [PMC free article: PMC1785320] [PubMed: 17160893]
- Vanniarajan A, Rajshekher GP, Joshi MB, Reddy AG, Singh L, Thangaraj K. Novel mitochondrial mutation in the ND4 gene associated with Leigh syndrome. Acta Neurol Scand. 2006;114:350–3. [PubMed: 17022785]
- van den Bosch BJ, Gerards M, Sluiter W, Stegmann AP, Jongen EL, Hellebrekers DM, Oegema R, Lambrichs EH, Prokisch H, Danhauser K, Schoonderwoerd K, de Coo IF, Smeets HJ. Defective NDUFA9 as a novel cause of neonatally fatal complex I disease. J Med Genet. 2012;49:10–5. [PubMed: 22114105]
- Van Hove JL, Saenz MS, Thomas JA, Gallagher RC, Lovell MA, Fenton LZ, Shanske S, Myers SM, Wanders RJ, Ruiter J, Turkenburg M, Waterham HR. Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr Res. 2010;68:159–64. [PMC free article: PMC2928220] [PubMed: 20453710]
- Vedrenne V, Gowher A, De Lonlay P, Nitschke P, Serre V, Boddaert N, Altuzarra C, Mager-Heckel AM, Chretien F, Entelis N, Munnich A, Tarassov I, Rötig A. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am J Hum Genet. 2012;91:912–8. [PMC free article: PMC3487136] [PubMed: 23084291]
- Veerapandiyan A, Chaudhari A, Traba CM, Ming X. Novel mutation in mitochondrial DNA in 2 siblings with Leigh syndrome. Neurol Genet. 2016;2:e99. [PMC free article: PMC4988465] [PubMed: 27574709]
- Vilarinho L, Santorelli FM, Rosas MJ, Tavares C, Melo-Pires M, DiMauro S. The mitochondrial A3243G mutation presenting as severe cardiomyopathy. J Med Genet. 1997;34:607–9. [PMC free article: PMC1051008] [PubMed: 9222976]
- Wang J, Brautbar A, Chan AK, Dzwiniel T, Li FY, Waters PJ, Graham BH, Wong LJ. Two mtDNA mutations 14487T>C (M63V, ND6) and 12297T>C (tRNA Leu) in a Leigh syndrome family. Mol Genet Metab. 2009;96:59–65. [PubMed: 19062322]
- Wang SB, Weng WC, Lee NC, Hwu WL, Fan PC, Lee WT. Mutation of mitochondrial DNA G13513A presenting with Leigh syndrome, Wolff-Parkinson-White syndrome and cardiomyopathy. Pediatr Neonatol. 2008;49:145–9. [PubMed: 19054921]
- Wedatilake Y, Brown RM, McFarland R, Yaplito-Lee J, Morris AA, Champion M, Jardine PE, Clarke A, Thorburn DR, Taylor RW, Land JM, Forrest K, Dobbie A, Simmons L, Aasheim ET, Ketteridge D, Hanrahan D, Chakrapani A, Brown GK, Rahman S. SURF1 deficiency: a multi-centre natural history study. Orphanet J Rare Dis. 2013;8:96. [PMC free article: PMC3706230] [PubMed: 23829769]
- Weraarpachai W, Antonicka H, Sasarman F, Seeger J, Schrank B, Kolesar JE, Lochmüller H, Chevrette M, Kaufman BA, Horvath R, Shoubridge EA. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet. 2009;41:833–7. [PubMed: 19503089]
- White SL, Collins VR, Wolfe R, Cleary MA, Shanske S, DiMauro S, Dahl HH, Thorburn DR. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet. 1999a;65:474–82. [PMC free article: PMC1377946] [PubMed: 10417290]
- White SL, Shanske S, Biros I, Warwick L, Dahl HM, Thorburn DR, Di Mauro S. Two cases of prenatal analysis for the pathogenic T to G substitution at nucleotide 8993 in mitochondrial DNA. Prenat Diagn. 1999b;19:1165–8. [PubMed: 10590437]
- White SL, Shanske S, McGill JJ, Mountain H, Geraghty MT, DiMauro S, Dahl HH, Thorburn DR. Mitochondrial DNA mutations at nucleotide 8993 show a lack of tissue or age-related variation. J Inherit Metab Dis. 1999c;22:899–914. [PubMed: 10604142]
