Clinical Description
Incontinentia pigmenti (IP) is a disorder of the skin and its appendages, eye, and central nervous system (CNS) that occurs primarily in females and on occasion in males.
The largest cohort of individuals with IP in whom the clinical and molecular diagnosis has been confirmed is reported in Fusco et al [2014].
Skin. See , , , and . IP manifests in stages that evolve sequentially. The onset and duration of each stage vary among individuals, and not all individuals experience all four stages. The skin abnormalities that define each stage occur along lines of embryonic and fetal skin development known as Blaschko lines (see ). Blaschko lines correspond with cell migration or growth pathways that are established during embryogenesis. Like dermatomes, they are linear on the limbs and circumferential on the trunk. Unlike dermatomes, Blaschko lines do not correspond to innervation patterns or spinal cord levels.
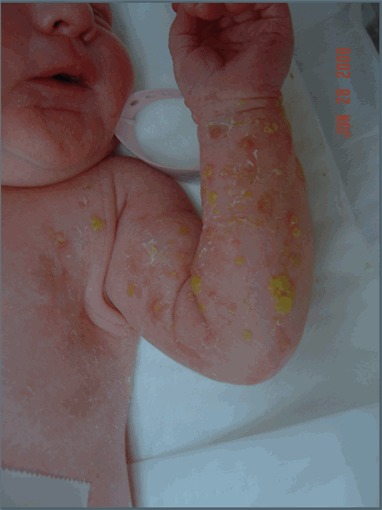
Stage I – bullous stage is characterized by blister-like bullous eruptions () that are linear on the extremities and/or circumferential on the trunk. The eruptions can be erythematous and may appear infectious. Stage I manifests within the first six to eight weeks and can be present at birth. The stage I rash generally disappears by age 18 months, although a vesicobullous eruption was reported in a girl age five years who was already manifesting the stage IV rash [
Darné & Carmichael 2007].
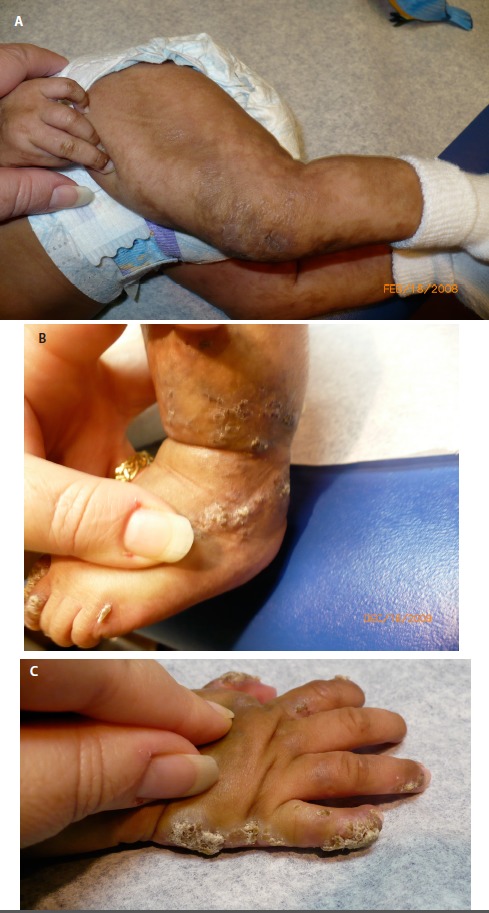
Stage II – verrucous stage is characterized by a hypertrophic, wart-like rash that is linear on the extremities and/or circumferential on the trunk (see ). This stage manifests within the first few months of life. It can occasionally be present at birth but typically arises as stage I begins to resolve. Stage II usually lasts for a few months, but it can last for years. Stage II can also include the appearance of dystrophic nails and abnormalities of tooth eruption.
