Holoprosencephaly Overview
Cedrik Tekendo-Ngongang, MD, Maximilian Muenke, MD, FACMG, and Paul Kruszka, MD, MPH.
Author Information and AffiliationsInitial Posting: December 27, 2000; Last Update: March 5, 2020.
Estimated reading time: 35 minutes
Summary
The purpose of this overview is to increase the awareness of clinicians regarding the genetic causes of holoprosencephaly and to inform genetic counseling of family members. The following are the goals of this overview.
Goal 3.
Provide an evaluation strategy to identify (when possible) the genetic cause of holoprosencephaly in a proband.
Goal 4.
Inform genetic counseling of family members of an individual with holoprosencephaly.
1. Clinical Characteristics of Holoprosencephaly
Holoprosencephaly (HPE), the most common malformation of the forebrain in humans, is a structural anomaly of the brain resulting from failed or incomplete forebrain division in the third to fourth weeks of gestation; the forebrain (prosencephalon) incompletely cleaves into right and left hemispheres, deep brain structures, and the olfactory and optic bulbs and tracts [Gropman & Muenke 2005, Dubourg et al 2007, Grinblat & Lipinski 2019].
While HPE is often first identified on prenatal ultrasound examination [Kousa et al 2018], it is most frequently diagnosed during the newborn period when abnormal facial findings and/or neurologic presentation prompt further evaluation. Infants with normal facies or only mildly abnormal facies and either mild or intermediate brain anomalies may not be diagnosed until the first year of life when neuroimaging studies obtained during evaluation for developmental delay and/or failure to thrive reveal HPE [Weiss et al 2018b].
Imaging of the brain by CT scan or MRI defines the type of HPE and identifies associated CNS anomalies [Hahn & Barnes 2010, Griffiths & Jarvis 2016, Kousa et al 2018]. The study of choice is cranial MRI examination, preferably obtained with adequate sedation at a pediatric center experienced in evaluating children for structural brain anomalies. Review of the study by a radiologist and/or other clinician familiar with the clinical subtypes of HPE is essential, as subtle midline anomalies may be missed, and non-HPE-related malformation findings may be mistaken for findings of HPE [Solomon et al 2009b].
Types of HPE
HPE, a continuum of brain malformations, is traditionally divided into the following types (in decreasing order of severity) (reviewed in Hahn & Barnes [2010]):
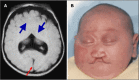
Alobar HPE, in which there is a single "monoventricle" and no separation of the cerebral hemispheres (), is the most severe form. The range of findings includes:
Alobar HPE A. MRI of alobar holoprosencephaly (HPE), the most severe form of HPE, characterized by an enlarged midline monoventricle (holoventricle, red/thin arrow) with fusion of the frontal lobes and the midline gray matter structures (thalami and basal (more...)
Cyclopia: single eye or partially divided eye in single orbit with a proboscis above the eye
Cyclopia without proboscis
Ethmocephaly: extremely closely spaced eyes but separate orbits with proboscis between the eyes
Cebocephaly: closely spaced eyes with single-nostril nose
Closely spaced eyes
Anophthalmia or microophthalmia
Premaxillary agenesis with median cleft lip, closely spaced eyes, depressed nasal ridge
Bilateral cleft lip
Relatively normal facial appearance (especially in persons with pathogenic variants in ZIC2)
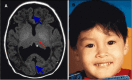
Semilobar HPE. The left and right frontal and parietal lobes are fused and the interhemispheric fissure is only present posteriorly (). The range of findings includes:
Semilobar HPE A. MRI showing semilobar HPE. Note fusion of the frontal lobes, but presence of some septation posteriorly with presence of a falx and interhemispheric fissure (red/thin arrow). The splenium of the corpus callosum is present but more anterior (more...)
Closely spaced eyes
Anophthalmia/microophthalmia
Depressed nasal ridge
Absent nasal septum
Flat nasal tip
Bilateral cleft lip with median process representing the philtrum-premaxilla anlage
Midline cleft (lip and/or palate)
Relatively normal facial appearance
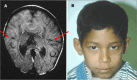
Lobar HPE. Most of the right and left cerebral hemispheres and lateral ventricles are separated but the frontal lobes, most rostral aspect of the telencephalon, are fused, especially ventrally (). The range of findings includes:
Lobar HPE A. MRI in axial plane depicting lobar HPE, the least severe of the major types of HPE. The cerebral hemispheres are separated (blue/thick arrows); the ventricles are misshapen as a result of absence of the septum pellucidum. The posterior portion (more...)
Middle interhemispheric fusion variant (MIHF/MIHV or syntelencephaly). The posterior frontal and parietal lobes fail to separate, with varying lack of cleavage of the basal ganglia and thalami and absence of the body of the corpus callosum but presence of the genu and splenium of the corpus callosum ().
Middle interhemispheric fusion (MIHF) A. MRI in axial plane depicting middle interhemispheric variant of HPE in which the anterior portions of the frontal lobes and the occipital lobes are well separated. The sylvian fissures are oriented nearly vertically (more...)
The range of findings includes:
Septopreoptic type. Nonseparation is restricted to the septal and/or preoptic regions; described in small case series [Hahn et al 2010].
Microforms of HPE (also termed "microform HPE") are clinical subtypes of HPE defined by the presence of HPE-related craniofacial anomalies without structural brain defects on imaging. They may occur in simplex HPE (i.e., a single occurrence of HPE in a family) or in relatives of probands with classic forms of HPE (). Their clinical spectrum includes the following:
Microforms of holoprosencephaly (HPE) spectrum with milder craniofacial anomalies in the absence of neurologic findings A. Premaxillary agenesis with repaired bilateral clefts of the lip
Other Structural CNS Findings
Other structural CNS findings that may occur with but are not specific to HPE:
Anomalies of midline structures: undivided thalami, agenesis of the corpus callosum (OMIM
217990), callosal dysgenesis [
Kidron et al 2016], absent septum pellucidum, and absent or hypoplastic olfactory bulbs and tracts (arrhinencephaly) and optic bulbs and tracts
Macrocephaly secondary to hydrocephalus
Dandy-Walker malformation
Neuronal migration anomalies
Abnormal circle of Willis
Caudal dysgenesis
Craniofacial Anomalies
The continuum of craniofacial anomalies, present in approximately 80% of individuals with HPE, includes cyclopia, synophthalmia, or a proboscis at the severe end and normal facies in individuals who have, but are not expressing, an HPE pathogenic variant inherited in an autosomal dominant manner. Common subtle facial features in individuals without obvious craniofacial findings include microcephaly (although hydrocephalus can result in macrocephaly), closely spaced eyes (also known as hypotelorism; potentially severe), depressed nasal ridge, and cleft lip and/or palate. A single maxillary central incisor may be present; although a nonspecific finding, it is a distinctive microform in autosomal dominant HPE [Lacbawan et al 2009, Richieri-Costa & Ribeiro 2010].
Of note, subtle facial anomalies in mildly affected family members can be easily overlooked [Lacbawan et al 2009, Solomon et al 2009a].
