Summary
Clinical characteristics.
HFE-related hemochromatosis (HFE HC) is characterized by increased intestinal iron absorption and increased recycling of iron derived from senescent red blood cells. The phenotypic spectrum of HFE HC includes clinical HFE HC (increased serum ferritin and transferrin saturation and end-organ damage secondary to iron overload), biochemical HFE HC (increased serum ferritin and transferrin saturation without end-organ damage), and non-penetrant HFE HC (neither clinical manifestations of HFE HC nor iron overload are present, although elevated transferrin saturation may occur). Clinical HFE HC is characterized by excessive iron in the liver, pancreas, heart, skin, joints, and anterior pituitary gland. In untreated individuals, early manifestations include weakness, chronic fatigue, abdominal pain, weight loss, arthralgias, and diabetes mellitus. Individuals with HFE HC have an increased risk of cirrhosis when their serum ferritin is higher than 1,000 µg/L. Other findings of severe iron overload include hypogonadism, congestive heart failure, arrhythmias, and progressive increase in skin pigmentation. Clinical HFE HC is more common in males than females.
Diagnosis/testing.
The diagnosis of HFE HC is established in most persons with characteristic laboratory and/or clinical features by identification of HFE p.Cys282Tyr homozygosity.
Management.
Targeted therapies: In individuals with clinical HFE HC, a major initial treatment goal is to remove excess iron by phlebotomy to achieve serum ferritin 50-100 µg/L. Erythrocytapheresis is sometimes used to remove excess iron. Iron chelation therapy may be used to treat individuals intolerant of phlebotomy or erythrocytapheresis or those with symptomatic anemia. In individuals with biochemical HFE HC, phlebotomy is indicated when serum ferritin exceeds 300 µg/L (males) and 200 µg/L (females).
Treatment of manifestations: Management of complications of HFE HC (arthropathy, diabetes mellitus, cirrhosis, hypogonadism, and cardiomyopathy) do not differ from the management of these conditions in persons without HFE HC. Treatment of arthropathy includes nonsteroidal anti-inflammatory drugs, physiotherapy, and joint replacement. Standard treatment for diabetes mellitus. Vaccination for hepatitis A and B. Treatment of hepatitis B or C with standard antiviral agents may reduce liver injury. In individuals with cirrhosis, evaluation and treatment of complications is warranted, including endoscopic surveillance of varices; prophylaxis with nonselective beta-blockers; salt restriction and diuretics for ascites, with paracentesis and portosystemic shunts as needed; antibiotics to decrease risk of spontaneous bacterial peritonitis; and low-protein diet for hepatic encephalopathy with lactulose and rifaximin as indicated. Orthotopic liver transplant for end-stage liver disease. Hormone replacement therapy for hypogonadism; gonadotropins for infertility. Standard treatments for heart failure and arrhythmias in individuals with cardiomyopathy.
Surveillance: In individuals with clinical HFE HC, after serum ferritin is ≤100 µg/L, monitor serum ferritin concentration every three to four months, and maintain serum ferritin <300 µg/L (males) and <200 µg/L (females) thereafter; joint radiographs as needed; in those with diabetes mellitus assess for complications every six to 12 months; liver enzyme tests every six to 12 months; standard evaluations for primary liver cancer in those with cirrhosis; assessment for hormonal deficiency in those with hypogonadism; cardiac assessment in those with cardiac siderosis annually or as needed. In individuals with biochemical HFE HC and non-penetrant p.Cys282Tyr homozygotes, monitor serum ferritin concentration every six to 12 months, and begin phlebotomy to achieve iron depletion when serum ferritin exceeds 300 µg/L (males) and 200 µg/L (females).
Agents/circumstances to avoid: Medicinal iron, mineral supplements containing iron, excess alcohol, excess vitamin C, uncooked seafood, and lifestyle behaviors that increase the risk of viral hepatitis infection; alcohol consumption should be avoided in those with hepatic fibrosis or cirrhosis.
Evaluation of relatives at risk: Offer molecular genetic testing to adult sibs of a proband to facilitate early diagnosis and surveillance.
Genetic counseling.
HFE HC is inherited in an autosomal recessive manner and has low clinical penetrance. (Note: Pseudodominance has been observed in HFE HC and is attributed to the relatively high prevalence of p.Cys282Tyr heterozygotes in persons of European ancestry.) Most parents of individuals with HFE HC are p.Cys282Tyr heterozygotes (i.e., have one copy of HFE p.Cys282Tyr). On occasion, one parent has p.Cys282Tyr homozygosity and may have clinical, biochemical, or non-penetrant HFE HC. If both parents are p.Cys282Tyr heterozygotes, each sib of an affected individual has a 25% chance of being homozygous for p.Cys282Tyr, a 50% chance of being heterozygous for p.Cys282Tyr, and a 25% chance of being neither homozygous nor heterozygous for p.Cys282Tyr. Sibs who are homozygous for p.Cys282Tyr are at risk for HFE-related iron overload, although the clinical penetrance of HFE HC is low.
Diagnosis
In 2022, a new classification of hemochromatosis based on genetic and clinical manifestations was proposed by an expert group, although this classification excludes persons who have loss-of-function SLC40A1 alleles ("ferroportin disease"). This classification also suggests clinical management when molecular genetic characterization of hemochromatosis is not available [Girelli et al 2022]. In 2022, the European Association for the Study of the Liver updated guidelines for diagnosis of hemochromatosis [European Association for the Study of the Liver 2022].
For the purposes of this GeneReview, the terms "male" and "female" are narrowly defined as the individual's biological sex at birth as it determines clinical care [Caughey et al 2021].
Suggestive Findings
HFE-related hemochromatosis (HFE HC) should be suspected in an individual with laboratory features of hemochromatosis, clinical manifestations of advanced iron overload, and/or a family history of HFE HC.
Laboratory features of hemochromatosis
- Elevated transferrin saturation. Elevated transferrin saturation in persons with HFE HC (≥60% in males, ≥50% in females) is an indicator of increased recycling of iron and increased risk for iron overload. Approximately 80% of individuals with HFE HC have a fasting serum transferrin saturation of ≥60% (in males) or ≥50% (in females) on two or more occasions in the absence of other known causes of elevated transferrin saturation. Transferrin saturation is not age related in adults with HFE HC and is not significantly associated with the presence or absence of clinical findings or increased serum ferritin levels. In some adults with inflammatory conditions, transferrin is a negative acute-phase reactant. Therefore, transferrin saturation may be spuriously elevated due to decreased production of transferrin by the liver.
- Elevated serum ferritin concentration. Normal serum ferritin values from the Hemochromatosis and Iron Overload Screening (HEIRS) Study are <300 µg/L in males and <200 µg/L in females [Adams et al 2005]. Serum ferritin concentration generally increases progressively over time in individuals with untreated clinical HFE HC. An elevated serum ferritin concentration alone is not specific for iron overload because serum ferritin is an acute-phase reactant and elevated serum ferritin levels may be caused by non-iron liver disorders or by inflammatory or neoplastic disorders. In these instances, transferrin saturation is usually not elevated. Serum ferritin values in individuals with HFE HC can range from below normal to much greater than 1,000 µg/L.
- Higher hemoglobin (Hb), mean corpuscular hemoglobin (MCH), and mean corpuscular volume (MCV). Mean values of Hb, MCH, and MCV are significantly higher in persons with HFE HC than controls, although most persons with HFE HC have neither erythrocytosis (Hb >18.0 g/L) nor macrocytosis (MCV >100 fL) [Barton et al 2000]. Higher values of Hb, MCH, and MCV are due in part to ample quantities of iron for Hb synthesis by erythroid cells due to elevated transferrin saturation [Barton et al 2000, McLaren et al 2007]. Liver disease, alcoholism, deficiencies of vitamin B12 or folate acid, or medications do not account for higher Hb, MCH, and MCV in most individuals with HFE HC. Accordingly, higher Hb, MCH, and MCV in persons with elevated transferrin saturation suggest the diagnosis of HFE HC. In one study, mean pre- and post-phlebotomy MCV and MCH values were always significantly higher than those of other groups and demonstrated excellent diagnostic utility for detection of HFE HC in males and females [Adris et al 2019]. Persons with hemochromatosis related to other HFE genotypes also have higher Hb, MCH, and MCV than controls.
