NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Entrez Programming Utilities Help [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.
Entrez Direct: E-utilities on the Unix Command Line
Authors
Jonathan Kans, PhD 1.
1.Affiliations
Corresponding author.Created: April 23, 2013; Last Update: April 4, 2024.
Estimated reading time: 1 hour
Getting Started
Introduction
Entrez Direct (EDirect) provides access to the NCBI's suite of interconnected databases (publication, sequence, structure, gene, variation, expression, etc.) from a Unix terminal window. Search terms are entered as command-line arguments. Individual operations are connected with Unix pipes to construct multi-step queries. Selected records can then be retrieved in a variety of formats.
Installation
EDirect will run on Unix and Macintosh computers, and under the Cygwin Unix-emulation environment on Windows PCs. To install the EDirect software, open a terminal window and execute one of the following two commands:
sh -c "$(curl -fsSL https://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh)"
sh -c "$(wget -q https://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/install-edirect.sh -O -)"
This will download a number of scripts and several precompiled programs into an "edirect" folder in the user's home directory. It may then print an additional command for updating the PATH environment variable in the user's configuration file. The editing instructions will look something like:
echo "export PATH=\$HOME/edirect:\$PATH" >> $HOME/.bash_profile
As a convenience, the installation process ends by offering to run the PATH update command for you. Answer "y" and press the Return key if you want it run. If the PATH is already set correctly, or if you prefer to make any editing changes manually, just press Return.
Once installation is complete, run:
export PATH=${HOME}/edirect:${PATH}to set the PATH for the current terminal session.
Quick Start
The readme.pdf file included in the edirect folder contains a highly-abridged version of this document. It is intended to convey the most important points in the least amount of time for the new user, while still presenting the minimal essential details. It also covers subtle issues in several Entrez biological databases, demonstrates integration of data from external sources, and has a brief introduction to scripting and programming.
The full documentation gives a much more in-depth exploration of the underlying topics, especially in the Complex Objects section, and in the Additional Examples web page, which is organized by Entrez database. This document also introduces other worthy topics, such as identifier lookup and sequence coordinate conversions, and has a more thorough treatment of automation.
Programmatic Access
EDirect connects to Entrez through the Entrez Programming Utilities interface. It supports searching by indexed terms, looking up precomputed neighbors or links, filtering results by date or category, and downloading record summaries or reports.
Navigation programs (esearch, elink, efilter, and efetch) communicate by means of a small structured message, which can be passed invisibly between operations with a Unix pipe. The message includes the current database, so it does not need to be given as an argument after the first step.
Accessory programs (nquire, transmute, and xtract) can help eliminate the need for writing custom software to answer ad hoc questions. Queries can move seamlessly between EDirect programs and Unix utilities or scripts to perform actions that cannot be accomplished entirely within Entrez.
All EDirect programs are designed to work on large sets of data. They handle many technical details behind the scenes (avoiding the learning curve normally required for E-utilities programming). Intermediate results are either saved on the Entrez history server or instantiated in the hidden message. For best performance, obtain an API Key from NCBI, and place the following line in your .bash_profile and .zshrc configuration files:
export NCBI_API_KEY=unique_api_key
Unix programs are run by typing the name of the program and then supplying any required or optional arguments on the command line. Argument names are letters or words that start with a dash ("‑") character.
Each program has a ‑help command that prints detailed information about available arguments.
Constructing Multi-Step Queries
EDirect allows individual operations to be described separately, combining them into a multi-step query by using the vertical bar ("|") Unix pipe symbol:
esearch -db pubmed -query "tn3 transposition immunity" | efetch -format medline
Writing Commands on Multiple Lines
A query can be continued on the next line by typing the backslash ("\") Unix escape character immediately before pressing the Return key.
esearch -db pubmed -query "opsin gene conversion" | \
Continuing the query looks up precomputed neighbors of the original papers, next links to all protein sequences published in the related articles, then limits those to the rodent division of GenBank, and finally retrieves the records in FASTA format:
elink -related | \
elink -target protein | \
efilter -division rod | \
efetch -format fasta
In most modern versions of Unix the vertical bar pipe symbol also allows the query to continue on the next line, without the need for an additional backslash.