Malformations of the nose include complete absence, agenesis of the nasal cartridge, and proboscis (flat nose with a single central nostril without nasal bones).
Palatal anomalies include various midline and lateral clefts, midline palatal ridge, bifid uvula, high-arched palate, and absence of the superior labial frenulum [Solomon et al 2010a].
The extremely variable phenotypic expression occurs both in simplex HPE (i.e., a single occurrence in a family) and among members of the same family with an inherited form of HPE.
Clinical Manifestations of HPE
Clinical manifestations (reviewed in Levey et al [2010] and Solomon et al [2010a]) commonly observed in children with HPE include the following:
Developmental delay is present in all individuals with the HPE spectrum of CNS anomalies. The degree of delay is variable, correlating with the severity of the brain malformation, but tends to be severe.
Seizures are common, and may be difficult to control.
Hydrocephalus can occur, and may result in macrocephaly, rather than the more commonly observed microcephaly.
Neural tube defects occur in a small proportion of individuals.
Hypothalamic and brain stem dysfunction may lead to swallowing difficulties and instability of temperature, heart rate, and respiration.
Pituitary dysfunction is manifest by partial or complete panhypopituitarism with abnormal function of any or all of the anterior and/or posterior pituitary hormones, though central diabetes insipidus is by far the most common finding in persons with nonchromosomal, nonsyndromic HPE [
Lacbawan et al 2009,
Solomon et al 2010a].
Short stature and failure to thrive are common, especially in more severely affected children. Growth hormone deficiency and/or chromosome anomalies may in part be responsible for poor growth in some individuals.
Feeding difficulties may be a major problem in children with HPE. At least part of the difficulty may derive from axial hypotonia, poor suck as a result of neurologic complications, lethargy, seizures and their effects, side effects of medications, and lack of interest. Often gastroesophageal reflux, choking, and gagging occur with feeds. Additional common problems include slowness in eating, frequent pauses, and frank vomiting with risk of aspiration. Oral-sensory dysfunction may affect feeding especially when associated with textural aversion and labial and lingual weakness. Children with cleft lip and/or palate often have additional difficulties with oral feeding.
Excessive intestinal gas/colic, irritability, and constipation frequently occur [
Levey et al 2010].
Aspiration pneumonia can be a complication of poor coordination of swallowing.
Erratic sleep patterns can occur.
Life expectancy. A common misperception is that children with HPE do not survive beyond early infancy. While this is the case for the most severely affected children, a significant proportion of more mildly affected children (as well as some severely affected children) survive past age 12 months. In fact, a proportion of individuals with HPE of various subtypes, including severe subtypes, survive until adulthood.
The longer survival may be on account of recent advances in diagnostic methods, including brain imaging methods that allow for early detection of both severe and mild malformations. Improvement in the management of HPE over time may have also contributed to longer survival [Levey et al 2010, Pineda-Alvarez et al 2010, Weiss et al 2018b].
The distribution of HPE subtypes appears to be similar among children, adolescents, and adults, with semilobar HPE representing approximately 50% of HPE in both children and adults. The exception is alobar HPE, which appears to be less frequent in adults than children [Weiss et al 2018a].
Among affected individuals with abnormal chromosome complement, an inverse relationship exists between the severity of the facial phenotype and length of survival.
Among affected adolescents and adults, sensorineural hearing loss is found in approximately 30% and cortical vision impairment in approximately 20% [Weiss et al 2018a].
Of note, 60% of adolescent and adult survivors have severe involvement: they are nonambulatory and nonverbal with minimal hand function, and full dependence on caregivers [Weiss et al 2018a]. There is a correlation between the degree of developmental delay and the severity of the brain malformation or HPE subtype [Levey et al 2010, Weiss et al 2018a].
2. Genetic Causes of Holoprosencephaly
Chromosome Abnormalities with Holoprosencephaly
Approximately 25%-50% of individuals with HPE have a chromosome abnormality. Chromosome abnormalities are nonspecific and either numeric or structural, and can involve any chromosome [Dubourg et al 2018].
Individuals with HPE and a normal chromosome complement cannot be distinguished from those with an abnormal chromosome complement based on craniofacial abnormality or HPE subtype; however, individuals with HPE caused by a chromosome abnormality are more likely to have other organ system involvement, resulting in a more severe clinical course in most.
Numeric chromosome abnormalities. Trisomy 13, the most common cause of HPE, is observed in 40%-60% of HPE of all causes and about 75% of HPE caused by chromosome abnormalities. Birth prevalence of trisomy 13 is 1:5000. Arrhinencephaly is seen in about 70% of individuals with trisomy 13.
The other common aneuploidies associated with HPE include trisomy 18 and triploidy. Various other aneuploidies have been reported [Kagan et al 2010, Solomon et al 2010c, Petracchi et al 2011, Toufaily et al 2016, Rosa et al 2017].
Structural chromosome abnormalities associated with HPE have been reported in virtually all chromosomes. The most frequent are deletions or duplications involving various regions of 13q, and del(18p), del(7)(q36), dup(3)(p24-pter), del(2)(p21), and del(21)(q22.3) [Hu et al 2018]. Many of these regions contain genes known to be associated with autosomal dominant nonsyndromic HPE (Tables 2a, 2b).
Pathogenic copy number variations (CNVs). Chromosomal microarray (CMA) includes array-based comparative genomic hybridization (array CGH) and SNP array. CMA has identified pathogenic CNVs (including loci already known to be associated with HPE) in 10% of all individuals with HPE. Note that CNV detection rates may vary by testing laboratory and methodology [Dubourg et al 2018, Hu et al 2018].
Monogenic Syndromes with Holoprosencephaly as an Occasional Finding
Approximately 18%-25% of individuals with HPE have a pathogenic variant in a single gene causing syndromic HPE. At least 25 different conditions in which HPE is an occasional finding have been described; the majority of these disorders are rare. Some of the more common are summarized in Table 1 [Kruszka & Muenke 2018].
Table 1.
Syndromes with Holoprosencephaly as an Occasional Finding: Monogenic Causes
View in own window
AD = autosomal dominant; AR = autosomal recessive; CHD = congenital heart disease; HPE = holoprosencephaly; MOI = mode of inheritance
Single-Gene Disorders with Isolated (Nonsyndromic) Holoprosencephaly
The nonsyndromic forms of HPE best understood at a molecular genetic level are inherited in an autosomal dominant manner (see Tables 2a, 2b).
The phenotype of individuals with pathogenic variants in genes associated with nonsyndromic HPE is extremely variable even within the same family, ranging from alobar HPE with cyclopia to clinically normal [Solomon et al 2010a, Solomon et al 2010b, Dubourg et al 2018].
In the majority of individuals with HPE, a correlation exists between the facial anomalies and the gene involved and/or type of pathogenic variant (see and Types of HPE). However, it is important to note that in many cases this correlation cannot be made.
Facial findings in holoprosencephaly (HPE) A. Alobar HPE with cyclopia and proboscis above the single eye
Table 2a.