Clinical manifestations of advanced iron overload
- Weakness, chronic fatigue
- Abdominal pain, weight loss
- Arthropathy (especially metacarpophalangeal joints, hips, knees)
- Diabetes
- Hepatomegaly
- Cirrhosis
- Primary liver cancer (hepatocellular carcinoma, cholangiocarcinoma)
- Hypogonadotropic hypogonadism (decreased libido and impotence in males, amenorrhea in females)
- Cardiomyopathy, arrhythmias
- Progressive increase in skin pigmentation
Family history is consistent with autosomal recessive inheritance, although penetrance is low. Absence of a family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of HFE HC is established in most persons with characteristic laboratory and/or clinical features by identification of HFE p.Cys282Tyr homozygosity (see Table 1).
Note: HFE HC is associated predominantly with p.Cys282Tyr homozygosity. The penetrance of other common HFE genotypes (e.g., p.Cys282Tyr/p.His63Asp compound heterozygosity and p.His63Asp homozygosity) is markedly lower than that of p.Cys282Tyr homozygosity. Most individuals with p.Cys282Tyr/p.His63Asp compound heterozygosity or p.His63Asp homozygosity who develop clinical or biochemical HC are believed to have a pathogenic allele in another HC-related gene (digenic inheritance) or a predisposing environmental factor. Approximately 100 other uncommon or rare pathogenic or likely pathogenic HFE alleles, many inherited in association with p.Cys282Tyr heterozygosity or another HFE allele, have been described in persons with and without clinical or biochemical HC. Penetrance of many of these uncommon or rare pathogenic or likely pathogenic HFE genotypes is uncertain. Almost all persons described to have HFE HC report European ancestry, although rarely HFE HC has been described in individuals who report African American or Chinese ancestry.
Molecular genetic testing approaches can include a combination of targeted testing and use of a multigene panel.
- Targeted analysis for HFE p.Cys282Tyr and p.His63Asp should be considered first in individuals of northern European ancestry.
- A hemochromatosis multigene panel that includes HFE and other genes of interest (see Differential Diagnosis) may be considered in individuals without northern European ancestry to identify the genetic cause of the condition while limiting identification of alleles of uncertain significance and pathogenic alleles in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 1.
Molecular Genetic Testing Used in HFE-Related Hemochromatosis
Clinical Characteristics
Clinical Description
HFE-related hemochromatosis (HFE HC) is composed of three phenotypes:
- Clinical HFE HC. Individuals with end-organ damage secondary to iron overload
- Biochemical HFE HC. Individuals with both elevated transferrin saturation and elevated serum ferritin concentration
- Non-penetrant HFE HC. Persons with, e.g., HFE p.Cys282Tyr homozygosity without clinical or laboratory features of iron overload (Transferrin saturation may be elevated in the absence of elevated serum ferritin.)
Clinical HFE HC
Individuals with clinical HFE HC absorb increased iron from a normal diet via small intestinal mucosa, resulting in excessive free parenchymal iron, which may cause target organ injury and, potentially, organ failure. They also have increased recycling of iron derived from senescent red cells, resulting in elevated transferrin saturation.
Onset. Clinical manifestations related to iron overload usually appear between age 40 and 60 years in males and after menopause in females. Occasionally, HFE HC manifests at an earlier age, but hepatic fibrosis or cirrhosis is rare before age 40 years. Initial manifestations of iron overload in persons with clinical HFE HC often include arthropathy of the metacarpophalangeal joints, diabetes mellitus, elevated serum concentrations of hepatic transaminases, and progressive increase in skin pigmentation. Weakness, chronic fatigue, abdominal pain, and weight loss are common nonspecific findings [Barton et al 2022, Barton & Parker 2023].
Arthropathy. The first sign of clinical HFE HC in many individuals is arthropathy (joint stiffness and pain), especially in metacarpophalangeal joints. Knee and hip arthropathy are also common and thus HFE HC may present at the time of joint arthroplasty.
Diabetes. The prevalence of diabetes mellitus type 2 in persons with HFE HC is similar to that in the general population and is the most common diabetes subtype in persons with HFE HC. Approximately 50% of individuals with cirrhosis or liver failure due to HFE HC also have diabetes mellitus that is often due to siderosis of pancreatic islets.
Liver disease. The Hemochromatosis and Iron Overload Screening (HEIRS) Study reported an odds ratio of 3.3 for liver disease among males homozygous for HFE p.Cys282Tyr compared to controls [Adams et al 2005]. Hepatomegaly may or may not be present on physical examination in individuals with HFE HC and hepatic iron overload. With progressive iron overload, cirrhosis may develop and be complicated by portal hypertension, primary liver cancer (e.g., hepatocellular carcinoma, cholangiocarcinoma), and end-stage liver disease [Kowdley et al 2005]. Alcohol consumption exacerbates manifestations of HFE HC [Scotet et al 2003]. Age, diabetes, alcohol consumption, and severity of iron overload increase the risk of cirrhosis, after adjustment for other factors [Barton et al 2018].
Hypogonadotropic hypogonadism. Some males, typically those with severe iron overload, have erectile dysfunction, hypotestosteronemia, loss of muscle mass, and osteoporosis due to hypogonadotropic hypogonadism. In some females with severe iron overload, hypogonadotropic hypogonadism causes diminished libido, amenorrhea, and infertility.
Cardiomyopathy. Heart disease, the result of iron deposition in cardiac parenchyma, is rare among persons with HFE HC. Approximately 15% of individuals with cirrhosis or liver failure also have congestive heart failure and/or cardiac arrhythmias.
Skin. Progressive increase in skin pigmentation resulting from deposits of melanin and iron may be observed.
Prognosis. Individuals diagnosed and treated prior to the development of cirrhosis have a normal life expectancy. Those diagnosed after the development of cirrhosis have a decreased life expectancy even with iron depletion therapy [Adams et al 2005], primarily due to the 10%-30% risk of primary liver cancer (e.g., hepatocellular carcinoma, cholangiocarcinoma). Failure to deplete iron stores after 18 months of treatment is a poor prognostic sign that reflects iron overload severity in most individuals and insufficient phlebotomy therapy in other individuals. With iron depletion, dysfunction of some organs (liver and heart) can improve. Arthropathy and endocrinopathy improve in ~20% of treated individuals. Death in individuals with clinical HFE HC is often caused by liver failure, primary liver cancer, extrahepatic cancers, congestive heart failure, or arrhythmia.
Biochemical HFE HC
Individuals with biochemical HFE HC have elevated transferrin saturation and elevated serum ferritin concentration in the absence of clinical features of iron overload. Approximately 75%-90% of these individuals are asymptomatic, although it is controversial whether the risk of developing clinical features of iron overload in individuals with biochemical HFE HC at presentation is increased. One prospective study found that iron overload does not progress in all individuals with HFE HC. Although serum ferritin concentration may rise in these individuals over time, end-organ damage is uncommon and is observed more frequently in males than females [Allen et al 2008, Gurrin et al 2008]. An Australian consortium concluded that the benefits of phlebotomy in individuals with HFE HC with mildly elevated serum ferritin remain unproven because a randomized study with long-term follow up that would clarify this possibility is lacking [Delatycki et al 2015]. In another study, individuals with HFE HC with a serum ferritin at diagnosis between the upper limit of normal and 1,000 μg/L who underwent phlebotomy had lower mortality than the general population [Bardou-Jacquet et al 2015a, Bardou-Jacquet et al 2015b, Bardou-Jacquet et al 2015c].