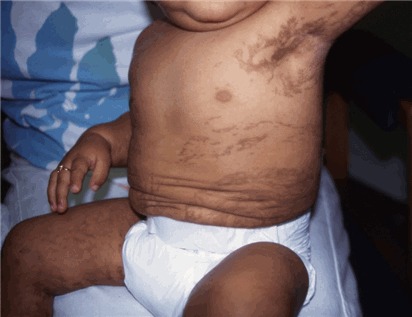
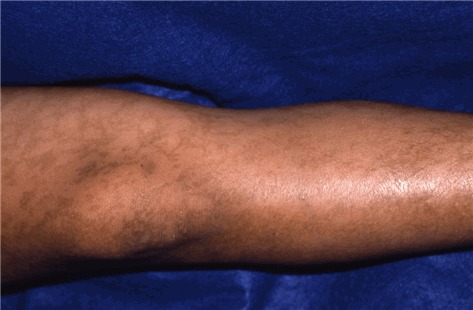
Stage III – hyperpigmentation stage is characterized by macular, slate gray, or brown hyperpigmentation that occurs in a "marble cake" or swirled pattern along Blaschko lines, usually circumferential on the trunk and linear on the extremities (see ). The hyperpigmentation stage is the most characteristic stage for IP. Not all women have extensive hyperpigmentation; it can be quite limited. The most frequently involved areas are the groin and axilla. The entire skin surface may need to be examined to find characteristic patterns. Hyperpigmentation begins between age six months and one year, usually as stage II begins to resolve. It is NOT present at birth. Stage III can persist into adulthood. The hyperpigmentation usually begins to fade in the teens and early twenties (see ). The pigmentation changes can be linear, swirled, or reticulated. A woman in her thirties or later may show no skin changes associated with IP.
Stage IV – atretic stage is characterized by linear hypopigmentation and alopecia, particularly noticeable on the extremities and, when it happens, on the scalp. The definition of stage IV remains open. There may not be true hypopigmentation, but rather a loss of hair and epidermal glands. As with the first three stages, the pattern follows Blaschko lines. Stage IV does not occur in all individuals. When present, it arises after the hyperpigmentation fades.
Hair. Alopecia may occur on the scalp and also on the trunk and extremities. Patchy alopecia of the scalp may correspond to areas of scarring left from blistering in stage I, but may also occur in individuals who have had no stage I or II lesions on the scalp. Alopecia occurs in areas of skin hypopigmentation as part of stage IV skin changes. Scalp hair may be thin or sparse in early childhood. Hair may also be lusterless, wiry, and coarse, often at the vertex in a "woolly-hair nevus." Areas of alopecia may be very small, unnoticed by the affected individual and difficult to find, particularly when covered by other scalp hair. Sparse eyelashes and eyebrows are also reported.
Teeth. Abnormalities include hypodontia (too few teeth), microdontia (small teeth), abnormally shaped teeth (e.g., conical teeth or accessory cusps), delayed eruption, or impaction. Enamel and tooth strength are normal. The tooth anomalies reported in individuals with IP are widely variable and may be said to encompass virtually any aberration in tooth shape and/or number.
Nails. Nails can be dystrophic (i.e., lined, pitted, or brittle). These changes often resemble fungal infections of the nails. Dystrophic nails are most commonly associated with stage II. The nail changes may be transient, but a single, chronic, longitudinal ridge in the nail was present in 28% of persons in one study [Phan et al 2005].
Ophthalmologic. Individuals with IP are at increased risk (20%-77%) for ophthalmologic abnormalities.
Central nervous system. Seizures, intellectual disability, and other CNS abnormalities have been reported in approximately 30% of individuals with IP [Minić et al 2014]. The actual incidence of neurocognitive disability is unclear because mildly affected individuals without neurocognitive problems may not come to medical attention [Phan et al 2005]. Neurocognitive disability is more common in simplex than in familial cases, presumably because mildly affected family members are identified. Males with IP are more likely than females to have neurologic abnormalities. In general, neurologic abnormalities in individuals with IP appear to be associated with underlying CNS vasculopathy [Meuwissen & Mancini 2012].
Seizures. Seizures in IP range from a single episode in a lifetime to chronic epilepsy. The type of seizure varies because the stroke etiology may involve any part of the cerebrum. In the review of well-documented individuals with IP who have neurocognitive disability,
Meuwissen & Mancini [2012] note that of seizure types reported, focal clonic seizures were the most frequently observed. Of all affected persons with neurocognitive problems, about 25% experience one or more seizures (i.e., ~7% of all individuals diagnosed with IP). The vast majority of seizures manifest within the first year of life (32 of 35 individuals with seizures where onset was reported). Fourteen of 25 individuals in whom recurrence was reported experienced only one seizure [
Meuwissen & Mancini 2012].