Nonsyndromic Holoprosencephaly: Most Common Monogenic Causes
View in own window
| Gene 1 | % of All
Nonsyndromic
HPE | Craniofacial
Findings | Other Key Features / Comment | Selected
OMIM Entries |
|---|
|
SHH
| 5.4%-5.9% 2 | Spectrum of HPE-related craniofacial features | Affected persons may be part of large kindreds segregating the variant; many of these families are not identified until ascertainment of the severely affected proband. 3 Renal/urinary anomalies may be more common than in those w/pathogenic variants in other HPE genes. 4
|
142945
|
|
ZIC2
| 4.8%-5.2% 2 | Bitemporal narrowing, upslanted palpebral fissures, large ears, short nose w/anteverted nares, & broad & deep philtrum 3 | Renal/urinary anomalies may be more common than in those w/pathogenic variants in other HPE genes. 4 Pathogenic variants are more frequently de novo than are variants in other HPE genes & appear to have high penetrance w/relatively few mildly manifesting persons. 5
|
609637
|
|
SIX3
| ~3% 6 | Spectrum of HPE-related craniofacial features | Affected persons may be part of large kindreds segregating the variant; many of these families are not identified until ascertainment of the severely affected proband. 3 May be assoc w/more severe types of HPE
|
157170
|
|
TGIF1
| <1% 2 | Spectrum of HPE-related craniofacial features | May demonstrate the entire spectrum of severity 7 |
142946
|
AD = autosomal dominant; AR = autosomal recessive; CHD = congenital heart disease; DD = developmental delay; HPE = holoprosencephaly; LOF = loss of function; MOI = mode of inheritance
- 1.
Genes are listed in order of most commonly involved.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
Table 2b.
Nonsyndromic Holoprosencephaly: Less Common Monogenic Causes
View in own window
| Gene 1 | % of All
Nonsyndromic
HPE | Craniofacial
Findings | Other Key Features / Comment | Selected
OMIM
Entries |
|---|
|
CDON
| Rare | Rare | Range of classic HPE-spectrum features described in several unrelated persons 2 |
614226
|
|
CNOT1
| <1.5% | Microtia, microcephaly, epicanthal folds, long philtrum | Semilobar HPE described in 2 unrelated persons Neonatal diabetes mellitus requiring insulin may be a feature. Pancreatic exocrine insufficiency may be present. May be assoc w/sensorineural & conductive hearing loss w/ossicle anomalies 3
| |
|
DISP1
| <1.2% | Bilateral cleft lip/palate, hypotelorism, single central maxillary incisor | Facial features may be consistent w/HPE-spectrum anomalies w/o corresponding brain anomalies. 4 |
607502
|
|
DLL1
| <1% | Rare | Facial features may be consistent w/HPE-spectrum anomalies. 5 |
606582
|
|
FGF8
| <2.2% | Spectrum of HPE-related craniofacial features | Range of classic HPE-spectrum features described in several unrelated persons 6 |
600483
|
|
FGFR1
| ~ 1.2% | Spectrum of HPE-related craniofacial features | Isolated HPE 7 | |
|
KMT2D
| Rare | Spectrum of HPE-related craniofacial features | Range of classic HPE-spectrum features described in 2 unrelated persons 8 | |
|
PPP1R12A
| Rare | Wide spectrum of craniofacial features | HPE, urogenital malformations, & DD 9 | |
|
RAD21
| Rare | Spectrum of HPE-related craniofacial features |
| |
|
SMC1A
| Rare | Spectrum of HPE-related craniofacial features | Range of classic HPE-spectrum features described in 5 unrelated females 10 | |
|
SMC3
| Rare | HPE-related craniofacial features | HPE-spectrum features described in 1 person w/semilobar HPE 10 | |
|
STAG2
| Rare | Spectrum of HPE-related craniofacial features | Range of classic HPE-spectrum features described in 6 unrelated females 10 | |
|
STIL
| Rare | Rare |
| |
AR = autosomal recessive; DD = developmental delay; HPE = holoprosencephaly
- 1.
Genes are listed in alphabetic order.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
Variants in other candidate genes including FOXH1, GAS1, NODAL, PTCH1, SUFU, and CRIPTO (formerly TDGF1) have seldom been reported in individuals with nonsyndromic HPE. However, because investigations of several large HPE cohorts have failed to reproduce these findings, more data may be needed to confirm the possible role of these genes in HPE pathogenesis.
Although GLI2 pathogenic variants were described as a cause of HPE, it has become clear that GLI2 variants do not cause HPE, but rather a distinct phenotype characterized by pituitary anomalies, polydactyly, and subtle facial features (sometimes similar to HPE facial features) consistent with Culler-Jones syndrome (OMIM 615849) [Bear et al 2014, Bear & Solomon 2015].
Holoprosencephaly of Unknown Cause
Autosomal dominant vs multifactorial. Microtia-anotia (OMIM 600674) and other anomalies
Unknown mode of inheritance [Kruszka & Muenke 2018]
Caudal dysgenesis (OMIM
600145)
Pseudotrisomy 13 (OMIM
264480)
Brachial amelia, cleft lip, and holoprosencephaly (OMIM
601357)
3. Evaluation Strategies to Identify the Genetic Cause of Holoprosencephaly in a Proband
Establishing a specific genetic cause of holoprosencephaly (HPE) can aid in discussions of prognosis (which are beyond the scope of this GeneReview) and genetic counseling.
Evaluations to determine a specific genetic cause of holoprosencephaly usually involve the following.
Prenatal history to identify possible environmental causes. The most common teratogen in humans known to cause HPE is diabetes mellitus. While both gestational and pre-gestational diabetes are risk factors for HPE, pre-gestational diabetes requiring insulin confers the highest (>10-fold increased) HPE risk [Johnson & Rasmussen 2010, Tinker et al 2019].
Physical examination. A detailed physical examination should be conducted with special emphasis on extracranial features, especially those associated with syndromic forms of HPE (see Table 1) [Kruszka & Muenke 2018]. Of note, it is not uncommon to find extracranial anomalies in individuals with nonsyndromic HPE (see Tables 2a, 2b) [Martinez et al 2018].
Family history. A three-generation family history should be taken with attention to pregnancy loss, neonatal deaths, and relatives with manifestations of holoprosencephaly and/or developmental delay with documentation of relevant findings through direct examination or review of medical records, including results of molecular genetic testing.
Focused examination of the parents and apparently normal sibs (whenever possible) to identify milder manifestations of HPE.
Molecular genetic testing. Approaches can include a combination of gene-targeted testing (multigene panel or single-gene testing) and comprehensive genomic testing (chromosomal microarray analysis, genome sequencing, exome sequencing, or exome array). Gene-targeted testing requires the clinician to hypothesize which gene(s) are likely involved, whereas genomic testing does not.
Chromosomal microarray analysis (CMA) using oligonucleotide or SNP arrays detects genome-wide large deletions/duplications in all forms of HPE, including apparently nonsyndromic HPE and HPE with associated anomalies. CMA has been successful in identifying pathogenic variants in up to 14% of individuals with HPE who have a normal karyotype and no causative gene identified on multigene panel testing.