Non-penetrant HFE HC
Normal serum ferritin concentration at diagnosis of HFE HC is usually associated with lack of development of clinical or laboratory features of iron overload. Females are more likely to have non-penetrant HFE HC than males [Barton & Parker 2023].
HFE p.Cys282Tyr or p.His63Asp Heterozygotes
Some individuals who are heterozygous for either HFE p.Cys282Tyr or p.His63Asp have elevated serum transferrin saturation and serum ferritin concentrations, although they rarely develop complications of iron overload [Barton & Parker 2023].
Genotype-Phenotype Correlations
p.Cys282Tyr. Most HFE p.Cys282Tyr homozygotes do not have clinical HFE HC. A significant proportion of individuals with p.Cys282Tyr homozygosity (especially males) have biochemical HFE HC. Three longitudinal population-based studies showed that 38%-50% of p.Cys282Tyr homozygotes develop elevated serum ferritin concentration and 10%-33% eventually develop clinical HFE HC [Whitlock et al 2006].
p.His63Asp. Some HFE p.Cys282Tyr/p.His63Asp compound heterozygotes and HFE p.His63Asp homozygotes identified in general population testing have elevated transferrin saturation and mild hyperferritinemia [Barton & Parker 2023]. Identification of serum ferritin >500 µg/L in persons with these genotypes should prompt a search for non-iron causes of hyperferritinemia.
In 58 screening program participants without HFE p.Cys282Tyr homozygosity who had both elevated transferrin saturation and elevated serum ferritin on two occasions in the absence of treatment for iron overload, anemia, erythrocyte transfusion >10 units, alcohol intake >30 g/day, hepatitis B or C, or pregnancy, relative risks of HFE genotypes p.Cys282Tyr/His63Asp, p.His63Asp/His63Asp, and p.Cys282Tyr heterozygosity were significantly higher (11.0-, 3.9-, and 1.9-fold, respectively) than those of 42,640 White screening participants with neither elevated transferrin saturation nor elevated serum ferritin. Serum ferritin levels were positively associated with mean corpuscular volume and swelling/tenderness of 2nd/3rd metacarpophalangeal joints. Elevated serum concentrations of hepatic transaminases were significantly associated with mean corpuscular volume and diabetes [Barton et al 2022].
Penetrance
Environmental factors. Some environmental factors modify penetrance of laboratory phenotypes of persons with HFE HC. Increased dietary consumption of heme iron was associated with higher serum ferritin levels in postmenopausal females [Cade et al 2005]. Nonalcoholic fatty liver in referred adults with HFE HC with iron overload was associated with significantly higher median serum ferritin and greater prevalence of diabetes mellitus type 2, after adjustment for other factors, although nonalcoholic fatty liver did not significantly influence the quantity of iron removed by phlebotomy to achieve iron depletion [Barton et al 2023]. Gastric acid suppression reduces phlebotomy requirements [Hutchinson et al 2007]. Nonetheless, effects of these factors on penetrance of HFE HC is probably small.
Consumption of more than two alcoholic drinks per day was associated with increased risk of elevated transferrin saturation and elevated serum ferritin in a general population cohort [Ioannou et al 2004]. This could explain in part observations that HFE HC is often associated with chronic alcoholism. Risk of liver injury or progression of hepatic fibrosis is increased in persons with HFE HC who also have alcoholism, nonalcoholic fatty liver, or viral hepatitis [Wood et al 2008].
Viral hepatitis accelerates liver injury in persons with HFE HC. Prevalence of viral hepatitis differs greatly across geographic regions. In 78 referred individuals with HFE HC in Italy, the prevalence of hepatitis B surface antigen (HBsAg) and anti-hepatitis C virus (HCV) were 5.1% and 20.5%, respectively [Piperno et al 1992]. Chronic hepatitis C infection prevalence in 961 referred individuals with HFE HC in the United States and Canada was 1.0% (10/961) [Barton et al 2019]. The anti-HBsAg antibody response to hepatitis B vaccination in persons with HFE HC is substantial. It is unknown whether hepatitis vaccinations reduce risk of cirrhosis or primary liver cancer or increase longevity.
Sex. The proportion of females with clinical HFE HC is lower than that of males (1% vs 28%, respectively) [Allen et al 2008]. Population studies demonstrate that the greatest sex-related factor that modifies penetrance of clinical HFE HC is the premenopausal state in females. It is plausible although unproven that physiologic blood (iron) loss in premenopausal females accounts for their lower penetrance of clinical HFE HC. Hepatic iron concentrations were higher in females who underwent menopause at age <50 years than in females who underwent menopause at an older age [Moirand et al 1997]. In contrast, the number of pregnancies was not significantly correlated with iron burden in females with HFE HC [Deugnier et al 2002]. Other undefined genetic factors may also contribute to reduced penetrance of clinical HFE HC in females [Wood et al 2008].
Genetic factors. Increased iron burden occurs in persons with HFE HC who are also heterozygous for HAMP or HJV pathogenic alleles. Some BMP alleles have been associated with variability of serum ferritin levels in persons with HFE HC [Wood et al 2008].
Nomenclature
HFE HC has been described as bronze diabetes with cirrhosis, primary hemochromatosis, genetic hemochromatosis, hereditary hemochromatosis, and hemochromatosis type 1. The current term is HFE-related hemochromatosis (HFE HC) [Girelli et al 2022].
Prevalence
Among most populations of European ancestry, the prevalence of individuals homozygous for HFE p.Cys282Tyr is 1:200 to 1:500 [Barton & Parker 2023]. In non-Hispanic Whites in North America, the prevalence of p.Cys282Tyr homozygotes is 1:200 to 1:400 [Adams et al 2005]. Among Hispanics in North America, the prevalence of p.Cys282Tyr homozygotes is 0.027% [Adams et al 2005]. Among Blacks in North America, the prevalence of p.Cys282Tyr homozygotes is 1:6,781 [Barton et al 2005]. Among Asians in North America, the prevalence of p.Cys282Tyr homozygotes is 1:25,000 [Adams et al 2005].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic alleles in HFE.
Differential Diagnosis
HFE-related hemochromatosis (HFE HC) differs from rarer primary iron overload disorders and secondary iron overload disorders.
Primary Iron Overload Disorders
Primary iron overload disorders (summarized in Table 2) are characterized by increased absorption of iron from a normal diet in subjects without severe anemia. Juvenile hemochromatosis and TFR2-related hemochromatosis result from hepcidin deficiency and thus the clinical manifestations of these disorders are similar to, but typically more severe than, those of HFE HC.
Table 2.
Primary Iron Overload Disorders in the Differential Diagnosis of HFE-Related Hemochromatosis
African iron overload (OMIM 601195), reported in sub-Saharan African populations, is associated with excessive intake of dietary iron in traditional beer brewed in non-galvanized steel drums. A genetic contribution to the pathogenesis of African iron overload, if any, is unknown.
Secondary Iron Overload Disorders
Liver diseases associated with secondary iron overload include alcoholic liver disease, acute viral hepatitis, chronic viral hepatitis C (uncommon), neoplasms, familial porphyria cutanea tarda, and inflammatory disorders such as rheumatoid arthritis.
Nonalcoholic fatty liver disease in persons with HFE HC does not increase amounts of iron removed by phlebotomy to achieve iron depletion, although serum ferritin levels are typically higher in persons with HFE HC and nonalcoholic fatty liver disease due to hepatic inflammation [Barton et al 2023].
In persons without HFE HC, nonalcoholic fatty liver disease is sometimes associated with elevated transferrin saturation or serum ferritin concentration but infrequently with increased hepatic iron deposition.
