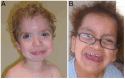
Clinical Description
HRAS-related Costello syndrome (Costello syndrome) affects multiple organ systems. The typical presentation is characterized by diffuse hypotonia and severe feeding difficulties in infancy; short stature; developmental delay or intellectual disability; characteristic facial features; curly or sparse, fine hair; loose, soft skin with deep palmar and plantar creases; papillomata of the face and perianal region; joint laxity with ulnar deviation of the wrists and fingers; tight Achilles tendons; and cardiac involvement (including hypertrophic cardiomyopathy [HCM], congenital heart defects, and arrhythmia). Postnatal cerebellar overgrowth can result in Chiari I malformation with associated hydrocephalus or syringomyelia. There is an approximate 15% lifetime risk for malignant tumors in individuals with Costello syndrome including rhabdomyosarcoma and neuroblastoma in young children and transitional cell carcinoma of the bladder in adolescents and young adults.
To date, more than 100 individuals have been identified with a pathogenic variant in HRAS [Aoki et al 2005, Estep et al 2006, Gripp et al 2006a, Kerr et al 2006, van Steensel et al 2006, Lin et al 2011, Weaver et al 2014]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Costello Syndrome: Frequency of Select Features
View in own window
| Feature | % or # of Persons w/Feature | Comment |
|---|
|
Growth
|
Failure to thrive
| >95% | Failure to thrive is typical in infancy. |
|
Short stature
| >95% | Stature typically remains short, even w/GH treatment. |
Gastrointestinal/
feeding
|
G-tube requirement
| 65% | Inability to eat by mouth is common throughout infancy & early childhood. Weight gain remains slow, even w/feeding tube use & high caloric intake.
|
|
Pyloric stenosis
| 5/58 |
|
Neurologic
|
Hypotonia
| 70% | |
|
Chiari I malformation
| 50% | Chiari I malformation may develop over time. |
|
Seizures
| 25% | Diagnosis of seizures should prompt investigation for hypoglycemia (see Management). |
|
Cardiac
|
Arrhythmia
| 55% | |
|
HCM
| 60% | |
|
Pulmonic valve stenosis
| 10% | |
|
Aortic dilatation
| 5%-10% | |
|
Atrial septal defects
| 5%-7% | |
|
Developmental delay
| >95% | |
|
Intellectual disability
| 80% | |
|
Anxiety
| 65% | |
|
Dermatologic
|
Papillomata
| 50%-60% | |
|
Callouses
| 65% | |
|
Musculoskeletal
|
Hip dysplasia
| 17%-45% | |
|
Osteopenia
| 45% | |
|
Scoliosis/kyphosis
| 17%-75% | Scoliosis & kyphosis may increase w/age. |
|
Ulnar deviation of fingers
| 82% | |
|
Elbow contractures
| 53% | |
|
Obstructive sleep apnea
| 7/10 | |
|
GH deficiency
| 45% | |
|
Solid tumors (overall risk incl rhabdomyosarcoma, neuroblastoma, & transitional cell carcinoma of bladder)
| 15% | See Genotype-Phenotype Correlations. |
|
Vision issues
|
Nystagmus
| 40% | |
|
Myopia
| 60% | |
|
Genitourinary
|
Cryptorchidism
| 50% of males | |
|
Renal anomalies
| 50% | |
GH = growth hormone; HCM = hypertrophic cardiomyopathy
Growth. Increased birth weight and head circumference (often >50th centile) for gestational age can lead to the categorization of Costello syndrome as a macrosomia disorder, which is misleading. Increased birth weight is instead related to fetal hydrops. Short stature is universal, delayed bone age is common [Johnson et al 1998], and testing may show partial or complete growth hormone deficiency.
Failure to thrive and severe feeding difficulties are almost universal and typically require a gastrostomy tube. Anecdotally, affected children have very high caloric needs. Even after nutrition is improved through supplemental feeding, growth restriction persists; therefore, aggressive (hypercaloric) feeding therapy is not effective. Children are typically able to take oral feeds beginning between ages two and four years.