Chromosome analysis. Diagnostic methods for detecting chromosome abnormalities in individuals with HPE are not different from those routinely used in the investigation of numeric and structural cytogenetic abnormalities in those with other genetic conditions or birth defects. Recommended methods include: G-banding karyotype and CMA [
Kruszka et al 2018]. If there is clinical suspicion for trisomy 13, a karyotype should be done first; otherwise, a CMA should be done first [
Pineda-Alvarez et al 2010,
Solomon et al 2010c], including trisomy 13.
Single-gene testing. When a specific syndromic cause of HPE is considered (
Table 1), sequence analysis of the gene of interest is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used and the gene involved, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to consider gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
A multigene panel that includes some or all of the genes listed in
Tables 1,
2a, and
2b is most likely to identify the genetic cause of HPE while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. Of note, given the rarity of some of the genes associated with HPE, some panels may not include all the genes mentioned in this overview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Comprehensive
genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is most commonly used; genome sequencing is also possible. If exome sequencing is not diagnostic – and particularly when evidence supports autosomal dominant or mendelian inheritance – exome array (when clinically available) may be considered to detect multiexon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click
here. More detailed information for clinicians ordering genomic testing can be found
here.
4. Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
If a genetic etiology (chromosome abnormality, monogenic condition, or pathogenic variant in a HPE-associated gene) is established in a proband with HPE, specific counseling for recurrence risk is indicated [Hadley et al 2018].
Chromosome Abnormality – Risk to Family Members
Parents of a proband
Parents of a child with a numeric chromosome abnormality (e.g., trisomy or triploidy) are expected to be chromosomally and phenotypically normal.
Parents of a child with a structural unbalanced chromosome rearrangement (e.g., deletion, duplication) are at risk of having a balanced chromosome rearrangement and should be offered chromosome analysis.
Sibs of a proband
Sibs of a child with a numeric chromosome abnormality are at a slightly increased risk of having a similar chromosome abnormality (depending on the specific abnormality and the age of the mother) with a similar or different phenotype.
The risk to the sibs of a child with a structural unbalanced chromosome rearrangement depends on the chromosome status of the parents:
If neither parent has a structural rearrangement, the risk to sibs is negligible.
If a parent has a balanced structural rearrangement, the risk is increased and depends on the specific rearrangement and possibly other variables.
Offspring of a proband. Individuals with HPE and a chromosome rearrangement are unlikely to reproduce.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has a chromosome rearrangement, the parent's family members are at risk and can be offered chromosome analysis.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Many individuals diagnosed with autosomal dominant nonsyndromic HPE (
Tables 2a,
2b) inherited a pathogenic variant from a heterozygous parent who may or may not have HPE-spectrum anomalies [
Mouden et al 2015].
Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant. Recommendations may also include evaluation of the parents for mild manifestations of HPE.
* Misattributed parentage can also be explored as an alternative explanation for an apparent de novo pathogenic variant.
The family history of some individuals diagnosed with HPE may appear to be negative because of reduced penetrance and failure to recognize the disorder in family members; this is particularly true for families with HPE caused by pathogenic variants in
SIX3 [
Stokes et al 2018]. In some cases, a single mild manifestation is the only clue that a given individual has autosomal dominant nonsyndromic HPE and thus is at increased risk of having affected offspring. Note, however, that none of the mild manifestations is pathognomonic for HPE and each can occur as an isolated finding apart from the HPE spectrum. Therefore, an apparently negative family history cannot be confirmed unless appropriate molecular genetic testing has been performed on the parents of the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the parents:
If a parent is affected or has an HPE pathogenic variant (with or without clinical manifestations), the risk to sibs of inheriting the variant is 50%. Empiric studies indicate that sibs who inherit a pathogenic variant have a 20% risk for HPE, 15% risk for an HPE microform, and a 15% likelihood of a normal phenotype.
If the parents have not been tested for the HPE pathogenic variant in the proband but are clinically unaffected and the family history is negative, the risk to the sibs of the proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for HPE because of the possibility of reduced penetrance in a parent or parental germline mosaicism.
Offspring of a proband
Every child of an individual with a pathogenic variant for autosomal dominant nonsyndromic HPE has a 50% chance of inheriting the pathogenic variant.
Although severely affected individuals do not reproduce, individuals with mild forms and microforms of autosomal dominant HPE may do so. The clinical manifestations and severity in offspring may range from mild to severe.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected or has a pathogenic variant, the parent's family members may be at risk.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
Sibs of a proband
If both parents are known to be heterozygous for a STIL pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic.
Offspring of a proband. To date, individuals with STIL-HPE are not known to reproduce.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a STIL pathogenic variant.
Carrier detection. Carrier testing for at-risk relatives requires prior identification of the STIL pathogenic variants in the family.
X-Linked Inheritance – Risk to Family Members
Parents of a female proband
A female proband with X-linked HPE caused by a pathogenic variant in STAG2 or SMC1A may have inherited a pathogenic variant from either her mother or her father, or the pathogenic variant may be de novo.
To date, the majority of individuals with X-linked HPE are female, represent simplex cases (i.e., a single occurrence in the family), and have the disorder as the result of a de novo pathogenic variant.
Molecular genetic testing is recommended for both parents of a female proband.
If the STAG2 or SMC1A pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the proband most likely has a de novo pathogenic variant. Another possible explanation is that the proband inherited a pathogenic variant from a parent with germline mosaicism.
Note: The mother of a proband who is found to be heterozygous for a STAG2 or SMC1A pathogenic variant may have favorable skewed X inactivation that results in her being mildly affected or unaffected.
Parents of a male proband
If a male is the only affected family member (i.e., a simplex case), the mother may be heterozygous or the affected male may have a
de novo pathogenic variant, in which case the mother is not heterozygous. To date, X-linked HPE has been reported in only one male; the affected male had the disorder as the result of a
de novo
STAG2 truncating variant [
Aoi et al 2019];
SMC1A-HPE has not been reported in a male proband.
Molecular genetic testing is recommended for the mother of a male proband.
The father of an affected male will not have the disorder nor will he be hemizygous for an X-linked HPE-causing pathogenic variant; therefore, he does not require further evaluation/testing.
Sibs of a female proband. The risk to sibs depends on the genetic status of the parents:
If the mother of the proband has a STAG2 or SMC1A pathogenic variant, the chance of transmitting it in each pregnancy is 50%.
If the father of the proband has a STAG2 or SMC1A pathogenic variant, he will transmit it to all his daughters and none of his sons.
If the proband represents a simplex case and if the STAG2 or SMC1A pathogenic variant cannot be detected in the leukocyte DNA of either parent, the risk to sibs is low but greater than that of the general population because of the possibility of parental germline mosaicism.
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
If the mother of the proband has a STAG2 or SMC1A pathogenic variant, the chance of transmitting it in each pregnancy is 50% (see Sibs of a female proband).
If the proband represents a simplex case (i.e., a single occurrence in a family) and if the STAG2 or SMC1A pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is greater than that of the general population because of the possibility of maternal germline mosaicism.
Offspring of a proband
Each child of a female proband with X-linked HPE has a 50% chance of inheriting the STAG2 or SMC1A pathogenic variant. Female probands with frank HPE are very unlikely to reproduce.