Iron overload can result from ingested iron in foods, cooking ware, and medicines, in addition to parenteral iron from iron injections or transfusions for anemia (e.g., beta-thalassemia, sickle cell disease, hereditary sideroblastic anemia [OMIM PS300751], pyruvate kinase deficiency [OMIM 266200], hereditary spherocytosis [see EPB42-Related Hereditary Spherocytosis], myelodysplastic syndrome with refractory anemia).
Erythroferrone is an inhibitor of hepcidin transcription. Due to bone marrow production of erythroferrone (and consequent hepcidin deficiency), iron absorption is increased in some heritable anemias (e.g., non-transfusion-dependent beta-thalassemia, pyruvate kinase deficiency, hereditary sideroblastic anemia).
Neonatal hemochromatosis develops in utero and is characterized by severe hepatic injury (marked loss of hepatocytes, coarsely granular hepatocyte siderosis) and extrahepatic siderosis (epithelia of thyroid, exocrine pancreas, and salivary glands; myocardium). Gestational alloimmune liver disease is the cause of fetal liver injury in most cases [Feldman & Whitington 2013]. Maternal alloimmunity accounts for the occurrence of neonatal hemochromatosis in two or more offspring of the same mother. Antenatal therapy with high-dose intravenous immunoglobulin G prevents poor outcome of pregnancies at risk for neonatal hemochromatosis [Whitington et al 2018]. There is little evidence that neonatal hemochromatosis is a heritable disorder attributed to an as-yet-unidentified gene. Some investigators have suggested that DGUOK pathogenic alleles lead to phenotypes that resemble those of neonatal hemochromatosis (see Deoxyguanosine Kinase Deficiency).
Management
In 2022, the European Association for the Study of the Liver updated guidelines for management of hemochromatosis, including the use of hepatic elastography, erythrocytapheresis, dietary recommendations, and management of persons with HFE p.Cys282Tyr/His63Asp compound heterozygosity [European Association for the Study of the Liver 2022]. Experts at the 2017 Hemochromatosis International meeting published an objective and practical set of recommendations on treatment of persons with HFE-related hemochromatosis (HFE HC) based on scientific studies and guidelines [Adams et al 2018].
Evaluations Following Initial Diagnosis
To establish the extent of iron overload and optimal management of persons diagnosed with HFE HC, the evaluations summarized in Table 3 are recommended if not performed as part of the evaluation that led to the diagnosis [Kowdley et al 2019, European Association for the Study of the Liver 2022].
Table 3.
HFE-Related Hemochromatosis: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
Targeted Therapies
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
Therapeutic phlebotomy is a standard of care for individuals with HFE HC who have either only biochemical evidence of iron overload (i.e., elevated serum ferritin) or clinical manifestations of iron overload (i.e., evidence of end-organ damage including cirrhosis, cardiac failure, skin hyperpigmentation, diabetes, or hypogonadotropic hypogonadism).
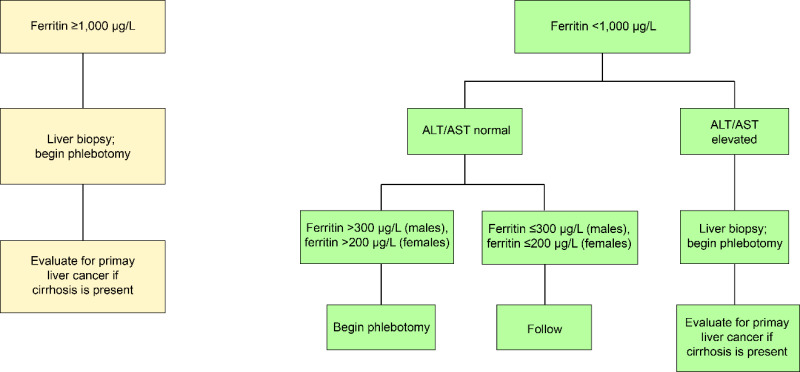
The European Association for the Study of the Liver [2022] suggests phlebotomy therapy for individuals with evidence of iron overload. The HEIRS Study suggested that phlebotomy therapy be started in males with serum ferritin >300 µg/L and in females with serum ferritin >200 µg/L [Adams et al 2005]. In a study of Australian individuals with HFE HC, there was evidence of subjective and objective improvement by reducing serum ferritin levels to <300 µg/L [Ong et al 2017].
Therapeutic phlebotomy (i.e., removal of a unit of blood) is a simple, inexpensive, safe, and effective way to remove excess iron. Each unit of blood (400-500 mL) with a hematocrit of 40% contains 160-200 mg of iron.
The usual therapy is phlebotomy weekly until the serum ferritin is ≤100 µg/L. Twice-weekly phlebotomy may be useful to accelerate iron depletion in individuals with serum ferritin >1,000 µg/L or those with evidence of end-organ damage due to iron overload. Hematocrit or hemoglobin (Hb) concentration should be assessed prior to each phlebotomy. If serum ferritin is very high, serum ferritin measurement should be performed approximately every five to ten phlebotomies. Some persons, especially females, tolerate phlebotomy poorly. In such instances, the interval between phlebotomies should be increased (e.g., every 10-14 days) or phlebotomy volume should be reduced (e.g., 250-300 mL).
Anemia is not a characteristic of HFE HC. In individuals with anemia due to other causes, the Hb concentration at which phlebotomy is not recommended or at which the phlebotomy regimen should be interrupted depends on individual factors including symptoms of anemia, comorbid conditions, age, and ability to perform ordinary tasks and carry out daily activities, while taking into account the relative importance of the procedure. For example, in individuals with severe iron overload, some degree of anemia is acceptable if phlebotomy is tolerated by the individual. In some causes of anemia (e.g., anemia of renal insufficiency), treatment of the underlying anemia with an erythropoiesis-stimulating agent may improve tolerability of phlebotomy. Increasing erythropoiesis will also enhance iron incorporation in erythrocytes.
The serum ferritin concentration is the most feasible and inexpensive way to monitor therapeutic phlebotomy. After serum ferritin is ≤100 µg/L, serum ferritin concentration should be quantified after each additional one or two treatments [Adams & Barton 2010]. On average, males require removal of twice as many units of blood to achieve iron depletion as females.
Maintenance phlebotomy to prevent reaccumulation of excess iron is indicated for males with serum ferritin ≥300 µg/L and for females with serum ferritin ≥200 µg/L [Adams & Barton 2010].
Phlebotomy may decrease fatigue, arthralgias, and hepatic enzyme levels in many individuals and induce regression of liver fibrosis and cirrhosis in some individuals. Conclusive data regarding the favorable influence of therapeutic phlebotomy on quality of life, diabetes, liver cancer risk, or cardiomyopathy is lacking [Prabhu et al 2020]. Therapeutic phlebotomy increases overall survival [Bomford & Williams 1976, Milman et al 2001].
Whether lowering transferrin saturation should be a target of phlebotomy therapy is debatable. Elevated transferrin saturation in HFE HC is caused by increased iron export from macrophages due to hepcidin deficiency and is not a marker of iron overload. Attempting to achieve and maintain low transferrin saturation in individuals with HFE HC may result in iron deficiency and associated adverse manifestations [Barton & Bottomley 2000].
Note: Hb, mean corpuscular hemoglobin (MCH), and mean corpuscular volume (MCV) levels are not targets of treatment in persons with HFE HC.
Erythrocytapheresis is an effective and safe alternative to phlebotomy therapy for individuals with HFE HC, although few facilities can provide erythrocytapheresis, whereas many can provide therapeutic phlebotomy. Fixed-volume erythrocytapheresis results in a faster initial decline in serum ferritin and a reduced number of procedures than phlebotomy therapy, although it does not achieve target serum ferritin levels sooner [Sundic et al 2014]. Reports of treatment duration and total costs of individualized erythrocytapheresis (based on sex, body weight, total blood volume, and hematocrit) in the induction phase of therapy, compared to those of phlebotomy, are variable [Rombout-Sestrienkova et al 2012, Sundic et al 2014]. In the maintenance phase of therapy, individualized erythrocytapheresis reduced the number of procedures required in comparison with phlebotomy therapy but not the total costs per treatment year [Rombout-Sestrienkova et al 2016]. Blood components removed during erythrocytapheresis may be suitable for transfusion. Mild citrate reactions are common with erythrocytapheresis.
