Parkinson Disease Overview
Lola Cook Shukla, MS, Jeanine Schulze, MS, Janice Farlow, BS, BA, Nathan D Pankratz, PhD, Joanne Wojcieszek, MD, and Tatiana Foroud, PhD.
Author Information and AffiliationsInitial Posting: May 25, 2004; Last Update: July 25, 2019.
Estimated reading time: 19 minutes
Summary
The purpose of this overview is to increase the awareness of clinicians regarding the genetics of Parkinson disease and related genetic counseling issues.
The following are the goals of this overview.
Goal 2.
Review the causes of Parkinson disease.
Goal 3.
Provide an evaluation strategy to identify the genetic cause of Parkinson disease in a proband.
Goal 4.
Inform genetic counseling for family members of an individual with Parkinson disease.
1. Clinical Characteristics of Parkinson Disease
Clinical Manifestations of Parkinson Disease
Parkinson disease, a neurodegenerative disorder, is characterized by rest tremor, muscle rigidity, slowed movement (bradykinesia), and often postural instability. Onset is typically unilateral and may include other abnormal movements such as postural or action tremor as well as limb dystonia. Common associated non-motor findings include insomnia, depression, anxiety, rapid-eye-movement (REM) sleep behavior disorder, fatigue, constipation, dysautonomia, and hyposmia. As part of a prodromal phase, non-motor features may predate formal diagnosis of Parkinson disease by years. With disease progression, bilateral manifestations, balance disturbances, and other complications such as levodopa-induced dyskinesia and motor fluctuations develop. Dementia and/or psychosis occur in 30%-40%.
The diagnosis of Parkinson disease is based on the clinical findings of bradykinesia plus rest tremor and/or rigidity. An important supportive diagnostic feature is an observable response to dopaminergic therapy. Dementia is not an exclusion criterion for diagnosis. While developed for use in a genetic research study, the diagnostic criteria summarized in the following citations are increasingly being used to guide diagnosis in a clinical setting [Postuma et al 2015, Postuma et al 2018].
In instances of diagnostic uncertainty, especially in early stages of disease, neuroimaging such as dopamine transporter single-photon emission computed tomography (DAT-SPECT) or fluoro-dopa positron emission tomography (PET) can be used to make a more definitive diagnosis of dopaminergic deficit [Ba & Martin 2015, Ibrahim et al 2016]. While these studies can document the presence of dopaminergic dysfunction, they cannot distinguish between Parkinson disease and other degenerative forms of parkinsonism such as those included in Parkinson plus syndromes (see ). DAT-SPECT is most commonly used to distinguish Parkinson disease from essential tremor; it can also be used to verify that a symptomatic individual has a non-degenerative form of parkinsonism, such as dopa-responsive dystonia. Laboratory or imaging studies such as brain MRI are useful only in excluding alternative diagnoses such as infarction, tumor, or normal-pressure hydrocephalus.
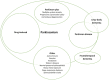
Parkinson disease: related terms in use in the clinical setting
The cardinal histopathologic feature of Parkinson disease is the loss of dopaminergic neurons in the substantia nigra with intracytoplasmic inclusions (alpha-synuclein- containing Lewy bodies) in the remaining intact nigral neurons.
Onset of Parkinson disease is most commonly around age 60 years but can vary. The following are generally accepted descriptions regarding age of onset:
Juvenile-onset Parkinson disease. Age <20 years
Early-onset adult Parkinson disease. Age 20-50 years
Late-onset adult Parkinson disease. Age >50 years
Various terms related to Parkinson disease used in the clinical setting are diagrammed in . Parkinsonism is the broadest term that refers to any disorder that includes bradykinesia, rigidity, tremor, and imbalance. "Parkinson plus" includes degenerative conditions in which parkinsonism is a major finding, such as multiple system atrophy, progressive supranuclear palsy, and corticobasal degeneration.
2. Causes of Parkinson Disease
Parkinson disease results from complex interplay of non-genetic and genetic factors. However, genetic factors are increasingly recognized as causative.