Each daughter of a male proband with X-linked HPE has a 50% chance of inheriting the STAG2 or SMC1A pathogenic variant. Surviving male probands with frank HPE are very unlikely to reproduce.
Other family members. The risk to other family members depends on the genetic status of the proband's parents: if a parent is affected or has a STAG2 or SMC1A pathogenic variant, the parent's family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
High-Risk Pregnancies
Molecular genetic testing. Once the HPE-causing pathogenic variant(s) have been identified in an affected family member, prenatal testing for a pregnancy at increased risk for HPE and preimplantation genetic testing are possible.
Fetal ultrasound examination. For families with nonsyndromic HPE and no identifiable etiology, alobar HPE can be diagnosed by prenatal ultrasound examination between ten to 14 weeks of gestation based on abnormal facial morphology (as seen in many cases) and absence of the normal configuration of the choroid plexuses within the lateral ventricles, called "butterfly sign" [Kousa et al 2018, Calloni et al 2019]. Milder degrees of HPE including semilobar or lobar HPE cannot reliably be detected by prenatal ultrasound examination.
Lobar HPE can be recognized in utero with ultrasound. However, a specific diagnosis is often difficult and relies on qualitative evaluation of the morphology of the ventricles. Though not specific, antenatal demonstration of an echogenic linear structure running anterior-posterior within the third ventricle is highly suggestive of lobar HPE, and can assist this difficult diagnosis.
Fetal MRI is routinely used as a second-line investigation in several centers to evaluate CNS structure when ultrasound studies have suggested the presence of an anomaly [Edwards & Hui 2018]. MRI is useful for the evaluation of the posterior fossa and the median telencephalon as well as for etiologic clarification of hydrocephalus. Fetal MRI is particularly valuable for clarifying the anatomic subtypes of HPE, and thus informing on its severity [Edwards & Hui 2018, Kousa et al 2018, Calloni et al 2019]. Ultrafast MRI minimizes artifacts caused by fetal motion. Because MRI involves no exposure to radiation, it appears to be safe.
Low-Risk Pregnancies
When HPE is found on routine prenatal ultrasound examination in a fetus not known to be at increased risk for HPE, an extensive fetal examination, preferably using high-resolution ultrasound examination (e.g., examination with 3D ultrasound) to determine the presence of additional structural anomalies is indicated [Edwards & Hui 2018]. Additional testing on amniotic fluid may be done to both establish the cause of HPE and assist in the management of the pregnancy and the recurrence risk counseling of the parents. Such testing can include the following [Kruszka et al 2018]:
Fetal karyotype to detect numeric and structural chromosome abnormalities. Fetal karyotype is usually the first test ordered in the context of low-risk pregnancies.
CMA if the karyotype is normal (or instead of karyotype) to detect pathogenic deletions or duplications (and aneuploidies if no previous karyotype) in the genes known to cause HPE (see
Tables 2a,
2b). Of note, alternative assays such as MLPA may be used for this purpose.
Multigene panel sequencing including at least the three following genes: SHH, ZIC2, and SIX3
It is essential to bear in mind that if the fetus has HPE identified by ultrasound examination, medical and parental decision making about the pregnancy may occur independent of a specific genetic diagnosis.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Families for Hope
1219 North Wittfield Street
Indianapolis IN 46229
Phone: 888-533-4443
Email: Info@FamiliesforHoPE.org
Genetic and Rare Diseases Information Center (GARD)
National Human Genome Research Institute (NHGRI)
National Institute of Neurological Disorders and Stroke (NINDS)
PO Box 5801
Bethesda MD 20824
Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
National Organization for Rare Disorders (NORD)
Chapter Notes
Author History
Andrea L Gropman, MD, FAAP, FACMG; Children's National Health System (2000-2020)
Paul Kruszka, MD, MPH (2020-present)
Maximilian Muenke, MD, FACMG (2000-present)
Benjamin D Solomon, MD; National Human Genome Research Institute (2010-2020)
Cedrik Tekendo-Ngongang, MD (2020-present)
Revision History
5 March 2020 (bp) Comprehensive update posted live
29 August 2013 (me) Comprehensive update posted live
3 November 2011 (cd) Revision: clinical testing available for FOXH1 and NODAL
30 March 2010 (me) Comprehensive update posted live
5 March 2008 (me) Comprehensive update posted live
11 March 2005 (me) Comprehensive update posted live
27 January 2003 (me) Comprehensive update posted live
27 December 2000 (pb) Overview posted live
August 2000 (mm) Original submission
References
Literature Cited
Alghamdi
M, Alkhamis
WH, Jamjoom
D, Al-Nafisah
G, Tahir
A, Abdouelhoda
M. Expanding the phenotype and the genotype of Stromme syndrome: A novel variant of the CENPF gene and literature review.
Eur J Med Genet.
2020;63:103844.
[
PubMed: 31953238]
Aoi
H, Lei
M, Mizuguchi
T, Nishioka
N, Goto
T, Miyama
S, Suzuki
T, Iwama
K, Uchiyama
Y, Mitsuhashi
S, Itakura
A, Takeda
S, Matsumoto
N.
Nonsense variants in STAG2 result in distinct sex-dependent phenotypes.
J Hum Genet.
2019;64:487-92.
[
PubMed: 30765867]
Arauz
RF, Solomon
BD, Pineda-Alvarez
DE, Gropman
AL, Parsons
JA, Roessler
E, Muenke
M. A Hypomorphic allele in the FGF8 gene contributes to holoprosencephaly and is allelic to gonadotropin-releasing hormone deficiency in humans.
Mol Syndromol.
2010;1:59-66.
[
PMC free article: PMC2941840] [
PubMed: 21045958]
Bae
GU, Domené
S, Roessler
E, Schachter
K, Kang
JS, Muenke
M, Krauss
RS. Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors.
Am J Hum Genet.
2011;89:231-40.
[
PMC free article: PMC3155179] [
PubMed: 21802063]
Bear
KA, Solomon
BD, Antonini
S, Arnhold
IJ, França
MM, Gerkes
EH, Grange
DK, Hadley
DW, Jääskeläinen
J, Paulo
SS, Rump
P, Stratakis
CA, Thompson
EM, Willis
M, Winder
TL, Jorge
AA, Roessler
E, Muenke
M. Pathogenic mutations in GLI2 cause a specific phenotype that is distinct from holoprosencephaly.
J Med Genet.
2014;51:413-8.
[
PMC free article: PMC6417429] [
PubMed: 24744436]
Bear
KA, Solomon
BD. GLI2 mutations typically result in pituitary anomalies with or without postaxial polydactyly.
Am J Med Genet A.
2015;167A:2491-2.
[
PubMed: 25974718]
Brown
LY, Odent
S, David
V, Blayau
M, Dubourg
C, Apacik
C, Delgado
MA, Hall
BD, Reynolds
JF, Sommer
A, Wieczorek
D, Brown
SA, Muenke
M. Holoprosencephaly due to mutations in ZIC2: alanine tract expansion mutations may be caused by parental somatic recombination.
Hum Mol Genet.