Iron chelation therapy to achieve iron depletion is a treatment alternative for individuals who have an elevated serum ferritin concentration and concomitant symptoms of anemia, inadequate venous access, or another circumstance that makes therapeutic phlebotomy or erythrocytapheresis inadvisable, although studies of chelation therapy in individuals with HFE HC are very limited. Oral deferasirox at 10 mg/kg/day was the starting dose for most individuals with HFE HC in two studies [Phatak et al 2010, Cançado et al 2015]. Deferasirox therapy is not appropriate for individuals with advanced liver disease. Subcutaneous deferoxamine was effective in achieving iron depletion in three individuals with HFE HC in whom phlebotomy therapy was not feasible [Nielsen et al 2003].
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, surveillance is based on guidelines proposed for individuals with HFE HC due to p.Cys282Tyr homozygosity in Europe [European Association for the Study of the Liver 2022] and North America [Kowdley et al 2019].
Table 5.
HFE-Related Hemochromatosis: Recommended Surveillance
Agents/Circumstances to Avoid
Avoid the following:
- Medicinal iron and nutritional supplements containing iron
- Excessive alcohol intake, which increases iron absorption and is toxic to hepatocytes. Individuals with cirrhosis should avoid alcohol consumption.
- Daily ingestion of more than 500 mg of supplemental vitamin C, which may enhance iron absorption
- Consumption of uncooked seafood, which increases the risk of infection from microorganisms that thrive in conditions of excess iron (Vibrio vulnificus and other Vibrio species)
- Lifestyle-related behaviors that increase the risk of viral hepatitis infection
Evaluation of Relatives at Risk
Evaluation of adult sibs and offspring of individuals with HFE HC identifies those who have HFE HC and might benefit from both treatment and preventive measures. The following strategy is recommended:
- 1.
Inform probands with HFE HC that HFE molecular genetic testing is recommended for their adult sibs (≥18 years)
- 2.
Measurement of transferrin saturation and serum ferritin concentration in sibs with HFE p.Cys282Tyr homozygosity (HFE HC)
- 3.
Phlebotomy therapy for sibs with HFE HC and elevated serum ferritin concentrations
Note: HFE molecular genetic testing has a low positive predictive value for clinical HC because few p.Cys282Tyr homozygotes and no p.Cys282Tyr/His63Asp compound heterozygotes develop clinically significant iron overload without other predisposing factors [El-Serag et al 2000, Beutler et al 2002].
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Inducing iron depletion during pregnancy or in females who anticipate pregnancy is undesirable. In the absence of severe iron-related liver disease or cardiomyopathy, it is prudent to withhold therapeutic phlebotomy during pregnancy [European Association for the Study of the Liver 2022].
Therapies Under Investigation
The oral iron chelator deferasirox (Exjade®) has been evaluated in Phase I/II studies of individuals with HFE HC, results of which suggest that deferasirox reduces iron burdens within an acceptable safety profile [Phatak et al 2010, Cançado et al 2015]. Subcutaneous deferoxamine was effective in achieving iron depletion in three individuals with HFE HC in whom phlebotomy therapy was not feasible [Nielsen et al 2003]. Rusfertide, a peptidic mimetic of hepcidin, reduced the mean number of maintenance phlebotomies in 12 individuals with HFE HC during a 24-week trial [Kowdley et al 2023]. Nonetheless, these studies are very limited and to date, the US Food and Drug Administration and the European Medicines Agency have not approved deferasirox, deferoxamine, deferiprone, or rusfertide for treatment of hemochromatosis.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
HFE-related hemochromatosis (HFE HC) is inherited in an autosomal recessive manner with low clinical penetrance.
Pseudodominance (the occurrence of an autosomal recessive disorder in two generations of a family without consanguinity) has been observed in HFE HC and is attributed to the high prevalence of HFE p.Cys282Tyr in persons of European ancestry (see Prevalence).
Risk to Family Members
Parents of a proband
- Most parents of individuals with HFE HC are HFE p.Cys282Tyr heterozygotes (i.e., have a single copy of p.Cys282Tyr). Individuals who are heterozygous for p.Cys282Tyr do not develop HFE-related iron overload, although some have abnormal serum iron measures (see Clinical Description, Heterozygotes).
- Infrequently, one parent of a proband with HFE HC may have HFE p.Cys282Tyr homozygosity and have clinical HFE HC (presence of significant end-organ damage including cirrhosis, cardiac failure, skin hyperpigmentation, diabetes, or hypogonadism). Thus, it is appropriate to recommend to probands that their parents be evaluated for HFE HC by molecular genetic testing or by measuring transferrin saturation and serum ferritin concentration.
Sibs of a proband
- If both parents of a proband with HFE HC are HFE p.Cys282Tyr heterozygotes, each sib of that proband has a 25% probability of being homozygous for p.Cys282Tyr, a 50% probability of being heterozygous for p.Cys282Tyr, and a 25% probability of being neither homozygous nor heterozygous for p.Cys282Tyr.
- If one parent of a proband with HFE HC is homozygous for HFE p.Cys282Tyr and the other parent is heterozygous for p.Cys282Tyr, each sib of that proband has a 50% chance of being homozygous for p.Cys282Tyr and a 50% chance of being heterozygous for p.Cys282Tyr.
- Sibs of probands with HFE HC who inherit biallelic p.Cys282Tyr alleles are at risk for HFE-related iron overload, although the clinical penetrance of HFE HC is low. Several large-scale screening studies of general populations have demonstrated that most individuals who are homozygous for HFE p.Cys282Tyr do not have or eventually develop clinical HFE HC (see Penetrance). Note: It is recommended that the proband's physician suggest that the proband notify their family members to undergo molecular genetic testing or measurement of transferrin saturation and serum ferritin to ascertain whether they also have HFE HC (see Management, Evaluation of Relatives at Risk).
- Sibs of probands with HFE HC who are p.Cys282Tyr heterozygotes are not at risk for HFE-related iron overload, although they may have abnormal serum iron studies (see Clinical Description, Heterozygotes).
Offspring of a proband
- Unless the reproductive partner of a proband with HFE HC also has HFE HC or is a p.Cys282Tyr heterozygote, all offspring will be obligate p.Cys282Tyr heterozygotes.
- It is sometimes recommended that reproductive partners of individuals with HFE HC undergo HFE molecular genetic testing or measurement of transferrin saturation and serum ferritin concentration to determine if their children are at risk for HFE HC.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults with HFE HC.
Molecular genetic testing for HFE p.Cys282Tyr can be offered to the reproductive partner of a person with HFE HC to determine if their offspring are at risk for HFE HC. Among most populations of European ancestry, prevalence of p.Cys282Tyr is approximately 1 in 9 persons. Therefore, offspring of an individual with HFE HC and an individual of European ancestry have an approximately 5% risk of inheriting biallelic HFE p.Cys282Tyr alleles.
Population Screening
Population screening has been performed due to the high prevalence of HFE HC, the lack of early clinical findings, the lack of specificity of clinical findings if they appear, the low cost of diagnosis, the availability of relatively simple and effective early treatment, and the high cost and low success rate of treatment when the diagnosis is established late.
- Molecular genetic testing-based population screening for HFE HC is not recommended because penetrance is low and the natural history of untreated individuals cannot be predicted. Some experts opine that available evidence is insufficient to make a decision regarding a recommendation to perform population screening for HFE HC [Schmidtke 2022].
- Biochemical-based screening (using transferrin saturation and serum ferritin) of males of European descent age >30 years may be considered [Phatak et al 2008, European Association for the Study of the Liver 2010, Bacon et al 2011].