Typically Parkinson disease occurs in only one family member (i.e., a simplex case); less commonly it occurs in several family members (i.e., familial Parkinson disease). Historically, about 15% of individuals with Parkinson disease have a positive family history of PD.
An estimated 5%-10% of all Parkinson disease is attributed to pathogenic variants in single genes (monogenic Parkinson disease) (Table 1, Table 2). In addition to these variants, other genetic and environmental factors – known and unknown – may contribute to overall risk [Kalinderi et al 2016, Kim & Alcalay 2017].
Monogenic Parkinson Disease
Monogenic Parkinson disease can be inherited in an autosomal dominant, autosomal recessive, or, uncommonly, X-linked manner. Age of onset in the proband can be useful in distinguishing autosomal dominant PD (typically age >50 years) from autosomal recessive PD (typically age <40 years) [Kasten et al 2018]; however, age of onset can vary greatly between individuals with the same genetic variant.
Adult-onset Parkinson disease.
Table 1 lists the genes associated with early-onset adult PD (age 20-50 years) and late-onset adult PD (age >50 years). Table 1 uses the recommended International Parkinson and Movement Disorder Society Task Force for Nomenclature of Genetic Movement Disorders, in which the phenotype prefix, PARK, is followed by the italicized gene symbol (e.g., PARK-LRRK2) [Marras et al 2016].
Table 1.
Early-Onset and Late-Onset Adult Parkinson Disease: Monogenic Causes
View in own window
Gene 1 | PD Designation 2 | MOI | % of Adult PD | Comments | GeneReview / Reference / OMIM Entry |
|---|
GBA1 3
(formerly GBA) | PARK-GBA | AD | 3%-7%
(20% in AJ ancestry) | Onset age may be <50 yrs. Higher likelihood of cognitive impairment, atypical motor findings & severe progression Associated w/dementia w/Lewy bodies Variable penetrance dependent on age, variant, & ethnicity 4
| OMIM 606463 |
|
LRRK2
| PARK-LRRK2 | AD | 1%-2%
(13%-30% in AJ ancestry; 41% in African Berber ancestry) | Classic manifestations w/less non-motor involvement Variable penetrance dependent on age, variant, & ethnicity 5
|
LRRK2-Related PD
|
PARK7
(DJ1) | PARK-DJ1 | AR | Rare | Phenotype similar to PARK-Parkin ID &/or seizures occasionally Risk to heterozygotes unknown
| OMIM 606324 |
|
PINK1
| PARK-PINK1 | AR | Rare
(3.7% of early-onset adult PD) | Phenotype similar to PARK-Parkin Non-motor manifestations incl psychiatric features more common Heterozygotes may have ↑ PD risk.
|
PINK1 Type of Young-Onset PD
|
|
PRKN
| PARK-Parkin | AR | 1%
(4.6%-10.5% of early-onset adult PD) | Slow progression Can have lower-limb dystonia, dyskinesias, hyperreflexia Mild non-motor manifestations Heterozygotes may have ↑ PD risk.
|
Parkin Type of Early-Onset PD
|
|
SNCA
| PARK-SNCA | AD | Rare |
| OMIM 168601, 605543 |
|
VPS13C
| Not assigned | AR | Rare | Early-onset PD w/very rapid progression; truncating variants cause severe disease. | Puschmann [2017], Reed et al [2019] |
|
VPS35
| PARK-VPS35 | AD | Rare |
|
VPS35-Related PD
|
AD = autosomal dominant; AJ = Ashkenazi Jewish; AR = autosomal recessive; ID = intellectual disability; MOI = mode of inheritance; PD = Parkinson disease
- 1.
Genes are listed in alphabetic order.
- 2.
- 3.
There is some disagreement among researchers as to whether GBA1 (formerly GBA) should be classified as a monogenic disorder or, alternatively, a risk factor – due to its low age-related penetrance. However, of note, some GBA1 variants (including the fairly common Leu444Pro variant) have estimated penetrance approaching that of LRRK2 variants [Gan-Or et al 2015, Lee et al 2017].
- 4.