Normative growth charts, derived from measurements of individuals who had not used growth hormone, document very slow weight gain in early infancy as well as short stature, with the 95th centile for individuals with Costello syndrome falling into the low-normal range of typical age-matched individuals [Sammon et al 2012]. The reported adult height range is 135-150 cm [Hennekam 2003].
Neurologic. Most infants have hypotonia, irritability, developmental delay, and nystagmus.
Hypotonia may be severe with low muscle mass and a skeletal myopathy phenotype [van der Burgt et al 2007, Tidyman et al 2011].
Progressive postnatal cerebellar overgrowth may result in the development of Chiari I malformation, syringomyelia, and hydrocephalus [Gripp et al 2010]. Cerebellar abnormalities include tonsillar ectopia or Chiari malformation, occasionally associated with syringomyelia [Gripp et al 2000, Gripp et al 2002, Calandrelli et al 2015].
EEG abnormalities are seen in approximately one third of individuals; between 20% and 50% have seizures [Delrue et al 2003, Kawame et al 2003].
Cardiac abnormalities, which typically present in infancy or early childhood, may be recognized at any age. In 146 individuals with molecularly confirmed Costello syndrome, 87% had some type of cardiovascular abnormality. A congenital heart defect was present in 44%, with non-progressive valvular pulmonic stenosis being the most common finding. Rarely, atrial septal defects are seen. Hypertrophic cardiomyopathy (HCM) comprising typical subaortic septal hypertrophy was noted in 61% and pathologic myocardial disarray was seen in 70% of those studied [Lin et al 2011].
A few neonates can present with very severe HCM that is lethal. In other infants, progressively severe HCM and/or severe multifocal atrial tachycardia can lead to death in the first two years of life. Use of the MEK inhibitor trametinib may be considered in these individuals [Geddes et al 2023] (see Targeted Therapy). Multifocal atrial tachycardia and other types pf atrial tachycardia may be very concerning but are usually self-limited with aggressive treatment.
Pulmonic valve stenosis is usually mild to moderate, and infrequently requires surgery or interventional catheterization.
Most children with HCM have either mild or moderate involvement. Of great interest are the few with moderate-to-severe involvement who appear to have "remodeling" over many years that gives the impression of disappearance of (or marked decrease in) left ventricular obstruction. Only a small number of these individuals are being followed, and their long-term natural history is incomplete [Lin et al 2011]. In addition to the rare severe lethal form, HCM can be chronic (persistent in its gradient severity) or progressive (increasing in gradient severity; 14/37 [37%]), stabilizing (without further increase in severity; 10/37 [27%]), or decreasing (resolving; 5/37 [14%]). Outcome was unavailable in 8/37 (22%) [Lin et al 2011], necessitating prudent surveillance.
Non-reentrant atrial tachycardias are generally self-limited but may persist or worsen in approximately one fourth of affected individuals. Non-reentrant atrial tachycardia occurs independently of HCM [Levin et al 2018].
Older individuals (ages 16 to 40 years) with moderate HCM or new-onset arrhythmia (both atrial and ventricular) represent the greatest challenge, and the outcomes in these individuals are not known. Hypertension is not uncommon.
Mild-to-moderate aortic dilatation not associated with bicuspid aortic valve is reported in approximately 5% of affected individuals [Lin et al 2011].
Primary vascular disease has rarely been reported.
Developmental delay or intellectual disability is present in all individuals [Axelrad et al 2004, Axelrad et al 2007, Axelrad et al 2009, Axelrad et al 2011].
Recognition memory in verbal memory functioning is relatively preserved compared to other cognitive tasks [Schwartz et al 2013].
While the underlying mechanism in Costello syndrome is not known, the skills necessary for swallowing and speech development are both affected and appear closely correlated. The onset of speech frequently coincides with the willingness to feed orally.