Intellect. Available studies of cognitive function in IP are limited. There is a range of function, including normal. Severe intellectual disability is not common. In males, co-occurrence of a 47,XXY karyotype may complicate the intellectual phenotype of IP.
Pizzamiglio et al [2014] reported a group of ten females with IP who were underwent cognitive assessment. Seven of the ten had deficits in calculation / arithmetic reasoning and reading but not writing skills. This evaluation makes it possible to place "learning disabilities" among the manifestations of IP. A follow-up study in 2017 [
Pizzamiglio et al 2017] showed that nine of 14 girls had normal development while five had intellectual disabilities ranging from mild to severe.
Evidence that
IKBKG pathogenic variants may cause abnormalities in microvasculature supports the theory that CNS dysfunction is secondary to vascular problems that result in transient ischemic attacks or full-blown hemorrhagic strokes [
Fiorillo et al 2003,
Hennel et al 2003,
Shah et al 2003]. Neurovascular abnormalities are most common in the first year of life, with only a handful of individuals reported after that, and only three after age four years [
Meuwissen & Mancini 2012].
Periventricular leukomalacia was identified on brain MRI in 27 of 43 individuals with IP who have neurocognitive disabilities, especially seizures, and subcortical white matter changes were also seen commonly. Some individuals have subsequent cystic changes. Myelination delays and ventricular dilatation have also been reported [
Meuwissen & Mancini 2012].
Spastic paresis. The frequency of this finding is unknown. It is difficult to interpret older literature findings. As with other neurologic abnormalities in IP, the risk and severity of spastic paresis appears to be related to CNS vasculopathy.
Breast. Abnormalities of mammary tissue ranging from aplasia of the breast to supernumerary nipples are variably present but are more common than in the general population [Minić et al 2014]. The recognized frequency of breast abnormalities may be limited because reports tend to focus on prepubertal children.
Other
Leukocytosis with up to 65% eosinophils may occur, particularly in stages I and II. The specific cause of the leukocytosis is unknown.
Zilberman-Rudenko et al [2016] note that pathogenic variants in
IKBKG impair suppression of NF-
KB, leading to hyperactive reactive inflammatory response. Eosinophilia is not consistently associated with any clinical manifestations and typically resolves spontaneously.
Primary pulmonary hypertension has been reported in some individuals and is presumably related to vasculopathy [
Alshenqiti et al 2017].
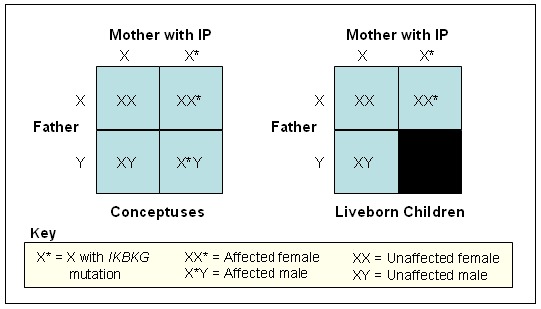
Males with IP. Although IP has been identified as a "male-lethal" disease, there are well-documented affected males. There are a few individual case reports published each year and intermittent reviews of the literature.
Survival in a male is mediated through one of two mechanisms:
The reasoning behind male lethality in IP is that male conceptuses that inherit an X chromosome with a mutated IKBKG gene lack the normal protein necessary for viability. The precise mechanism of male lethality is unknown [Hatchwell 1996], although mouse models suggest that liver failure plays a role [Rudolph et al 2000].
Pathogenic variants that produce a milder form of the condition are always associated with immunodeficiency, known as X-linked hypohidrotic ectodermal dysplasia and immunodeficiency (HED-ID), in males [Fusco et al 2008]. Only one male has been reported with HED-ID and also clinical findings of IP in association with the c.1167dupC
IKBKG variant [Chang et al 2008].
Life expectancy. For persons without significant neonatal or infantile complications, life expectancy is considered to be normal.
Reproductive fitness. Women with IP are at increased risk for pregnancy loss, presumably related to low viability of male fetuses. It is common for women with IP to experience multiple miscarriages, often around the third or fourth month of gestation. Fertility does not otherwise appear to be impaired; conception of an unaffected fetus would be expected to result in an uncomplicated pregnancy and delivery.