The estimated penetrance of the c.1226A>G (p.Asn409Ser) GBA1 variant is 6%-14% for all populations. Higher penetrance is reported for severe GBA1 variants [Gan-Or et al 2015].
- 5.
The estimated penetrance of the c.6055G>A (p.Gly2019Ser) LRRK2 variant is 25%-43% for all populations [Marder et al 2015, Lee et al 2017]. The estimated penetrance for other LRRK2 variants may be higher or lower, possibly related to the location of the variant within the gene [Trinh et al 2014].
Juvenile-onset Parkinson disease. Variants in several genes have been associated with juvenile-onset Parkinson disease (i.e., onset age generally <20 years but can vary) (Table 2). Inheritance of juvenile-onset PD is usually autosomal recessive; the clinical presentation often includes additional signs such as dystonia, spasticity, and dementia.
Table 2.
Juvenile-Onset Parkinson Disease: Monogenic Causes
View in own window
| Gene 1 | PD Designation 2 | MOI | % of Juvenile-Onset PD | Comments | GeneReview / Reference / OMIM Entry |
|---|
|
ATP13A2
| PARK-ATP13A2 | AR | Rare | Triad of spasticity, supranuclear gaze palsy, & dementia Wide variability Also referred to as Kufor-Rakeb syndrome or juvenile-onset atypical PD
| Park et al [2015], OMIM 606693 |
|
DNAJC6
| PARK-DNAJC6 | AR | Rare | 2 subtypes identified:
Slowly progressing, levodopa-responsive parkinsonism; onset in 3rd-4th decade Rare juvenile onset w/rapid disease progression; atypical features of hyperreflexia, seizures, & ID
Variants w/milder effect on protein function may cause adult early-onset PD w/few other symptoms. 3 |
DNAJC6 Parkinson Disease
|
|
FBXO7
| PARK-FBXO7 | AR | Rare | Juvenile or early-onset adult, rapidly progressive, may have corticospinal signs Early-onset parkinsonism w/bradykinesia in some families
| OMIM 260300 |
|
PODXL
| Not assigned | AR | 1 family reported | Levodopa-responsive PD w/no atypical features 4 |
Puschmann [2017]
|
|
SLC6A3
| DYT/PARK-
SLC6A3 | AR | Rare |
|
SLC6A3-Related Dopamine Transporter Deficiency Syndrome
|
| SYNJ1 5 | PARK-SYNJ1 | AR | Rare |
| OMIM 617389 |
AR = autosomal recessive; ID = intellectual disability; MOI = mode of inheritance; PD = Parkinson disease
- 1.
Genes are listed in alphabetic order.
- 2.
- 3.
Variants that are deleterious or have a large effect on protein function may lead to more severe, juvenile-onset phenotype, whereas those with a milder effect on protein function may lead to "adult early-onset PD with few or no additional signs and symptoms" [Puschmann 2017].
- 4.
Atypical features are neurologic manifestations including early cognitive decline, severe dystonia, corticospinal signs (hyperreflexia), spasticity, seizures, and oculomotor limitations – in addition to the rigidity, tremor, and bradykinesia that characterize PD.
- 5.
Other suspected genetic risk factors for Parkinson disease. Evidence that variants in other genes may increase the risk for Parkinson disease is less well supported [Kalia & Lang 2015, Ruiz-Martinez et al 2015, Zhang et al 2015, Lee & Hsu 2017, Puschmann 2017, Youn et al 2019]:
Genetic variants reported in single families include RIC3, TMEM230, and UCHL1.
Evidence is conflicting for EIF4G1 and DNAJC13.
Further confirmation is needed to clarify the risk associated with CHCHD2, GIGYF2, and HTRA2.
Additional studies are needed to confirm and clarify the role of variants in these genes in Parkinson disease causation [Kim & Alcalay 2017]. While these genes may appear on Parkinson disease multigene testing panels, it is suggested that they not be included in diagnostic testing because of their limited clinical utility (see Evaluation Strategies).