2001;10:791–6.
[
PubMed: 11285244]
Calloni, SF, Caschera
L, Triulzi
FM. Disorders of ventral induction/spectrum of holoprosencephaly.
Neuroimaging Clin N Am.
2019;29:411-21.
[
PubMed: 31256862]
Dubourg
C, Carre
W, Hamdi-Roze
H, Mouden
C, Roume
J, Abdelmajid
B, Amram
D, Baumann
C, Chassaing
N, Coubes
C, Faivre-Olivier
L, Ginglinger
E, Gonzales
M, Levy-Mozziconacci
A, Lynch
S-A, Naudion
S, Pasquier
L, Poidvin
A, Prieur
F, Sarda
P, Toutain
A, Dupé
V, Akloul
L, Odent
S, de Tayrac
M, David, V. Mutational spectrum in holoprosencephaly shows that FGF is a new major signaling pathway.
Hum Mut.
2016;37:1329-39.
[
PubMed: 27363716]
Dubourg
C, Kim
A, Watrin
E, de Tayrac
M, Odent
S, David
V, Dupé
V. Recent advances in understanding inheritance of holoprosencephaly.
Am J Med Genet.
2018;178:258-69.
[
PubMed: 29785796]
Dupé
V, Rochard
L, Mercier
S, Le Pétillon
Y, Gicquel
I, Bendavid
C, Bourrouillou
G, Kini
U, Thauvin-Robinet
C, Bohan
TP, Odent
S, Dubourg
C, David
V. NOTCH, a new signaling pathway implicated in holoprosencephaly.
Hum Mol Genet.
2011;20:1122-31.
[
PMC free article: PMC3390777] [
PubMed: 21196490]
Edwards
L, Hui
L. First and second trimester screening for fetal structural anomalies.
Semin Fetal Neonatal Med.
2018;23:102-11.
[
PubMed: 29233624]
Grinblat
Y, Lipinski
RJ. A forebrain undivided: Unleashing model organisms to solve the mysteries of holoprosencephaly.
Developmental Dynamics.
2019;248:626-33.
[
PubMed: 30993762]
Gropman AL, Muenke M. Holoprosencephaly. In: Cassidy SB, Allanson JE, eds. Management of Genetic Syndromes. New York, NY: Wiley-Liss, Inc; 2005:279-96.
Hadley
DW, Kruszka
P, Muenke
M. Challenging issues arising in counseling families experiencing holoprosencephaly.
Am J Med Genet.
2018;178:238-45.
[
PubMed: 30182441]
Hahn
JS, Barnes
PD, Clegg
NJ, Stashinko
EE. Septopreoptic holoprosencephaly: a mild subtype associated with midline craniofacial anomalies.
AJNR Am J Neuroradiol.
2010;31:1596-601.
[
PMC free article: PMC7965016] [
PubMed: 20488907]
Hahn
JS, Barnes
PD. Neuroimaging advances in holoprosencephaly: refining the spectrum of the midline malformation.
Am J Med Genet Part C Semin Med Genet.
2010;154C:120-32.
[
PubMed: 20104607]
Hong
S, Hu
P, Marino
J, Hufnagel
SB, Hopkin
RJ, Toromanović
A, Richieri-Costa
A, Ribeiro-Bicudo
LA, Kruszka
P, Roessler
E, Muenke
M. Dominant-negative kinase domain mutations in
FGFR1 can explain the clinical severity of Hartsfield syndrome.
Hum Mol Genet.
2016;25:1912-22.
[
PMC free article: PMC5062582] [
PubMed: 26931467]
Hu
T, Kruszka
P, Martinez
AF, Ming
JE, Shabason
EK, Raam
MS, Shaikh
TH, Pineda-Alvarez
DE, Muenke
M. Cytogenetics and holoprosencephaly: A chromosomal microarray study of 222 individuals with holoprosencephaly.
Am J Med Genet C Semin Med Genet.
2018;178:175-86.
[
PMC free article: PMC6127867] [
PubMed: 30182442]
Hughes
JJ, Alkhunaizi
E, Kruszka
P, Pyle
LC, Grange
DK, Berger
SI, Payne
KK, Masser-Frye
D, Hu
T, Christie
MC, Clegg
NJ, Everson
JL, Martinez
AF, Walsh
LE, Bedoukian
E, Jones
MC, Harris
CJ, Riedhammer
KM, Choukair
D, Fechner
PY, Rutter
MM, Hufnagel
SB, Roifman
M, Kletter
GB, Delot
E, Vilain
E, Lipinski
RJ, Vezina
CM, Muenke
M, Chitaya
D. Loss-of-function variants in
PPP1R12A: from isolated sex reversal to holoprosencephaly spectrum and urogenital malformations.
Am J Hum Genet.
2020;106:121-8.
[
PMC free article: PMC7042489] [
PubMed: 31883643]
Johnson
CY, Rasmussen
SA. Non-genetic risk factors for holoprosencephaly.
Am J Med Genet C Semin Med Genet.
2010;154C:73-85.
[
PubMed: 20104598]
Jones
GE, Robertson
L, Maniyar
A, Shammas
C, Phelan
MM, Vasudevan
PC, Tanteles
GA. Microform holoprosencephaly with bilateral congenital elbow dislocation; increasing the phenotypic spectrum of Steinfeld syndrome.
Am J Med Genet A.
2016;170:754-9.
[
PubMed: 26728615]
Jónsson
H, Sulem
P, Kehr
B, Kristmundsdottir
S, Zink
F, Hjartarson
E, Hardarson
MT, Hjorleifsson
KE, Eggertsson
HP, Gudjonsson
SA, Ward
LD, Arnadottir
GA, Helgason
EA, Helgason
H, Gylfason
A, Jonasdottir
A, Jonasdottir
A, Rafnar
T, Frigge
M, Stacey
SN, Th Magnusson
O, Thorsteinsdottir
U, Masson
G, Kong
A, Halldorsson
BV, Helgason
A, Gudbjartsson
DF, Stefansson
K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland.
Nature.
2017;549:519-22.
[
PubMed: 28959963]
Kagan
KO, Staboulidou
I, Syngelaki
A, Cruz
J, Nicolaides
KH. The 11–13-week scan: Diagnosis and outcome of holoprosencephaly, exomphalos and megacystis.
Ultrasound in Obstetrics and Gynecology.
2010;36:10-4.
[
PubMed: 20564304]
Kakar
N, Ahmad
J, Morris-Rosendahl
DJ, Altmüller
A, Friedrich
K, Barbi
G, Nürnberg
P, Kubisch
C, Dobyns
WB, Borck
G.
STIL mutation causes autosomal recessive microcephalic lobar holoprosencephaly.
Hum Genet.
2015;134:45-51.
[
PubMed: 25218063]
Keaton
AA, Solomon
BD, Kauvar
EF, El-Jaick
KB, Gropman
AL, Zafer
Y, Meck
JM, Bale
SJ, Grange
DK, Haddad
BR, Gowans
GC, Clegg
NJ, Delgado
MR, Hahn
JS, Pineda-Alvarez
DE, Lacbawan
F, Vélez
JI, Roessler
E, Muenke
M. TGIF Mutations in Human Holoprosencephaly: Correlation between Genotype and Phenotype.
Mol Syndromol.