Accessory Programs
Nquire retrieves data from remote servers with URLs constructed from command line arguments:
nquire -get https://icite.od.nih.gov api/pubs -pmids 2539356 |
Transmute converts a concatenated stream of JSON objects or other structured formats into XML:
transmute -j2x |
Xtract can use waypoints to navigate a complex XML hierarchy and obtain data values by field name:
xtract -pattern data -element cited_by |
The resulting output can be post-processed by Unix utilities or scripts:
fmt -w 1 | sort -V | uniq
Retrieving PubMed Reports
Piping PubMed query results to efetch and specifying the "abstract" format:
esearch -db pubmed -query "lycopene cyclase" |
efetch -format abstract
returns a set of reports that can be read by a person:
...
85. PLoS One. 2013;8(3):e58144. doi: 10.1371/journal.pone.0058144. Epub ...
Levels of lycopene β-cyclase 1 modulate carotenoid gene expression and
accumulation in Daucus carota.
Moreno JC(1), Pizarro L, Fuentes P, Handford M, Cifuentes V, Stange C.
Author information:
(1)Departamento de Biología, Facultad de Ciencias, Universidad de Chile,
Santiago, Chile.
Plant carotenoids are synthesized and accumulated in plastids through a
highly regulated pathway. Lycopene β-cyclase (LCYB) is a key enzyme
involved directly in the synthesis of α-carotene and β-carotene through
...
If "medline" format is used instead:
esearch -db pubmed -query "lycopene cyclase" |
efetch -format medline
the output can be entered into common bibliographic management software packages:
...
PMID- 23555569
OWN - NLM
STAT- MEDLINE
DA - 20130404
DCOM- 20130930
LR - 20131121
IS - 1932-6203 (Electronic)
IS - 1932-6203 (Linking)
VI - 8
IP - 3
DP - 2013
TI - Levels of lycopene beta-cyclase 1 modulate carotenoid gene expression
and accumulation in Daucus carota.
PG - e58144
LID - 10.1371/journal.pone.0058144 [doi]
AB - Plant carotenoids are synthesized and accumulated in plastids
through a highly regulated pathway. Lycopene beta-cyclase (LCYB) is a
key enzyme involved directly in the synthesis of alpha-carotene and
...
Retrieving Sequence Reports
Nucleotide and protein records can be downloaded in FASTA format:
esearch -db protein -query "lycopene cyclase" |
efetch -format fasta
which consists of a definition line followed by the sequence:
...
>gi|735882|gb|AAA81880.1| lycopene cyclase [Arabidopsis thaliana]
MDTLLKTPNKLDFFIPQFHGFERLCSNNPYPSRVRLGVKKRAIKIVSSVVSGSAALLDLVPETKKENLDF
ELPLYDTSKSQVVDLAIVGGGPAGLAVAQQVSEAGLSVCSIDPSPKLIWPNNYGVWVDEFEAMDLLDCLD
TTWSGAVVYVDEGVKKDLSRPYGRVNRKQLKSKMLQKCITNGVKFHQSKVTNVVHEEANSTVVCSDGVKI
QASVVLDATGFSRCLVQYDKPYNPGYQVAYGIIAEVDGHPFDVDKMVFMDWRDKHLDSYPELKERNSKIP
TFLYAMPFSSNRIFLEETSLVARPGLRMEDIQERMAARLKHLGINVKRIEEDERCVIPMGGPLPVLPQRV
VGIGGTAGMVHPSTGYMVARTLAAAPIVANAIVRYLGSPSSNSLRGDQLSAEVWRDLWPIERRRQREFFC
FGMDILLKLDLDATRRFFDAFFDLQPHYWHGFLSSRLFLPELLVFGLSLFSHASNTSRLEIMTKGTVPLA
KMINNLVQDRD
...
Sequence records can also be obtained as GenBank or GenPept flatfiles:
esearch -db protein -query "lycopene cyclase" |
efetch -format gp
which have features annotating particular regions of the sequence:
...
LOCUS AAA81880 501 aa linear PLN ...
DEFINITION lycopene cyclase [Arabidopsis thaliana].
ACCESSION AAA81880
VERSION AAA81880.1 GI:735882
DBSOURCE locus ATHLYC accession L40176.1
KEYWORDS .