Susceptibility Genes for Parkinson Disease
In addition to the major known monogenic causes of Parkinson disease (Table 1, Table 2), additional genes and susceptibility loci have been identified through genome-wide association studies (GWAS) and other research. Furthermore, genetic modifiers may influence lifetime risk for PD or clinical aspects of PD such as age of onset and progression of motor and non-motor features [Davis et al 2016]. While these genetic modifiers may contribute in small ways to PD risk [Reed et al 2019], most are not yet clinically applicable and more research is needed to clarify their role in clinical diagnosis and risk assessment.
Variants in such susceptibility genes differ from variants in genes known to cause Parkinson disease: no variant in any of the susceptibility genes "causes" PD. Thus, such genes should not be included in diagnostic testing (see Evaluation Strategies).
Environmental Risk Factors for Parkinson Disease
One of the most important non-genetic factors contributing to Parkinson disease risk is advancing age. Epidemiologic studies have shown possible association of Parkinson disease with environmental factors including pesticide exposure, head injury, rural living, and infectious agents.
Environmental factors that may be associated with a lower risk for PD include tobacco smoking, caffeine consumption, use of nonsteroidal anti-inflammatory drugs (NSAID), high blood urate levels, and physical activity [Kalia & Lang 2015, Ascherio & Schwarzschild 2016].
3. Evaluation Strategies to Identify the Genetic Cause of Parkinson Disease in a Proband
Establishing a specific genetic cause of Parkinson disease:
Can aid in discussions of causation, recurrence risks, and research eligibility (see
Goal 5).
May provide some information about phenotype including prognosis of a particular monogenic cause of Parkinson disease (
Table 1,
Table 2).
Usually involves evaluation of medical and family histories, and molecular genetic testing. Physical examination may be less helpful in suggesting a specific genetic cause because of the overlap of clinical features [
Kasten et al 2018,
Trinh et al 2019].
Medical history. Age of onset and age at diagnosis may help distinguish autosomal dominant monogenic causes of PD and autosomal recessive monogenic causes of PD (Table 1, Table 2). History of another related diagnosis or overlapping condition may aid in diagnosis and guide genetic testing. For example, if dystonia is a predominant feature of the condition, this may suggest alternative testing. (See Dystonia Overview.)
Physical and mental examination. Typically, findings on a physical or mental examination will not entirely implicate a monogenic cause of Parkinson disease. However, the presence of certain clinical features can provide clues, such as significant cognitive impairment (Table 1, Table 2).
Family history. A three-generation family history should be obtained, with particular attention to any individual with a movement disorder and/or neurodegenerative disorder, with the following also noted:
Age of onset/diagnosis of disease, which may help distinguish autosomal dominant from autosomal recessive monogenic PD if genetic (
Table 1,
Table 2)
Age (or age at death), which will help distinguish unaffected individuals from individuals whose clinical status cannot be determined as they are younger than the typical age of onset for PD
Details and results of genetic testing of relatives
Ethnicity, as some populations are more likely to have certain causative pathogenic variants (see
Table 1)
Evaluation by a neurologist (preferably one specializing in movement disorders) of first-degree relatives who have findings concerning for PD
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel or single-gene testing) and less commonly, comprehensive genomic testing (exome sequencing, exome array, or genomic sequencing). Gene-targeted testing requires the clinician to hypothesize which gene(s) are likely involved, whereas genomic testing does not.
Single-gene or targeted variant testing. In a family with an established molecular diagnosis of PD (i.e., a PD-related pathogenic variant has been identified in an affected family member), targeted testing for the familial variant would be indicated. Gene-targeted testing may be appropriate in other limited circumstances (e.g., a family history of
Gaucher disease indicates an increased likelihood of
GBA1 (formerly
GBA) involvement and an African Berber ancestry indicates a higher likelihood of
LRRK2 involvement). However, single-gene testing is rarely useful and typically not recommended because of the significant overlap in clinical manifestations and age of onset in PD.