Prenatal Testing and Preimplantation Genetic Testing
Prenatal testing is not usually performed because HFE HC is an adult-onset, treatable disorder with low clinical penetrance, although preimplantation genetic testing and prenatal testing for a pregnancy at increased risk are feasible once biallelic HFE p.Cys282Tyr alleles have been identified in a family member.
Differences in perspective exist among medical professionals and within families regarding the use of prenatal testing. Although most centers consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Canadian Hemochromatosis SocietyCanadaPhone: 877-223-4766; 604-279-7135Email: office@toomuchiron.ca
- EFAPH: European Federation of Associations of Patients with HaemochromatosisPhone: 32 2 280 23 34Email: info@eu-patient.eu
- Haemochromatosis AustraliaAustraliaPhone: 1300 019 028
- Haemochromatosis UKUnited KingdomPhone: 03030 401 101Email: office@huk.org.uk
- MedlinePlus
- National Human Genome Research Institute
- National Institute of Diabetes and Digestive and Kidney DiseasesPhone: 800-860-8747Email: nddic@info.niddk.nih.gov
- NCBI Genes and Disease
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
HFE-Related Hemochromatosis: Genes and Databases
Table B.
OMIM Entries for HFE-Related Hemochromatosis (View All in OMIM)
Molecular Pathogenesis
HFE encodes hereditary hemochromatosis protein (HFE). The largest predicted primary translation product is 348 amino acids, which gives rise to a mature protein of approximately 321 amino acids after cleavage of the signal sequence. HFE is similar to HLA class I molecules at the level of both primary [Feder et al 1996] and tertiary structure [Lebrón et al 1998]. The mature protein is expressed on the cell surface as a heterodimer with beta-2-microglobulin, and this interaction is necessary for normal presentation on the cell surface. HFE binds to transferrin receptor protein 1 on the cell surface and interacts with the trimolecular complex of transferrin receptor protein 2, bone morphogenic protein receptor, and hemojuvelin.
HFE allele c.845G>A (p.Cys282Tyr) disrupts a disulfide bond and alters the conformation of HFE, affecting its interaction with beta-2-microglobulin, transferrin receptor protein 2, bone morphogenic protein receptor, and hemojuvelin on hepatocytes. Consequently, hepatocytes lose the capacity to upregulate hepcidin, the master regulator of iron metabolism. The action of hepcidin is mediated by binding to ferroportin, the only known exporter of intracellular iron. Following binding of hepcidin to ferroportin, the complex is degraded intracellularly. Therefore, the end result of hepcidin binding to ferroportin (which is present primarily on hepatocytes, enterocytes, and reticuloendothelial cells) is reduction of expression of ferroportin. When hepcidin is low (as in HFE HC), ferroportin activity is concomitantly increased, resulting in greater iron absorption by enterocytes and greater iron recycling by reticuloendothelial cells [Olynyk & Ramm 2022].
Mechanism of disease causation. Loss of function
Table 6.
HFE Pathogenic Alleles Referenced in This GeneReview
Chapter Notes
Author Notes
James C Barton, MD, is a Clinical Professor of Medicine in the Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, and Medical Director of Southern Iron Disorders Center, Birmingham, AL. As a hematologist, his interests include population screening, diagnosis, and management of hemochromatosis, iron deficiency, and other iron-related disorders. Dr Barton received his MD from the University of Tennessee College of Medicine, Memphis, TN. He received training in internal medicine, hematology, and medical oncology at the University of Tennessee College of Medicine, Memphis, TN, and the University of Alabama at Birmingham, Birmingham, AL. Dr Barton has edited two textbooks on hemochromatosis. Email: moc.liamg@633semajnotrab
Charles J Parker, MD, is a Professor of Medicine in the Division of Hematology and Hematologic Malignancies, Department of Internal Medicine, at the University of Utah and the Huntsman Cancer Institute, Salt Lake City, UT. As a hematologist, his clinical interests include paroxysmal nocturnal hemoglobinuria (PNH), hemochromatosis, and porphyria. Dr Parker received his MD from The University of North Carolina, Chapel Hill, NC. He received training in medicine and hematology from the North Carolina Memorial Hospital, Chapel Hill, NC, and Duke University, Durham, NC, respectively. In the past, Dr Parker served as President of the International Paroxysmal Nocturnal Hemoglobinuria Interest Group and Editor-in-Chief of The Hematologist. Web page: medicine.utah.edu/internal-medicine/hematology; email: ude.hatu.csh@rekrap.selrahc
Acknowledgments
Dr Barton acknowledges Drs Marcel E Conrad, William H Crosby, Ernest Beutler, Corwin Q Edwards, and Ronald T Acton for sharing their experience, collaborations, and enthusiasm about studying hemochromatosis and other iron-related disorders.
Author History
James C Barton, MD (2018-present)
Robin L Bennett, MS; University of Washington (2000-2015)
Corwin Q Edwards, MD; University of Utah School of Medicine (2018-2024)
Kris V Kowdley, MD; Virginia Mason Medical Center (2000-2015)
Arno G Motulsky, MD; University of Washington (2000-2015)
Charles J Parker, MD (2024-present)
Lawrie Powell, AC, MD, PhD; Royal Brisbane & Women's Hospital (2015-2018)
Rebecca Seckington; University of Queensland (2015-2018)
Jonathan F Tait, MD, PhD; University of Washington School of Medicine (2000-2011)
Revision History
- 11 April 2024 (sw) Comprehensive update posted live
- 6 December 2018 (sw) Comprehensive update posted live
- 17 September 2015 (me) Comprehensive update posted live
- 19 April 2012 (me) Comprehensive update posted live
- 4 December 2006 (me) Comprehensive update posted live
- 7 October 2003 (me) Comprehensive update posted live
- 3 April 2000 (me) Review posted live
- October 1998 (kk) Original submission
References
Published Guidelines / Consensus Statements
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS, et al. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328–43. [PubMed]
- Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available online. 2013. Accessed 4-2-24.
- European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53:3–22. [PubMed]
- European Association for the Study of the Liver. EASL clinical practice guidelines on haemochromatosis. J Hepatol. 2022;77:479-502. [PubMed]
- National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset conditions. Available online. 2018. Accessed 4-2-24.