Behavioral/social issues. Many children younger than age four years meet criteria for autism spectrum disorder (ASD). In contrast, none of the children older than age four years met criteria for ASD, suggesting that early signs consistent with ASD tend to resolve by age four years [Schwartz et al 2017].
Separation anxiety, seen in 39% of individuals with Costello syndrome, is more common in males than in females [Axelrad et al 2011]. In older individuals, anxiety was reported in 65% [Shikany et al 2020].
Limited detailed information is available on the quality of life in older individuals with Costello syndrome. Quality of life in individuals aged 16-34 years is compromised by four factors: limited relationships outside of the immediate circle of friends and family, lack of independence, male sex, and the presence of major medical issues [Hopkins et al 2010]. Functional limitations from orthopedic issues related to mobility, as well as limitations in social and cognitive domains, were documented using normative scales [Johnson et al 2015]. A query of 20 individuals aged 16 years or older found one individual living independently, three living in a residential setting, and 16 living with family [Shikany et al 2020]. A history of seizures was reported in five (25%) and Chiari I malformation in 10 (50%). Mobility issues included the use of a walker in one (5%), wheelchair or stroller for distance only in nine (45%), wheelchair reliance in three (15%), and no mobility issues in seven individuals (35%) [Shikany et al 2020].
Dermatologic. Papillomata, absent in infancy, appear in young children, usually in the perinasal region and less commonly in the perianal region, torso, and extremities. While papillomata are mostly of cosmetic concern, they can become noticeable and at times bothersome.
Palmoplantar keratoderma is common and can affect function in severe cases [Marukian et al 2017]. Additional findings include acanthosis nigricans and thick toenails.
Musculoskeletal. Individuals with Costello syndrome have very loose joints, particularly involving the fingers. Ulnar deviation of the wrists and fingers is also common. Developmental hip dysplasia may result in severe pain and prevent ambulation. Tight Achilles tendons may develop and were either present or previously surgically corrected in 11 of 17 individuals (65%) [Leoni et al 2021].
More than half of a cohort of 43 individuals examined by an orthopedic surgeon with review of available radiographs showed ligamentous laxity, scoliosis, kyphosis, characteristic hand and wrist deformities, shoulder and elbow contractures, tight Achilles tendons, and flat feet [Detweiler et al 2013]. Hip dysplasia, seen in 45% of individuals, was not universally congenital but acquired in some.
Osteoporosis is common in young adults with Costello syndrome [White et al 2005, Detweiler et al 2013]. In adults ranging in age from 16 to 40 years, all eight individuals who had bone density measurements had abnormal results that suggested osteoporosis or osteopenia; three had bone pain, vertebral fractures, and height loss [White et al 2005]. In a study of nine individuals with Costello syndrome who had dual-energy x-ray absorptiometry scans performed, all showed significantly decreased bone mineral density compared to age-matched controls [Leoni et al 2014]. A review of older individuals identified osteoporosis or osteopenia in 9/20 (45%) and kyphosis or scoliosis in 15/20 (75%) [Shikany et al 2020].
Respiratory. Seven of ten individuals ages three to 29 years who underwent polysomnography had obstructive sleep apnea [Della Marca et al 2006]. A literature review showed respiratory complications in 78% of neonates, with the majority resolving over time [Gomez-Ospina et al 2016].
More severe complications (intrauterine hydrops, postnatal pulmonary effusions with respiratory compromise, and severe progressive HCM) have been reported in a few individuals with HRAS pathogenic variants associated with the severe Costello syndrome phenotype [Gomez-Ospina et al 2016] (see Genotype-Phenotype Correlations).
Upper airway obstruction was seen more often in older children and young adults [Gomez-Ospina et al 2016].
Endocrine. Neonatal hyperinsulinism has been reported [Alexander et al 2005, Sheffield et al 2015] and in one case was correlated with focal uniparental disomy for 11p within the pancreatic nodule [Gripp et al 2016].