A multigene panel that includes some or all of the genes listed in
Table 1 and
Table 2 is most likely to identify a genetic cause of Parkinson disease while limiting identification of variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. Of note, given the rarity of some of the genes associated with Parkinson disease, some panels may not include all the genes included in
Table 1 and
Table 2. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Comprehensive
genomic testing (which does not require the clinician to determine which gene[s] are likely involved) may be considered; however, genomic testing in Parkinson disease is still being evaluated, and yield and utility remain to be determined [
Schormair et al 2018,
Trinh et al 2019].
For an introduction to comprehensive genomic testing click
here. More detailed information for clinicians ordering genomic testing can be found
here.
4. Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Typical, Late-Onset Parkinson Disease of Unknown Cause
Typical, late-onset Parkinson disease of unknown cause is generally presumed to be multifactorial in origin (i.e., the result of combined contributions of genetic and environmental factors). Recurrence risk counseling for family members of individuals with presumed multifactorial Parkinson disease (often referred to as "idiopathic" or "sporadic" in the Parkinson disease literature; see Note) and their family members must be empiric and relatively nonspecific. PD is fairly common in the general population and the cumulative lifetime risk (i.e., to age 85 years) of developing PD approaches 4% [Tysnes & Storstein 2017, Deng et al 2018, Licher et al 2019]. Taking into account family history, the cumulative lifetime risk to first-degree relatives (parents, sibs, and offspring) of an individual with typical, late-onset Parkinson disease of unknown cause who represents a simplex case (i.e., the only affected family member) is estimated be about twofold greater (i.e., 8%) [Gaare et al 2017, Liu et al 2018].
Note: "Idiopathic Parkinson disease" and "sporadic Parkinson disease" are terms used in the Parkinson disease medical literature to describe Parkinson disease of unknown cause diagnosed in an individual with a negative family history. Because future advances in the understanding of genetic risk factors are likely to identify genetic causes / risk factors for some Parkinson disease currently considered "idiopathic" or "sporadic," these terms are generally not used in this GeneReview. Instead, the term "Parkinson disease of unknown cause representing a simplex case" is preferred because "simplex case" refers specifically to an individual with no family history of Parkinson disease without implying presence or absence of a genetic factor or a specific recurrence risk.
Monogenic (Mendelian) Parkinson Disease
Mode of Inheritance
Monogenic Parkinson disease is inherited in an autosomal dominant, autosomal recessive, or X-linked manner. (See X-Linked Dystonia-Parkinsonism for a review of X-linked inheritance.)
Autosomal Dominant Inheritance – Risk to Family Members
Parkinson disease caused by a pathogenic variant in one of four genes – GBA1 (formerly GBA), LRRK2, SNCA, or VPS35 – is inherited in an autosomal dominant manner (Table 1). This section refers only to families having a known Parkinson disease-causing pathogenic variant in one of these four genes.
Parents of a proband
Almost all individuals diagnosed with autosomal dominant Parkinson disease inherited a pathogenic variant from a parent, who may or not be affected.
In very rare instances, a proband diagnosed with autosomal dominant Parkinson disease has the disorder as the result of a de novo pathogenic variant.
If the parents of a proband undergo molecular genetic testing for the pathogenic variant found in the proband and the pathogenic variant cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or the theoretic possibility of germline mosaicism in a parent.
The family history of some individuals diagnosed with autosomal dominant Parkinson disease may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, late onset of the disease in the affected parent, variable expression, or (especially in GBA1- and LRRK2-related Parkinson disease) reduced penetrance. Therefore, an apparently negative family history can only be confirmed with parental molecular genetic testing.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the proband is known to have the pathogenic variant identified in the proband, the risk to each sib of having inherited the variant is 50%. While all sibs who inherit a familial pathogenic variant will be at increased risk for Parkinson disease, not all heterozygous sibs will manifest the disease; both reduced, age-related penetrance and intrafamilial clinical variability are observed in autosomal dominant Parkinson disease.
If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
If the parents have not been tested for the pathogenic variant identified in the proband but are clinically unaffected, sibs are still presumed to be at increased risk for Parkinson disease because of the possibility of reduced penetrance in a heterozygous parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband
Autosomal Recessive Inheritance – Risk to Family Members
Parkinson disease caused by biallelic pathogenic variants in ATP13A2, DJ1, DNAJC6, FBXO7, PINK1, PODXL, PRKN, SLC6A3, SYNJ1, or VPS13C is inherited in an autosomal recessive manner (Table 1, Table 2).