Literature Cited
- Adams P, Altes A, Brissot P, Butzeck B, Cabantchik I, Cançado R, Distante S, Evans P, Evans R, Ganz T, Girelli D, Hultcrantz R, McLaren G, Marris B, Milman N, Nemeth E, Nielsen P, Pineau B, Piperno A, Porto G, Prince D, Ryan J, Sanchez M, Santos P, Swinkels D, Teixeira E, Toska K, Vanclooster A, White D, et al. Therapeutic recommendations in HFE hemochromatosis for p.Cys282Tyr (C282Y/C282Y) homozygous genotype. Hepatol Int. 2018;12:83–6. [PMC free article: PMC5904234] [PubMed: 29589198]
- Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010;116:317–25. [PubMed: 20308595]
- Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, Dawkins FW, Acton RT, Harris EL, Gordeuk VR, Leiendecker-Foster C, Speechley M, Snively BM, Holup JL, Thomson E, Sholinsky P. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769–78. [PubMed: 15858186]
- Adris N, Hazeldine S, Bentley P, Trinder D, Chua ACG, Powell LW, Ramm LE, Ramm GA, Olynyk JK. Detection of HFE haemochromatosis in the clinic and community using standard erythrocyte tests. Blood Cells Mol Dis. 2019;74:18-24. [PubMed: 30340937]
- Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, McLaren CE, Bahlo M, Nisselle AE, Vulpe CD, Anderson GJ, Southey MC, Giles GG, English DR, Hopper JL, Olynyk JK, Powell LW, Gertig DM. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221–30. [PubMed: 18199861]
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS, et al. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328–43. [PMC free article: PMC3149125] [PubMed: 21452290]
- Bardou-Jacquet E, de Tayrac M, Mosser J, Deugnier Y. GNPAT variant associated with severe iron overload in HFE hemochromatosis. Hepatology. 2015a;62:1917–8. [PubMed: 25891252]
- Bardou-Jacquet E, Lainé F, Deugnier Y. Reply to: "Reduced mortality due to phlebotomy in moderately iron-loaded HFE haemochromatosis? The need for clinical trials." J Hepatol. 2015b;63:283–4. [PubMed: 25841362]
- Bardou-Jacquet E, Morcet J, Manet G, Lainé F, Perrin M, Jouanolle AM, Guyader D, Moirand R, Viel JF, Deugnier Y. Decreased cardiovascular and extrahepatic cancer-related mortality in treated patients with mild HFE hemochromatosis. J Hepatol. 2015c;62:682–9. [PubMed: 25450707]
- Barton JC, Acton RT, Dawkins FW, Adams PC, Lovato L, Leiendecker-Foster C, McLaren CE, Reboussin DM, Speechley MR, Gordeuk VR, McLaren GD, Sholinsky P, Harris EL. Initial screening transferrin saturation values, serum ferritin concentrations, and HFE genotypes in whites and blacks in the Hemochromatosis and Iron Overload Screening Study. Genet Test. 2005;9:231-41. [PubMed: 16225403]
- Barton JC, Barton JC, Acton RT. Iron overload phenotypes and HFE genotypes in white Hemochromatosis and Iron Overload Screening Study participants without HFE p.C282Y/p.C282Y. PLoS One. 2022;27;17:e0271973. [PMC free article: PMC9328571] [PubMed: 35895739]
- Barton JC, Barton JC, Acton RT. Non-alcoholic fatty liver disease in hemochromatosis probands with iron overload and HFE p.C282Y/p.C282Y. BMC Gastroenterology. 2023;23:137. [PMC free article: PMC10148383] [PubMed: 37118679]
- Barton JC, Barton JC, Adams PC. Prevalence and characteristics of anti-HCV positivity and chronic hepatitis C virus infection in HFE p.C282Y homozygotes. Ann Hepatol. 2019;18:354-359. [PubMed: 31056361]
- Barton JC, Bertoli LF, Rothenberg BE. Peripheral blood erythrocyte parameters in hemochromatosis: evidence for increased erythrocyte hemoglobin content. J Lab Clin Med 2000;135:96-104. [PubMed: 10638700]
- Barton JC, Bottomley SS. Iron deficiency due to excessive therapeutic phlebotomy in hemochromatosis. Am J Hematol. 2000;65:223–6. [PubMed: 11074539]
- Barton JC, Edwards CQ, Acton RT. HFE gene: structure, function, mutations, and associated iron abnormalities. Gene. 2015;574:179–92. [PMC free article: PMC6660136] [PubMed: 26456104]
- Barton JC, McLaren CE, Chen WP, Ramm GA, Anderson GJ, Powell LW, Subramaniam VN, Adams PC, Phatak PD, Gurrin LC, Phillips JD, Parker CJ, Emond MJ, McLaren GD. Cirrhosis in hemochromatosis: independent risk factors in 368 HFE p.C282Y homozygotes. Ann Hepatol. 2018;17:871–9. [PMC free article: PMC6368858] [PubMed: 30145563]
- Barton JC, Parker CJ. Hemochromatosis. In: Means RT Jr, Roders GM, Glader B, Arber DA, Appelbaum FR, Disperenzieri A, Fehniger TA, Michaelis LC, Leonard JPl, eds. Wintrobe's Clinical Hematology. 15 ed. Philadelphia, PA: Wolters-Kluwer/Lippincott Williams and Wilkins; 2023:ch 27.
- Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845G-->A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211–8. [PubMed: 11812557]
- Bomford A, Williams R. Long term results of venesection therapy in idiopathic haemochromatosis. Q J Med 1976;45:611–623. [PubMed: 188063]
- Cade JE, Moreton JA, O'Hara B, Greenwood DC, Moor J, Burley VJ, Kukalizch K, Bishop DT, Worwood M. Diet and genetic factors associated with iron status in middle-aged women. Am J Clin Nutr. 2005;82:813-820. [PubMed: 16210711]
- Cançado R, Melo MR, de Moraes Bastos, Santos PCJL, Guerra-Shinohara EM, Chiattone C, Ballas SK. Deferasirox in patients with iron overload secondary to hereditary hemochromatosis: results of a 1-yr Phase 2 study. Eur J Haematol. 2015;95:545-50. [PubMed: 25684349]
- Caughey AB, Krist AH, Wolff TA, Barry MJ, Henderson JT, Owens DK, Davidson KW, Simon MA, Mangione CM. USPSTF approach to addressing sex and gender when making recommendations for clinical preventive services. JAMA. 2021;326:1953-61. [PubMed: 34694343]
- Delatycki MB, Gurrin LC, Ong SY, Ramm GA, Anderson GJ, Olynyk JK, Allen KJ, Nicoll AJ, Powell LW. Reduced mortality due to phlebotomy in moderately iron-loaded HFE haemochromatosis? The need for clinical trials. J Hepatol. 2015;63:282–3. [PubMed: 25839407]
- Deugnier Y, Jouanolle A-M, Chaperon J, Moirand R, Pithois C, Meyer J-F, Pouchard M, Lafraise B, Brigand A, Caserio-Schoenemann C, Mosser J, Adams P, Le Gall J-Y, David V. Gender-specific phenotypic expression and screening strategies in C282Y-linked haemochromatosis: a study of 9396 French people. Br J Haematol. 2002;118:1170-8. [PubMed: 12199803]
- Dobrindt EM, Keshi E, Neulichedl J, Schöning W, Öllinger R, Pratschke J, Eurich D. Long-term outcome of orthotopic liver transplantation in patients With hemochromatosis: a summary of a 30-year transplant program. Transplant Direct. 2020;6:e560. [PMC free article: PMC7531770] [PubMed: 33062844]
- El-Serag HB, Inadomi JM, Kowdley KV. Screening for hereditary hemochromatosis in siblings and children of affected patients. A cost-effectiveness analysis. Ann Intern Med. 2000;132:261–9. [PubMed: 10681280]
- European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53:3–22. [PubMed: 20471131]
- European Association for the Study of the Liver. EASL clinical practice guidelines on haemochromatosis. J Hepatol. 2022;77:479-502. [PubMed: 35662478]
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, Wolff RK. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. [PubMed: 8696333]
- Feldman AG, Whitington PF. Neonatal hemochromatosis. J Clin Exp Hepatol. 2013;3:313–20. [PMC free article: PMC3940210] [PubMed: 25755519]
- Girelli D, Busti F, Brissot P, Cabantchik I, Muckenthaler MU, Porto G. Hemochromatosis classification: update and recommendations by the BIOIRON Society. Blood. 2022;139;3018-3029. [PMC free article: PMC11022970] [PubMed: 34601591]
