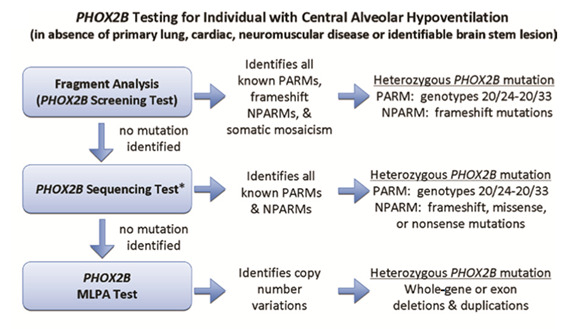
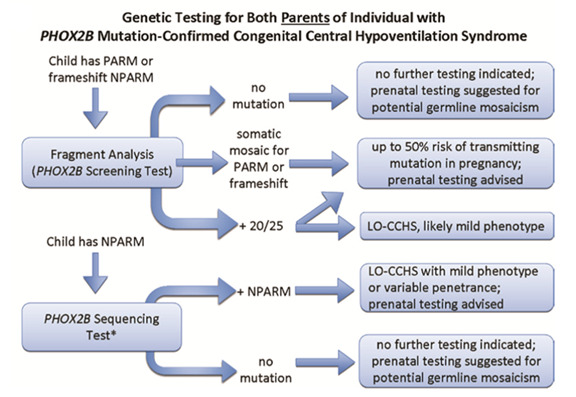
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with CCHS, the evaluations by phenotype summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) and text that follows are recommended to be performed every six months until age three years, then annually thereafter at a minimum.
Table 4.
Congenital Central Hypoventilation Syndrome: Recommended Evaluations by Genotype Following Initial Diagnosis
View in own window
| PHOX2B Variant | Comprehensive Physiologic Testing 1 | Neuro-
cognitive
Assessment | 72-hr Holter
& Echo-
cardiogram | Hirschsprung Disease 2, 3 | Neural
Crest
Tumors |
|---|
|
PARM genotypes
|
20/24-20/25
| X | X | X | | |
|
20/26
| X | X | X | X | |
|
20/27
| X | X | X | X | |
|
20/28-20/33
| X | X | X | X | X 4 |
|
NPARM
| X | X | X | X | X 4 |
|
Deletion/duplication 5
| X | X | X | X | X 4 |
NPARM = non-polyalanine repeat expansion mutation (i.e., missense, nonsense, frameshift, stop codon, splice site); PARM = polyalanine repeat expansion mutation with number of repeats on each allele (i.e., 20/24-20/33)
- 1.
Evaluations asleep and awake during age-appropriate activities of daily living with exogenous and endogenous ventilatory challenges and autonomic testing
- 2.
Evaluation as per standard protocols
- 3.
A punch biopsy in the neonatal period or a full thickness biopsy later should be considered based on symptoms and PHOX2B variant/genotype (see Table 2).
- 4.
Chest and abdominal imaging to detect ganglioneuroblastomas and ganglioneuroma; chest and abdominal imaging and urine catecholamines to detect neuroblastomas
- 5.
Exon or whole-gene deletion or duplication
Evaluations should also include assessment in a pediatric respiratory physiology laboratory experienced in the diagnosis and care of individuals with CCHS, including the following:
Clinical study of spontaneous breathing awake and asleep including (at a minimum) tidal volume, respiratory inductance plethysmography of the chest and abdomen, hemoglobin saturation with pulse waveform, end-tidal carbon dioxide level with visible waveform, electrocardiogram, blood pressure, cerebral regional blood flow/oxygenation, and appropriate sleep-state staging measures
Evaluation of the awake and asleep physiologic responses to exogenous and endogenous challenges of hypercarbia and/or hypoxemia in varied age-appropriate activities of daily living
Venous or arterial blood gas or serum bicarbonate level to look for elevated carbon dioxide content at the time of presentation
Hemoglobin, hematocrit, and reticulocyte count to assess for polycythemia
72-hour Holter recording to assess for abrupt, prolonged asystoles
Echocardiogram to assess for changes consistent with right ventricular hypertrophy and cor pulmonale
Neurocognitive assessment to determine baseline function
Comprehensive autonomic testing of all organ systems regulated by the autonomic nervous system, including but not limited to pupillometry, head up-tilt testing, thermoregulatory chamber sweat testing, Q-Sweat testing, heart rate deep breathing, Valsalva maneuver, and measures of cerebral regional blood flow and blood pressure in activities of daily living as well as orthostatic testing
Consultation with a genetics professional (e.g., medical geneticist, certified genetic counselor, or certified advanced genetic nurse) is recommended to inform affected individuals and their families about the nature, mode of inheritance, and implications of CCHS in order to facilitate medical and personal decision making.
Treatment of Manifestations
Management by multidisciplinary specialists including pediatric pulmonology, sleep medicine, cardiology, oncology, ophthalmology, gastroenterology, neurodevelopmental psychology, and neurology is recommended.