Parents of a proband
Sibs of a proband
At conception, each sib of an individual with autosomal recessive Parkinson disease has a 25% chance of inheriting both pathogenic variants and being affected, a 50% chance of inheriting one pathogenic variant and being heterozygous, and a 25% chance of inheriting neither pathogenic variant.
Heterozygotes for a pathogenic variant associated with adult-onset Parkinson disease may be at increased the risk for Parkinson disease (see
AR Inheritance,
Parents of a proband).
Offspring of a proband. The offspring of an individual with an autosomal recessive Parkinson disease are obligate heterozygotes for a pathogenic variant. Heterozygotes for a pathogenic variant associated with adult-onset Parkinson disease may be at increased the risk of Parkinson disease (see AR Inheritance, Parents of a proband).
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
American Parkinson Disease Association (APDA)
Phone: 800-223-2732
Fax: 718-981-4399
Email: apda@apdaparkinson.org
Fox Trial Finder
MedlinePlus
Michael J. Fox Foundation for Parkinson's Research
Phone: 800-708-7644 (toll-free)
Email: info@michaeljfox.org
National Institute of Neurological Disorders and Stroke (NINDS)
Parkinson’s Disease Society (UK)
United Kingdom
Phone: 0808 800 0303
Email: hello@parkinsons.org.uk
Parkinson's Foundation
Phone: 800-4PD-INFO (473-4636)
Email: contact@parkinson.org
5. Participation in Targeted Therapeutic Clinical Trials
Several gene-targeted therapies for Parkinson disease are currently in development [Sardi et al 2018]. Individuals may be candidates for a given Parkinson disease clinical trial if they undergo molecular genetic testing and a pathogenic variant in the gene under study is identified.
Clinical trials for individuals with Parkinson disease and GBA1 (formerly GBA) or LRRK2 pathogenic variants are now ongoing (www.clinicaltrials.gov). Testing for common GBA1 or LRRK2 variants is available through direct-to-consumer (DTC) testing or, more formally, through a health care professional. See GeneReviews
Resources for Genetics Professionals for an overview of DTC testing.
Chapter Notes
Revision History
25 July 2019 (bp) Comprehensive update posted live
27 February 2014 (me) Comprehensive update posted live
16 October 2006 (me) Comprehensive update posted live
25 May 2004 (me) Overview posted live
12 November 2003 (tmf) Original submission
References
Literature Cited
Ascherio
A, Schwarzschild
MA. The epidemiology of Parkinson's disease: risk factors and prevention.
Lancet Neurol.
2016;15:1257-72.
[
PubMed: 27751556]
Ba
F, Martin
WRW. Dopamine transporter imaging as a diagnostic tool for parkinsonism and related disorders in clinical practice.
Parkinsonism Relat Disord
2015;21:87-94.
[
PubMed: 25487733]
Davis
AA, Andruska
KM, Benitez
BA, Racette
BA, Perlmutter
JS, Cruchaga
C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression.
Neurobiol Aging.
2016;37:209.e1-e7.
[
PMC free article: PMC4688052] [
PubMed: 26601739]
Deng
H, Wang
P, Jankovic
J.
The genetics of Parkinson disease.
Ageing Res Rev.
2018;42:72-85.
[
PubMed: 29288112]
Gaare
JJ, Skeie
GO, Tzoulis
C, Larsen
JP, Tysnes
OB. Familial aggregation of Parkinson’s disease may affect progression of motor symptoms and dementia.
Mov Disord.
2017;32:241-5.
[
PubMed: 27862270]
Gan-Or
Z, Amshalom
I, Kilarski
LL, Bar-Shira
A, Gana-Weisz
M, Mirelman
A, Marder
K, Bressman
S, Giladi
N, Orr-Urtreger
A. Differential effects of severe vs mild GBA mutations on Parkinson disease.
Neurology, 2015;84:880-7.