In older individuals, hypoglycemia may be related to growth hormone deficiency. Growth hormone deficiency is common (30%-50%) [Estep et al 2006, Gripp et al 2010].
Several affected individuals have been diagnosed with hypothyroidism requiring thyroid hormone replacement.
Other endocrine issues may include delayed or dysregulated puberty including precocious puberty.
Solid tumors. Benign and malignant solid tumors occur with far greater frequency in individuals with Costello syndrome than the general population. The overall tumor incidence is approximately 15% over the lifetime of individuals with HRAS pathogenic variants [Gripp et al 2006a]. Kratz et al [2011] reviewed published cases and confirmed the 15% cumulative incidence of cancer in individuals with Costello syndrome by age 20 years. Rhabdomyosarcoma occurs most frequently, followed by neuroblastoma, transitional cell carcinoma of the bladder, and other solid tumors [Gripp 2005].
In a meta-analysis of 234 publications reporting 621 individuals from 35 countries, more than 9% had cancer, including rhabdomyosarcoma, transitional cell carcinoma of the bladder, and neuroblastoma. Cumulative incidence by age 20 years was 13% for cancer and 11% for cancer-free death. Death rate was 3%-4% until age three years. Survival after cancer appeared reduced [Astiazaran-Symonds et al 2023].
Rhabdomyosarcoma and neuroblastoma, tumors of early childhood, present in Costello syndrome at ages comparable to the general population. In contrast, transitional cell carcinoma of the bladder, which occurs in older adults (70% occurs in adults age >65 years) in the general population, may be found in adolescents with Costello syndrome. The ages at presentation in the three individuals with Costello syndrome with transitional cell carcinoma of the bladder were ten, 11, and 16 years. A review of cystoscopy findings in 13 individuals aged ten years or older found a macroscopic bladder lesion in 10/13 on first exam. Histology showed low-grade epithelial dysplasia in 7/10 (70%) and papillary urothelial neoplasm of low-malignant potential (PUNLMP) or low-grade bladder cancer in 3/10 (30%) [Leoni et al 2022].
Other
Neuroimaging. A systematic review of brain and spinal cord MRI studies revealed posterior fossa crowding with cerebellar tonsillar herniation in 27/28 (96%) individuals [Gripp et al 2010]. In a majority of those with serial studies, posterior fossa crowding progressed. Due to the progressive nature of the cerebellar overgrowth – which likely results from abnormal cell differentiation as reported by Paquin et al [2009] – the sequelae of posterior fossa crowding included hydrocephalus requiring shunt placement or ventriculostomy (7/28), Chiari I malformation (9/28), and syringomyelia (7/28). In individuals age 16 years or older, a Chiari I malformation was reported in 10/20 (50%) individuals [Shikany et al 2020].
Tethered cord is relatively common.
Life expectancy. Causes of death in the literature, including an analysis of cardiovascular findings [Lin et al 2011], were reported in 10% of affected individuals and include HCM, coronary artery fibromuscular dysplasia, multifocal tachycardia, neoplasia, pulmonary causes, and multiorgan failure.
Mosaic Costello syndrome. Individuals with somatic mosaicism for a pathogenic variant in HRAS may show patchy skin findings only (as reported in the father of an individual with typical Costello syndrome [Sol-Church et al 2009, Bertola et al 2017]) or have findings indistinguishable from Costello syndrome caused by a germline pathogenic variant [Girisha et al 2010].
One individual with somatic mosaicism (20%-30% of DNA derived from buccal cells had the pathogenic HRAS variant p.Gly12Ser, which was not detected in DNA derived from peripheral blood cells) had typical findings attributed to Costello syndrome (intellectual disability, short stature, sparse hair, coarse facial features, nasal papillomata, and tight Achilles tendons) as well as several atypical findings attributed to mosaicism (microcephaly, streaky areas of skin hypo- and hyperpigmentation, and normal menarche with subsequent regular menses) [Gripp et al 2006b].