- White SL, Thorburn DR, Christodoulou J, Dahl HH. Novel mitochondrial DNA variant that may give a false positive diagnosis for the T8993C mutation. Mol Diagn. 1998;3:113–7. [PubMed: 10029662]
- Wortmann SB, van Hasselt PM, Barić I, Burlina A, Darin N, Hörster F, Coker M, Ucar SK, Krumina Z, Naess K, Ngu LH, Pronicka E, Riordan G, Santer R, Wassmer E, Zschocke J, Schiff M, de Meirleir L, Alowain MA, Smeitink JA, Morava E, Kozicz T, Wevers RA, Wolf NI, Willemsen MA. Eyes on MEGDEL: distinctive basal ganglia involvement in dystonia deafness syndrome. Neuropediatrics. 2015;46:98–103. [PubMed: 25642805]
- Wortmann SB, Vaz FM, Gardeitchik T, Vissers LE, Renkema GH, Schuurs-Hoeijmakers JH, Kulik W, Lammens M, Christin C, Kluijtmans LA, Rodenburg RJ, Nijtmans LG, Grünewald A, Klein C, Gerhold JM, Kozicz T, van Hasselt PM, Harakalova M, Kloosterman W, Barić I, Pronicka E, Ucar SK, Naess K, Singhal KK, Krumina Z, Gilissen C, van Bokhoven H, Veltman JA, Smeitink JA, Lefeber DJ, Spelbrink JN, Wevers RA, Morava E, de Brouwer AP. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. 2012;44:797–802. [PubMed: 22683713]
- Wray CD, Friederich MW, du Sart D, Pantaleo S, Smet J, Kucera C, Fenton L, Scharer G, Van Coster R, Van Hove JL. A new mutation in MT-ND1 m.3928G>C p.V208L causes Leigh disease with infantile spasms. Mitochondrion. 2013;13:656–61. [PubMed: 24063851]
- Xu B, Li X, Du M, Zhou C, Fang H, Lyu J, Yang Y. Novel mutation of ND4 gene identified by targeted next-generation sequencing in patient with Leigh syndrome. J Hum Genet. 2017;62:291–7. [PubMed: 27761019]
- Yamashita S, Nishino I, Nonaka I, Goto Y. Genotype and phenotype analyses in 136 patients with single large-scale mitochondrial DNA deletions. J Hum Genet. 2008;53:598–606. [PubMed: 18414780]
- Zaha K, Matsumoto H, Itoh M, Saitsu H, Kato K, Kato M, Ogata S, Murayama K, Kishita Y, Mizuno Y, Kohda M, Nishino I, Ohtake A, Okazaki Y, Matsumoto N, Nonoyama S. DNM1L-related encephalopathy in infancy with Leigh syndrome-like phenotype and suppression-burst. Clin Genet. 2016;90:472–4. [PubMed: 27301544]
- Zeviani M, Amati P, Bresolin N, Antozzi C, Piccolo G, Toscano A, DiDonato S. Rapid detection of the A----G(8344) mutation of mtDNA in Italian families with myoclonus epilepsy and ragged-red fibers (MERRF). Am J Hum Genet. 1991;48:203–11. [PMC free article: PMC1683006] [PubMed: 1899320]
- Zhadanov SI, Grechanina EY, Grechanina YB, Gusar VA, Fedoseeva NP, Lebon S, Münnich A, Schurr TG. Fatal manifestation of a de novo ND5 mutation: insights into the pathogenetic mechanisms of mtDNA ND5 gene defects. Mitochondrion. 2007;7:260–6. [PubMed: 17317336]
- Zhang J, Liu H, Luo S, Lu Z, Chávez-Badiola A, Liu Z, Yang M, Merhi Z, Silber SJ, Munné S, Konstantinidis M, Wells D, Tan JJ, Huang T. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online. 2017;34:361–8. [PubMed: 28385334]
Publication Details
Author Information and Affiliations
Royal Children's Hospital
Victoria, Australia
University of Melbourne
Melbourne, Australia
UCL Great Ormond Street Institute of Child Health
London, United Kingdom
UCL Great Ormond Street Institute of Child Health
London, United Kingdom
Publication History
Initial Posting: October 30, 2003; Last Revision: May 4, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. 2003 Oct 30 [Updated 2023 May 4]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.