[
PMC free article: PMC4351661] [
PubMed: 25653295]
Ibrahim
N, Kusmirek
J, Struck
A, Floberg
JM, Perlman
SB, Gallagher
C, Hall
LT. The sensitivity and specificity of F-DOPA PET in a movement disorder clinic.
Am J Nucl Med Mol Imaging.
2016;6:102-9.
[
PMC free article: PMC4749509] [
PubMed: 27069770]
Kalia
LV, Lang
AE. Parkinson’s disease.
Lancet.
2015;386:896-912.
[
PubMed: 25904081]
Kalinderi
K, Bostantjopoulou
S, Fidani
L. The genetic background of Parkinson’s disease: current progress and future prospects.
Acta Neurol Scand.
2016;134:314-26.
[
PubMed: 26869347]
Kasten
M, Hartmann
C, Hampf
J, Schaake
S, Westenberger
A, Vollstedt
EJ, Balck
A, Domingo
A, Vulinovic
F, Dulovic
M, Zorn
I, Madoev
H, Zehnle
H, Lembeck
CM, Schawe
L, Reginold
J, Huang
J, König
IR, Bertram
L, Marras
C, Lohmann
K, Lill
CM, Klein
C. Genotype-phenotype relations for the Parkinson's disease genes Parkin, PINK1, DJ1: MDSGene Systematic Review.
Mov Disord.
2018;33:730-41.
[
PubMed: 29644727]
Kim
CY, Alcalay
RN. Genetic forms of Parkinson’s disease.
Semin Neurol.
2017;37:135-46.
[
PubMed: 28511254]
Lee
AJ, Wang
Y, Alcalay
RN, Mejia-Santana
H, Saunders-Pullman
R, Bressman
S, Corvol
JC, Brice
A, Lesage
S, Mangone
G, Tolosa
E, Pont-Sunyer
C, Vilas
D, Schüle
B, Kausar
F, Foroud
T, Berg
D, Brockmann
K, Goldwurm
S, Siri
C, Asselta
R, Ruiz-Martinez
J, Mondragón
E, Marras
C, Ghate
T, Giladi
N, Mirelman
A, Marder
K; Michael
J. Fox LRRK2 Cohort Consortium. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry.
Mov Disord.
2017;32:1432–8.
[
PMC free article: PMC5656509] [
PubMed: 28639421]
Lee
YC, Hsu
SD. Familial mutations and post-translational modifications of UCH-L1 in Parkinson's disease and neurodegenerative disorders.
Curr Protein Pept Sci.
2017;18:733-45.
[
PubMed: 26899237]
Licher
S, Darweesh
SKL, Wolters
FJ, Fani
L, Heshmatollah
A, Mutlu
U, Koudstaal
PJ, Heeringa
J, Leening
MJG, Ikram
MK, Ikram
MA. Lifetime risk of common neurological diseases in the elderly population.
J Neurol Neurosurg Psychiatry.
2019;90:148-56.
[
PubMed: 30279211]
Liu
FC, Lin
HT, Kuo
CF, Hsieh
MY, See
LC, Yu
HP. Familial aggregation of Parkinson's disease and coaggregation with neuropsychiatric diseases: a population-based cohort study.
Clin Epidemiol.
2018;10:631-41.
[
PMC free article: PMC5985793] [
PubMed: 29881310]
Marder
K, Wang
Y, Alcalay
RN, Mejia-Santana
H, Tang
MX, Lee
A, Raymond
D, Mirelman
A, Saunders-Pullman
R, Clark
L, Ozelius
L, Orr-Urtreger
A, Giladi
N, Bressman
S, et al.
Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium.
Neurology.
2015;85:89-95.
[
PMC free article: PMC4501942] [
PubMed: 26062626]
Marras
C, Lang
A, van de Warrenburg
BP, Sue
CM, Tabrizi
SJ, Bertram
L, Mercimek-Mahmutoglu
S, Ebrahimi-Fakhari
D, Warner
TT, Durr
A, Assmann
B, Lohmann
K, Kostic
V, Klein
C. Nomenclature of genetic movement disorders: recommendations of the International Parkinson and Movement Disorder Society task force.