Ventilatory support. The treatment goals for neonatal-onset CCHS are to secure the airway and to use chronic artificial ventilatory support at home to compensate for the hypoventilation and the altered/absent ventilatory responses to hypoxemia and hypercarbia. Of note, although oxygen administration without artificial ventilation improves the PaO2 (partial pressure of oxygen in arterial blood) and relieves cyanosis, it may worsen the hypoventilation. Supplemental oxygen alone is NOT a treatment of hypoventilation.
Because individuals with CCHS may experience severe hypoventilation or complete respiratory arrest and, thus, the sequelae of hypoxemia, they require monitoring of objective measures of oxygenation (i.e., peripheral pulse oximeter SpO2) and ventilation (i.e., PETCO2 monitor) continuously during sleep and at regular intervals while awake regardless of mode of ventilatory support in-hospital and in-home. They also require observation and continuous care – especially during all sleep – by an RN trained and experienced in ventilator management and life support.
For each of the options listed below, the goal is to provide the affected individual with the technology optimal for the individual's lifestyle needs.
Typically, the infant needing ventilatory support 24 hours/day is most safely and effectively supported via tracheostomy and use of a home mechanical ventilator. Tracheostomy is also recommended for children and adults who require artificial ventilation during sleep only, though transition to mask ventilation is a consideration in a subset of individuals after maturation of the facial configuration.
As toddlers who require continuous ventilatory support become ambulatory, diaphragm pacing by phrenic nerve stimulation can be considered to allow for increased mobility and improved quality of life. Diaphragm pacing is not typically recommended for the young child who requires only nighttime ventilatory support because the benefits do not outweigh the risks; however, for a subset of older adolescents and young adults, this could be an appropriate consideration. Tracheal decannulation is not assured in affected individuals who use diaphragm pacing during sleep, especially young children [Valika et al 2019].
Diaphragm pacers for the active child with CCHS should be implanted at each phrenic nerve in the chest, ideally by thoracoscopic technique by highly experienced surgeons and phrenic nerve-diaphragm pacer teams [
Weese-Mayer et al 1996,
Shaul et al 2002,
Chin et al 2012].
Older infants, toddlers, and children with diaphragm pacers should be assessed for use of a speaking valve (Tracoe or Passy-Muir one-way speaking valve) while awake, allowing for vocalization and use of the upper airway on exhalation.
Children with diaphragm pacers may be assessed for capping of the tracheostomy tube while awake and paced, thereby allowing for inspiration and exhalation via the upper airway; tracheostomy is typically still required for mechanical ventilation during sleep to avoid upper-airway obstruction and physiologic compromise.
Although not yet accomplished, the older child with an entirely normal airway may be able to eliminate the need for a tracheostomy by relying on diaphragm pacing while awake and on mask ventilation while asleep; however, such a child may require interim endotracheal intubation to allow for optimal oxygenation and ventilation during acute illness that requires more aggressive ventilatory management.
Cooperative older children who consistently require ventilatory support only while sleeping may be candidates for noninvasive support with either mask ventilation or negative-pressure ventilation; however, this must be done with careful consideration of the individual child's needs. If successful, tracheal decannulation can be considered (with the caveat that in the event of severe illness, interim endotracheal intubation may be required in a pediatric intensive care unit).
The child who normally requires ventilatory support during sleep only may, during an intercurrent illness, also require artificial ventilation both awake and asleep.
Note: While Straus et al [2010] reported that the ventilatory response to hypercarbia appeared to improve with the use of oral contraceptives in two young women heterozygous for PARM genotypes 20/25 and 20/26, ongoing studies have not confirmed this report.
Cardiac. Prolonged transient asystoles may be asymptomatic or present as syncope and/or staring spells and may be of such significant duration (≥3 seconds) as to warrant placement of a cardiac pacemaker for management [Silvestri et al 2000, Gronli et al 2008].
Hirschsprung disease, altered esophageal motility/dysphagia, and severe constipation are treated in the standard manner.
Tumors of neural crest origin. Neuroblastomas are removed surgically and followed by chemotherapy if they have advanced beyond Stage 1. Other tumors of neural crest origin are treated individually by location and type, though surgical removal is typically recommended even for benign tumors.
Abnormal pupillary reactivity. With so much light exposure in daily life from LED lights and screen time in educational settings, computer-based work environments, and mobile devices, abnormal pupillary reactivity may necessitate protective eye wear.
Because individuals with CCHS may receive medications expected to alter pupillary light response or because of potential use of illicit drugs, it is essential that medical personnel appreciate the innate abnormalities of pupillary reactivity to light. Use of pupillometry at the time of each clinical evaluation allows reliable objective documentation of the individual's signature pupillary light response relative to an established clinical baseline and published norms.
Neurocognitive. The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life.
Agents/Circumstances to Avoid
Ideally, children with CCHS should not go swimming. If they do, they should be carefully supervised, regardless of the presence or absence of a tracheostomy.
Children with CCHS should not compete in underwater swimming contests as they cannot perceive the asphyxia that occurs with drowning and breath-holding and, therefore, are likely to swim longer and farther than children without CCHS, thereby increasing the risk of drowning.
Breath-holding contests may lead to asphyxia and/or death.
Alcohol (respiratory depression), recreational drugs (varied effects), and prescribed as well as non-prescribed medications/sedatives/anesthetics that could induce respiratory depression should be avoided [Chen et al 2006].