Clinical Description
Neonatal respiratory compromise. Neonates with diastrophic dysplasia (DTD) may experience respiratory insufficiency because of the small rib cage and tracheal instability and collapsibility. Mechanical ventilation is required in a significant proportion of infants. Mortality in the first months of life is increased, mainly because of respiratory complications such as pneumonia, including aspiration pneumonia.
Musculoskeletal manifestations. The tendons, ligaments, and joint capsules are tighter and shorter than normal, causing restricted joint mobility. Peltonen et al [2003] reported a high prevalence of congenital aplasia of menisci and cruciate ligaments within the knee joints. Pretibial dimples may be present, possibly a consequence of reduced intrauterine movement.
Joint contractures and spine deformity tend to worsen with age. Painful degenerative arthrosis of the hip is common in young adults. Anterior tilting of the pelvis may occur as a consequence and exacerbate the lumbar lordosis. The spine frequently develops excessive lumbar lordosis, thoracolumbar kyphosis, and scoliosis. In anteroposterior radiographs of the lumbar spine, a decrease of the vertebral interpedicular distance is almost invariably observed; however, related neurologic symptoms are only rarely observed [Remes et al 2004, Shafi et al 2023].
The knee may be unstable in childhood; flexion contractures develop with progressive valgus deformity and lateral positioning of the patella. The development and position of the patella may determine whether contraction of the quadriceps muscle results in extension of the knee or paradoxic flexion of the knee. If paradoxic flexion occurs, severe difficulty with walking results [Remes et al 2004].
Because of foot deformities (clubfoot) and shortened tendons, many adults with DTD are unable to place their heels on the ground, and thus stand solely on their metatarsals and toes.
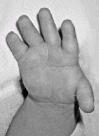
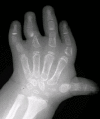
Brachydactyly, ulnar deviation, phalangeal synostosis, and ankylosis of the fingers with significant disability may be observed. Phalangeal synostosis, usually between proximal and middle phalanges, develops in those fingers that have an abnormal phalangeal patterning at birth, including so-called delta-shaped phalanges that usually lack a proper joint space. Often, newborns with DTD lack phalangeal flexion creases (see ), a sign of marked reduction of joint motion already present at early developmental stages. The thumb may be placed more proximally than usual and may be hypotonic and thus weak. As a consequence, some individuals may have difficulty opposing the thumb and the index finger to accomplish a pincer grasp. In older children and adults, ulnar deviation of the second finger frequently occurs together with radial deviation of the fifth finger (clinodactyly), giving a characteristic "brackets" appearance.
In addition to the skeletal abnormalities, a mild degree of muscular hypoplasia of the thighs and legs is common.
Facial features. The forehead is broad with a high anterior hairline; the palpebral fissures may be downslanting; the nose is long and thin with hypoplastic alae nasi; the facial tissues are tight; the mouth is small, and the mandible normally developed. Cystic ear swelling is frequent and can be associated with inflammation and pain [Cushing et al 2011].
Adult stature ranged between 100 and 140 cm in an early review of Americans and Europeans with DTD. A 1982 study reported a mean adult height of 118 cm [Horton et al 1982]; while a study of Finnish individuals with DTD (who are genetically homogeneous at the SLC26A2 locus) revealed a mean adult height of 136 cm for males and 129 cm for females [Mäkitie & Kaitila 1997]. The discrepancy in mean height between the older studies and the later Finnish study may be the result of variant heterogeneity or may reflect bias of ascertainment of more severely affected individuals in the older studies. It must be noted that the usefulness of such growth curves in predicting adult height is limited by the occurrence of many different allelic combinations.
Neurologic complications may occur, particularly in the cervical region. Cervical kyphosis is seen in lateral radiographs in most newborns; in most children, the kyphosis becomes less severe over the first three to five years of life, but in some instances severe cervical kyphosis may lead to spinal cord compression, either spontaneously or during endotracheal intubation, which requires hyperextension of the neck. A newborn with DTD and severe cervical kyphosis died immediately after birth of respiratory insufficiency; autopsy revealed neuronal degeneration and gliosis of the cervical spinal cord that had developed before birth.
MRI findings have confirmed that in individuals with DTD, the foramen magnum is of normal size but the cervical spinal canal is narrowed. Individual cervical vertebral bodies (usually C3 to C5) may be hypoplastic, but the frequently observed kyphosis is not explained by changes of the vertebral bodies and may thus be the consequence of abnormal intervertebral disks. The rate of spontaneous correction of cervical kyphosis is rather high. MRI studies have shown a peculiar signal anomaly of intervertebral disks, suggesting reduced water content. This anomaly may be the consequence of reduced proteoglycan sulfation.
Cervical spina bifida occulta has been frequently reported in individuals with DTD.
Mental development and intelligence are normal; numerous individuals affected by DTD attain high academic and social recognition or success in the arts.
Hearing loss is unusual in individuals with DTD, although it may be overestimated if studies are based on small cohorts [Tunkel et al 2012].
Vision defects are seldom observed, although a tendency toward myopia has been reported.
Genotype-Phenotype Correlations
Genotype-phenotype correlations indicate that the amount of residual activity of the sulfate transporter modulates the phenotype in this spectrum of disorders, which extends from lethal achondrogenesis type 1B (ACG1B) to SLC26A2-related multiple epiphyseal dysplasia (SLC26A2-MED). Homozygosity or compound heterozygosity for pathogenic variants predicting stop codons or structural pathogenic variants in transmembrane domains of the sulfate transporter are associated with ACG1B, while pathogenic variants located in extracellular loops, in the cytoplasmic tail of the protein, or in the regulatory 5'-flanking region of the gene result in less severe phenotypes [Superti-Furga et al 1996, Karniski 2001, Maeda et al 2006].
The pathogenic variant p.Arg279Trp is the most common SLC26A2 variant found outside of Finland (45% of alleles); it results in the mild SLC26A2-MED phenotype when homozygous and mostly in the DTD and SLC26A2-related
atelosteogenesis phenotypes when found in the compound heterozygous state [Barbosa et al 2011].
Pathogenic variant p.Arg178Ter is the second most common variant (9% of alleles) and is associated with a more severe DTD phenotype or even the perinatal-lethal SLC26A2-related atelosteogenesis phenotype, particularly when combined in trans with the p.Arg279Trp variant.
Pathogenic variants p.Cys653Ser and c.-26+2T>C are the third most common variants (8% of alleles).
Pathogenic variant p.Cys653Ser results in SLC26A2-MED when homozygous and in SLC26A2-MED or DTD when present in trans with other pathogenic variants [Czarny-Ratajczak et al 2010].
Pathogenic variant c.-26+2T>C is sometimes referred to as the "Finnish" variant because it is much more frequent in Finland than in the remainder of the world population. It produces low levels of correctly spliced mRNA and results in DTD when homozygous. It is the only variant that has been identified in all four SLC26A2-related dysplasias, in compound heterozygosity with mild (SLC26A2-MED and DTD) or severe (SLC26A2-related atelosteogenesis and ACG1B) alleles [Dwyer et al 2010].
The same pathogenic variants found in the ACG1B phenotype can also be found in the milder phenotypes (SLC26A2-related atelosteogenesis and DTD) if the second allele is a relatively mild variant. Indeed, missense variants located outside of the transmembrane domain of the sulfate transporter are often associated with a residual activity that can "rescue" the effect of a null allele [Rossi & Superti-Furga 2001].