Mov Disord.
2016;31:436-57.
[
PubMed: 27079681]
Park
J-S, Blair
NF, Sue
CM. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms.
Mov Disord.
2015;30:770-9.
[
PubMed: 25900096]
Postuma
RB, Berg
D, Stern
M, Poewe
W, Olanow
CW, Oertel
W, Obeso
J, Marek
K, Litvan
I, Lang
AE, Halliday
G, Goetz
CG, Gasser
T, Dubois
B, Chan
P, Bloem
BR, Adler
CH, Deuschl
G. MDS clinical diagnostic criteria for Parkinson's disease.
Mov Disord.
2015;30:1591-601.
[
PubMed: 26474316]
Postuma
RB, Poewe
W, Litvan
I, Lewis
S, Lang
AE, Halliday
G, Goetz
CG, Chan
P, Slow
E, Seppi
K, Schaffer
E, Rios-Romenets
S, Mi
T, Maetzler
C, Li
Y, Heim
B, Bledsoe
IO, Berg
D. Validation of the MDS clinical diagnostic criteria for Parkinson's disease.
Mov Disord.
2018;33:1601-8.
[
PubMed: 30145797]
Rahbari
R, Wuster
A, Lindsay
SJ, Hardwick
RJ, Alexandrov
LB, Turki
SA, Dominiczak
A, Morris
A, Porteous
D, Smith
B, Stratton
MR, Hurles
ME, et al.
Timing, rates, and spectra of human germline mutation.
Nat Genet.
2016;48:126-33.
[
PMC free article: PMC4731925] [
PubMed: 26656846]
Ruiz-Martinez
J, Krebs
CE, Makarov
V, Gorostidi
A, Martí-Massó
JF, Paisán-Ruiz
C. GIGYF2 mutation in late-onset Parkinson’s disease with cognitive impairment.
J Hum Genet.
2015;60:637-40.
[
PMC free article: PMC4624020] [
PubMed: 26134514]
Schormair
B, Kemlink
D, Mollenhauer
B, Fiala
O, Machetanz
G, Roth
J, Berutti
R, Strom
TM, Haslinger
B, Trenkwalder
C, Zahorakova
D, Martasek
P, Ruzicka
E, Winkelmann
J. Diagnostic exome sequencing in early-onset Parkinson's disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson's disease.
Clin Genet.
2018;93:603-12.
[
PubMed: 28862745]
Trinh J, Guelle I, Farrer MJ. Disease penetrance of late-onset parkinsonism: a meta-analysis. 2014;71:1535-9. [
PubMed: 25330418]
Trinh
J, Lohmann
K, Baumann
H, Balck
A, Borsche
M, Brüggemann
N, Dure
L, Dean
M, Volkmann
J, Tunc
S, Prasuhn
J, Pawlack
H, Imhoff
S, Lill
CM, Kasten
M, Bauer
P, Rolfs
A; International Parkinson's Disease Genomics Consortium (IPDGC), Klein C. Utility and implications of exome sequencing in early-onset Parkinson's disease.
Mov Disord.
2019;34:133-7.
[
PMC free article: PMC8950081] [
PubMed: 30537300]
Tysnes
OB, Storstein
A. Epidemiology of Parkinson’s disease.
J Neural Transm (Vienna). 2017;124:901-5.
[
PubMed: 28150045]
Youn
J, Lee
C, Oh
E, Park
J, Kim
JS, Kim
HT, Cho
JW, Park
WY, Jang
W, Ki
CS. Genetic variants of PARK genes in Korean patients with early-onset Parkinson’s disease.
Neurobiol Aging.
2019; 75:224.e9-224.e15
[
PubMed: 30502028]
Zhang
Y, Sun
Q-Y, Yu
R-H, Guo
J-F, Tang
B-S, Yan
X-X. The contribution of GIGYF2 to Parkinson’s disease: a meta-analysis.
Neurol Sci.
2015;36:2073-9.
[
PubMed: 26152800]