2010;1:211-22.
[
PMC free article: PMC3214944] [
PubMed: 22125506]
Kidron
D, Shapira
D, Ben Sira
L, Malinger
G, Lev
D, Cioca
A, Sharony
R, Lerman
ST. Agenesis of the corpus callosum. An autopsy study in fetuses.
Virchows Arch.
2016;468:219-30.
[
PubMed: 26573426]
Kousa
YA, du Plessis
AJ, Vezina
G. Prenatal diagnosis of holoprosencephaly.
Am J Med Genet C Semin Med Genet.
2018;178:206-13.
[
PubMed: 29770996]
Kruszka
P, Berger
SI, Weiss
K, Everson
JL, Martinez
AF, Hong
S, Anyane-Yeboa
K, Lipinski
RJ, Muenke
M. A
CCR4-NOT transcription complex, subunit 1,
CNOT1, variant associated with holoprosencephaly.
Am J Hum Genet.
2019a;104:990-3.
[
PMC free article: PMC6506867] [
PubMed: 31006510]
Kruszka
P, Berger
SI, Casa
V, Dekker
MR, Gaesser
J, Weiss
K, Martinez
AF, Murdock
DR, Louie
RJ, Prijoles
EJ, Lichty
AW, Brouwer
OF, Zonneveld-Huijssoon
E, Stephan
MJ, Hogue
J, Hu
P, Tanima-Nagai
M, Everson
JL, Prasad
C, Cereda
A, Iascone
M, Schreiber
A, Zurcher
V, Corsten-Janssen
N, Escobar
L, Clegg
NJ, Delgado
MR, Hajirnis
O, Balasubramanian
M, Kayserili
H, Deardorff
M, Poot
RA, Wendt
KS, Lipinski
RJ, Muenke
M. Cohesin complex-associated holoprosencephaly.
Brain.
2019b;142:2631-43.
[
PMC free article: PMC7245359] [
PubMed: 31334757]
Lacbawan
F, Solomon
BD, Roessler
E, El-Jaick
K, Domené
S, Vélez
JI, Zhou
N, Hadley
D, Balog
JZ, Long
R, Fryer
A, Smith
W, Omar
S, McLean
SD, Clarkson
K, Lichty
A, Clegg
NJ, Delgado
MR, Levey
E, Stashinko
E, Potocki
L, Vanallen
MI, Clayton-Smith
J, Donnai
D, Bianchi
DW, Juliusson
PB, Njølstad
PR, Brunner
HG, Carey
JC, Hehr
U, Müsebeck
J, Wieacker
PF, Postra
A, Hennekam
RC, van den Boogaard
MJ, van Haeringen
A, Paulussen
A, Herbergs
J, Schrander-Stumpel
CT, Janecke
AR, Chitayat
D, Hahn
J, McDonald-McGinn
DM, Zackai
EH, Dobyns
WB, Muenke
M. Clinical spectrum of SIX3-associated mutations in holoprosencephaly: correlation between genotype, phenotype and function.
J Med Genet.
2009;46:389-98.
[
PMC free article: PMC3510661] [
PubMed: 19346217]
Levey
EB, Stashinko
E, Clegg
NJ, Delgado
MR. Management of children with holoprosencephaly.
Am J Med Genet C Semin Med Genet.
2010;154C:183-90.
[
PubMed: 20104615]
Mercier
S, Dubourg
C, Garcelon
N, Campillo-Gimenez
B, Gicquel
I, Belleguic
M, Ratié
L, Pasquier
L, Loget
P, Bendavid
C, Jaillard
S, Rochard
L, Quélin
C, Dupé
V, David
V, Odent
S.
New findings for phenotype-genotype correlations in a large European series of holoprosencephaly cases.
J Med Genet.
2011;48:752-60.
[
PMC free article: PMC3386902] [
PubMed: 21940735]
Mouden
C, de Tayrac
M, Dubourg
C, Rose
S, Carré
W, Hamdi-Rozé
H, Babron
MC, Akloul
L, Héron-Longe
B, Odent
S, Dupé
V, Giet
R, David
V. Homozygous STIL mutation causes holoprosencephaly and microcephaly in two siblings.
PLoS One.
2015;10:e0117418.
[
PMC free article: PMC4319975] [
PubMed: 25658757]
Nanni
L, Ming
JE, Bocian
M, Steinhaus
K, Bianchi
DW, Die-Smulders
C, Giannotti
A, Imaizumi
K, Jones
KL, Campo
MD, Martin
RA, Meinecke
P, Pierpont
ME, Robin
NH, Young
ID, Roessler
E, Muenke
M. The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly.
Hum Mol Genet.
1999;8:2479–88
[
PubMed: 10556296]
Palumbo
P, Petracca
A, Maggi
R, Biagini
T, Nardella
G, Sacco
MC, Di Schiavi
E, Carella
M, Micale
L, Castori
M. A novel dominant-negative FGFR1 variant causes Hartsfield syndrome by deregulating RAS/ERK1/2 pathway.
Eur J Hum Genet.
2019;27:1113-20.
[
PMC free article: PMC6777633] [
PubMed: 30787447]
Petracchi
F, Crespo
L, Michia
C, Igarzabal
L, Gadow
E. Holoprosencephaly at prenatal diagnosis: Analysis of 28 cases regarding etiopathogenic diagnoses.
Prenatal Diagnosis.
2011;31:887–891.
[
PubMed: 21706511]
Pineda-Alvarez
DE, Dubourg
C, David
V, Roessler
E, Muenke
M. Current recommendations for the molecular evaluation of newly diagnosed holoprosencephaly patients.
Am J Med Genet C Semin Med Genet.
2010;154C:93-101.
[
PMC free article: PMC2815008] [
PubMed: 20104604]
Pineda-Alvarez
DE, Solomon
BD, Roessler
E, Balog
JZ, Hadley
DW, Zein
WM, Hadsall
CK, Brooks
BP, Muenke
M. A broad range of ophthalmologic anomalies is part of the holoprosencephaly spectrum.
Am J Med Genet A.
2011;155A:2713-20.
[
PMC free article: PMC3200498] [
PubMed: 21976454]
Richieri-Costa
A, Ribeiro
LA. Holoprosencephaly and holoprosencephaly-like phenotypes: Review of facial and molecular findings in patients from a craniofacial hospital in Brazil.
Am J Med Genet C Semin Med Genet.
2010;154C:149-57.
[
PubMed: 20104612]
Roessler
E, Ma
Y, Ouspenskaia
MV, Lacbawan
F, Bendavid
C, Dubourg
C, Beachy
PA, Muenke
M. Truncating loss-of-function mutations of DISP1 contribute to holoprosencephaly-like microform features in humans.
Hum Genet.
2009;125:393-400.
[
PMC free article: PMC2774849] [
PubMed: 19184110]
Roessler
E, Vélez
JI, Zhou
N, Muenke
M. Utilizing prospective sequence analysis of SHH, ZIC2, SIX3 and TGIF in holoprosencephaly probands to describe the parameters limiting the observed frequency of mutant gene×gene interactions.