- Gurrin LC, Osborne NJ, Constantine CC, McLaren CE, English DR, Gertig DM, Delatycki MB, Southey MC, Hopper JL, Giles GG, Anderson GJ, Olynyk JK, Powell LW, Allen KJ, et al. The natural history of serum iron indices for HFE C282Y homozygosity associated with hereditary hemochromatosis. Gastroenterology. 2008;135:1945–52. [PubMed: 18848943]
- Hutchinson C, Geissler C, Powell J, Bomford A. Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. Gut. 2007;56:1291-5. [PMC free article: PMC1954964] [PubMed: 17344278]
- Ioannou GN, Dominitz JA, Weiss NS, Heagerty PJ, Kowdley K. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology. 2004;126:1293-301. [PubMed: 15131790]
- Kowdley KV, Brandhagen DJ, Gish RG, Bass NM, Weinstein J, Schilsky ML, Fontana RJ, McCashland T, Cotler SJ, Bacon BR, Keeffe EB, Gordon F, Polissar N. Survival after liver transplantation in patients with hepatic iron overload: the national hemochromatosis transplant registry. Gastroenterology. 2005;129:494–503. [PubMed: 16083706]
- Kowdley KV, Brown KE, Ahn J, Sundaram V. ACG clinical guideline: hereditary hemochromatosis. Am J Gastroenterol. 2019;114:1202-18. [PubMed: 31335359]
- Kowdley KV, Modi NB, Peltekian PK, Vierling JM, Ferris C, Valone FH, Gupta A. Rusfertide for the treatment of iron overload in HFE-related haemochromatosis: an open-label, multicentre, proof-of-concept phase 2 trial. Lancet Gastroenterol Hepatol. 2023;8:1118-28. [PubMed: 37863080]
- Lebrón JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, Feder JN, Bjorkman PJ. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93:111–23. [PubMed: 9546397]
- Le Gac G, Congiu R, Gourlaouen I, Cau M, Férec C, Melis MA. Homozygous deletion of HFE is the common cause of hemochromatosis in Sardinia. Haematologica. 2010;95:685–7. [PMC free article: PMC2857203] [PubMed: 20007136]
- McLaren CE, Barton JC, Gordeuk VR, Wu L, Adams PC, Reboussin DM, Speechley M, Chang H, Acton RT, Harris EL, Ruggiero AM, Castro O, Hemochromatosis and Iron Overload Screen Study Research Investigators. Determinants and characteristics of mean corpuscular volume and hemoglobin concentration in white HFE C282Y homozygotes in the hemochromatosis and iron overload screening study. Am J Hematol. 2007;82:898-905 [PubMed: 17597476]
- Milman N, Pedersen P, á Steig T, Byg KE, Graudal N, Fenger K. Clinically overt hereditary hemochromatosis in Denmark 1948–1985: epidemiology, factors of significance for long-term survival, and causes of death in 179 patients. Ann Hematol. 2001;80:737–44. [PubMed: 11797115]
- Moirand R, Adams PC, Bicheler V, Brissot P, Deugnier Y. Clinical features of genetic hemochromatosis in women compared with men. Ann Intern Med. 1997;127:105-10. [PubMed: 9229998]
- Morrison ED, Brandhagen DJ, Phatak PD, Barton JC, Krawitt EL, El-Serag HB, Gordon SC, Galan MV, Tung BY, Ioannou GN, Kowdley KV. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med. 2003;138:627–33. [PubMed: 12693884]
- Nielsen P, Fischer R, Buggisch P, Janka-Schaub G. Effective treatment of hereditary haemochromatosis with desferrioxamine in selected cases. Br J Haematol. 2003;123:952-3. [PubMed: 14632789]
- Olynyk JK, Ramm GA. Hemochromatosis. N Engl J Med. 2022;387:2159-70. [PubMed: 36477033]
- Ong SY, Gurrin LC, Dolling L, Dixon J, Nicoll AJ, Wolthuizen M, Wood EM, Anderson GJ, Ramm GA, Allen KJ, Olynyk JK, Crawford D, Ramm LE, Gow P, Durrant S, Powell LW, Delatycki MB. Reduction of body iron in HFE-related haemochromatosis and moderate iron overload (Mi-Iron): a multicentre, participant-blinded, randomised controlled trial. Lancet Haematol. 2017;4:e607–e614. [PubMed: 29195602]
- Phatak PD, Bonkovsky HL, Kowdley KV. Hereditary hemochromatosis: time for targeted screening. Ann Intern Med. 2008;149:270–2. [PubMed: 18711158]
- Phatak P, Brissot P, Wurster M, Adams PC, Bonkovsky HL, Gross J, Malfertheiner P, McLaren GD, Niederau C, Piperno A, Powell LW, Russo MW, Stoelzel U, Stremmel W, Griffel L, Lynch N, Zhang Y, Pietrangelo A. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology. 2010;52:1671-779. [PMC free article: PMC3034044] [PubMed: 20814896]
- Piperno A, Arosio C, Fossati L, Viganò M, Trombini P, Vergani A, Mancia G. Two novel nonsense mutations of HFE gene in five unrelated Italian patients with hemochromatosis. Gastroenterology. 2000;119:441–5. [PubMed: 10930379]
- Piperno A, Fargio S, D'Alba R, Roffi L, Fracanzani AL, Vecchi L, Failla M, Fiorelli G. Liver damage in Italian patients with hereditary hemochromatosis is highly influenced by hepatitis B and C virus infection. J Hepatol 1992;16:364-8. [PubMed: 1487615]
- Prabhu A, Cargill T, Roberts N, Ryan JD. Systematic review of the clinical outcomes of iron reduction in hereditary hemochromatosis. Hepatology 2020;72:1469–82. [PubMed: 32500577]
- Rombout-Sestrienkova E, Nieman FH, Essers BA, van Noord PA, Janssen MC, van Deursen CT, Bos LP, Rombout F, van der Braak R, de Leeuw PW, Koek GH. Erythrocytapheresis versus phlebotomy in the initial treatment of HFE hemochromatosis patients: results from a randomized trial. Transfusion 2012;52:470–7. [PubMed: 21848963]
- Rombout-Sestrienkova E, Winkens B, Essers BA, Nieman FH, Noord PA, Janssen MC, van Deursen CTBM, Boonen A, Reuser-Kaasengrood EPJM, Heeremans J, van Kraaij M, Masclee A, Koek GH. Erythrocytapheresis versus phlebotomy in the maintenance treatment of HFE hemochromatosis patients: results from a randomized crossover trial. Transfusion 2016;56:261–70. [PubMed: 26358375]
- Schmidtke J. Twenty-five years of contemplating genotype-based hereditary hemochromatosis population screening. Genes (Basel). 2022;13:1622. [PMC free article: PMC9498654] [PubMed: 36140790]
- Scotet V, Mérour MC, Mercier AY, Chanu B, Le Faou T, Raguénes O, Le Gac G, Mura C, Nousbaum JB, Férec C. Hereditary hemochromatosis: effect of excessive alcohol consumption on disease expression in patients homozygous for the C282Y mutation. Am J Epidemiol. 2003;158:129–34. [PubMed: 12851225]
- Sundic T, Hervig T, Hannisdal S, Assmus J, Ulvik RJ, Olaussen RW, Berentson S. Erythrocytapheresis compared with whole blood phlebotomy for the treatment of hereditary haemochromatosis. Blood Transfus 2014;12:s84–s89. [PMC free article: PMC3934269] [PubMed: 24333062]
- Whitington PF, Kelly S, Taylor SA, Nóbrega S, Schreiber RA, Sokal EM, Hibbard JU. Antenatal treatment with intravenous immunoglobulin to prevent gestational alloimmune liver disease: comparative effectiveness of 14-week versus 18-week initiation. Fetal Diagn Ther. 2018;43:218–25. [PubMed: 28787718]
- Whitlock EP, Garlitz BA, Harris EL, Beil TL, Smith PR. Screening for hereditary hemochromatosis: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2006;145:209–23. [PubMed: 16880463]
- Wood MJ, Powell LW, Ramm GA. Environmental and genetic modifiers of the progression to fibrosis and cirrhosis in hemochromatosis. Blood. 2008;111;4456-62. [PubMed: 18316631]
Figure 1.
Use of serum ferritin concentration to direct management of persons with HFE-related hemochromatosis ALT =alanine transaminase; AST =aspartate transaminase
Publication Details
Author Information and Affiliations
Southern Iron Disorders Center
Birmingham, Alabama
University of Utah School of Medicine
Salt Lake City, Utah
Publication History
Initial Posting: April 3, 2000; Last Update: April 11, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Barton JC, Parker CJ. HFE-Related Hemochromatosis. 2000 Apr 3 [Updated 2024 Apr 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.