Mol Genet Metab.
2012;105:658-64.
[
PMC free article: PMC3309119] [
PubMed: 22310223]
Rosa
RFM, Correia
EPE, Bastos
CS, da Silva
GS, Correia
JD, da Rosa
EB, Silveira
DB, Targa
LV, da Cunha
AC, Zen
PRG. Trisomy 18 and holoprosencephaly.
Am J Med Genet A.
2017;173:1985-7.
[
PubMed: 28449414]
Ruda
J, Grischkan
J, Allarakhia
Z. Radiologic, genetic, and endocrine findings in isolated congenital nasal pyriform aperture stenosis patients.
Int J Pediatr Otorhinolaryngol.
2020;128:109705.
[
PubMed: 31606685]
Simonis
N, Migeotte
I, Lambert
N, Perazzolo
C, de Silva
DC, Dimitrov
B, Heinrichs
C, Janssens
S, Kerr
B, Mortier
G, Van Vliet
G, Lepage
P, Casimir
G, Abramowicz
M, Smits
G, Vilain
C. FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly.
J Med Genet.
2013;50:585-92.
[
PMC free article: PMC3756455] [
PubMed: 23812909]
Solomon
BD, Bear
KA, Wyllie
A, Keaton
AA, Dubourg
C, David
V, Mercier
S, Odent
S, Hehr
U, Paulussen
A, Clegg
NJ, Delgado
MR, Bale
SJ, Lacbawan
F, Ardinger
HH, Aylsworth
AS, Bhengu
NL, Braddock
S, Brookhyser
K, Burton
B, Gaspar
H, Grix
A, Horovitz
D, Kanetzke
E, Kayserili
H, Lev
D, Nikkel
SM, Norton
M, Roberts
R, Saal
H, Schaefer
GB, Schneider
A, Smith
EK, Sowry
E, Spence
MA, Shalev
SA, Steiner
CE, Thompson
EM, Winder
TL, Balog
JZ, Hadley
DW, Zhou
N, Pineda-Alvarez
DE, Roessler
E, Muenke
M. Genotypic and phenotypic analysis of 396 individuals with mutations in Sonic Hedgehog.
J Med Genet.
2012a;49:473-9.
[
PubMed: 22791840]
Solomon
BD, Lacbawan
F, Jain
M, Domené
S, Roessler
E, Moore
C, Dobyns
WB, Muenke
M. A novel SIX3 mutation segregates with holoprosencephaly in a large family.
Am J Med Genet A.
2009a;149A:919-25.
[
PMC free article: PMC2737713] [
PubMed: 19353631]
Solomon
BD, Lacbawan
F, Mercier
S, Clegg
NJ, Delgado
MR, Rosenbaum
K, Dubourg
C, David
V, Olney
AH, Wehner
LE, Hehr
U, Bale
S, Paulussen
A, Smeets
HJ, Hardisty
E, Tylki-Szymanska
A, Pronicka
E, Clemens
M, McPherson
E, Hennekam
RC, Hahn
J, Stashinko
E, Levey
E, Wieczorek
D, Roeder
E, Schell-Apacik
CC, Booth
CW, Thomas
RL, Kenwrick
S, Keaton
A, Balog
JZ, Hadley
D, Zhou
N, Long
R, Velez
JI, Pineda-Alvarez
DE, Odent
S, Roessler
E, Muenke
M. Mutations in ZIC2 in human holoprosencephaly: description of a novel ZIC2-specific phenotype and comprehensive analysis of 157 individuals.
J Med Genet.
2010a;47:513-24.
[
PMC free article: PMC3208626] [
PubMed: 19955556]
Solomon
BD, Mercier
S, Vélez
JI, Pineda-Alvarez
DE, Wyllie
A, Zhou
N, Dubourg
C, David
V, Odent
S, Roessler
E, Muenke
M. Analysis of genotype-phenotype correlations in human holoprosencephaly.
Am J Med Genet C Semin Med Genet.
2010b;154C:133-41.
[
PMC free article: PMC2815217] [
PubMed: 20104608]
Solomon
BD, Pineda-Alvarez
DE, Balog
JZ, Hadley
D, Gropman
AL, Nandagopal
R, Han
JC, Hahn
JS, Blain
D, Brooks
B, Muenke
M. Compound heterozygosity for mutations in PAX6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia.
Am J Med Genet A.
2009b;149A:2543-6.
[
PMC free article: PMC2783496] [
PubMed: 19876904]
Solomon
BD, Pineda-Alvarez
DE, Gropman
AL, Willis
MJ, Hadley
DW, Muenke
M. High intellectual function in individuals with mutation-positive microform holoprosencephaly.
Mol Syndromol.
2012b;3:140-2.
[
PMC free article: PMC3473351] [
PubMed: 23112757]
Stokes, B, Berger
SI, Hall, BA, Weiss
K, Martinez
AF, Hadley
DW, Murdock
DR, Ramanathan
S, Clark
RD, Roessler
E, Kruszka
P, Muenke
M. SIX3 deletions and incomplete penetrance in families affected by holoprosencephaly.
Congenit Anom (Kyoto). 2018;58:29-32.
[
PMC free article: PMC5750110] [
PubMed: 28670735]
Tinker SC, Gilboa SM, Moore CA, Waller DK, Simeone RM, Kim SY, Jamieson DJ, Botto LD, Reefhuis J, et al. Specific birth defects in pregnancies of women with diabetes: National Birth Defects Prevention Study, 1997-2011. Am J Obstet Gynecol. 2019;S0002-9378:31030-0. [
PMC free article: PMC7186569] [
PubMed: 31454511]
Toufaily
MH, Roberts
DJ, Westgate
MN, Holmes
LB. Triploidy: Variation of Phenotype.
Am J Clin Pathol.
2016;145:86-95.
[
PubMed: 26712875]
Weaver
DD, Solomon
BD, Akin-Samson
K, Kelley
RI, Muenke
M. Cyclopia (synophthalmia) in Smith-Lemli-Opitz Syndrome - first reported case and consideration of mechanism.
Am J Med Genet C Semin Med Genet.
2010;154C:142-5.
[
PMC free article: PMC2815131] [
PubMed: 20104611]
Weiss
K, Kruszka
P, Sacoto
MJG, Addissie
YA, Hadley
DW, Hadsall
CK, Stokes
B, Hu
Ping, Roessler
E, Solomon
B, Wiggs
E, Thurm
A, Hufnagel
RB, Zein
WM, Hahn
JS, Stashinko
E, Levey
E, Baldwin
D, Clegg
NJ, Delgado
MR, Muenke
M. In-depth investigations of adolescents and adults with holoprosencephaly identify unique characteristics.
Genet Med.
2018a;20:14-23.
[
PMC free article: PMC5763157] [
PubMed: 28640243]
Weiss
K, Kruszka
PS, Levey
E, Muenke
M. Holoprosencephaly from conception to adulthood.
Am J Med Genet.
2018b;178:122-7.
[
PubMed: 30182446]