SOURCE Arabidopsis thaliana (thale cress)
ORGANISM Arabidopsis thaliana
Eukaryota; Viridiplantae; Streptophyta; Embryophyta;
Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons;
Brassicales; Brassicaceae; Camelineae; Arabidopsis.
REFERENCE 1 (residues 1 to 501)
AUTHORS Scolnik,P.A. and Bartley,G.E.
TITLE Nucleotide sequence of lycopene cyclase (GenBank L40176) from
Arabidopsis (PGR95-019)
JOURNAL Plant Physiol. 108 (3), 1343 (1995)
...
FEATURES Location/Qualifiers
source 1..501
/organism="Arabidopsis thaliana"
/db_xref="taxon:3702"
Protein 1..501
/product="lycopene cyclase"
transit_peptide 1..80
mat_peptide 81..501
/product="lycopene cyclase"
CDS 1..501
/gene="LYC"
/coded_by="L40176.1:2..1507"
ORIGIN
1 mdtllktpnk ldffipqfhg ferlcsnnpy psrvrlgvkk raikivssvv sgsaalldlv
61 petkkenldf elplydtsks qvvdlaivgg gpaglavaqq vseaglsvcs idpspkliwp
121 nnygvwvdef eamdlldcld ttwsgavvyv degvkkdlsr pygrvnrkql kskmlqkcit
181 ngvkfhqskv tnvvheeans tvvcsdgvki qasvvldatg fsrclvqydk pynpgyqvay
241 giiaevdghp fdvdkmvfmd wrdkhldsyp elkernskip tflyampfss nrifleetsl
301 varpglrmed iqermaarlk hlginvkrie edercvipmg gplpvlpqrv vgiggtagmv
361 hpstgymvar tlaaapivan aivrylgsps snslrgdqls aevwrdlwpi errrqreffc
421 fgmdillkld ldatrrffda ffdlqphywh gflssrlflp ellvfglslf shasntsrle
481 imtkgtvpla kminnlvqdr d
//
...
Searching and Filtering
Restricting Query Results
The current results can be refined by further term searching in Entrez (useful in the protein database for limiting BLAST neighbors to a taxonomic subset):
esearch -db pubmed -query "opsin gene conversion" |
elink -related |
efilter -query "tetrachromacy"
Limiting by Date
Results can also be filtered by date. For example, the following statements:
efilter -days 60 -datetype PDAT
efilter -mindate 2000
efilter -maxdate 1985
efilter -mindate 1990 -maxdate 1999
restrict results to articles published in the previous two months, since the beginning of 2000, through the end of 1985, or in the 1990s, respectively. YYYY/MM and YYYY/MM/DD date formats are also accepted.
Fetch by Identifier
Efetch and elink can take a list of numeric identifiers or accessions in an ‑id argument:
efetch -db pubmed -id 7252148,1937004 -format xml
efetch -db nuccore -id 1121073309 -format acc
efetch -db protein -id 3OQZ_a -format fasta
efetch -db bioproject -id PRJNA257197 -format docsum
efetch -db pmc -id PMC209839 -format medline
elink -db pubmed -id 2539356 -cites
without the need for a preceding esearch command.
Non-integer accessions will be looked up with an internal search, using the appropriate field for the database:
esearch -db bioproject -query "PRJNA257197 [PRJA]" |
efetch -format uid | ...
Most databases use the [ACCN] field for identifier lookup, but there are a few exceptions:
annotinfo [ASAC]
assembly [ASAC]
bioproject [PRJA]
books [AID]
clinvar [VACC]
gds [ALL]
genome [PRJA]
geoprofiles [NAME]
gtr [GTRACC]
mesh [MHUI]
nuccore [ACCN] or [PACC]
pcsubstance [SRID]
snp [RS] or [SS]
(For ‑db pmc it merely removes any "PMC" prefix from the integer identifier.)
For backward compatibility, esummary is a shortcut for esearch ‑format docsum:
esummary -db bioproject -id PRJNA257197
esummary -db sra -id SRR5437876
Reading Large Lists of Identifiers
Efetch and elink can also read a large list of identifiers or accessions piped in through stdin:
cat "file_of_identifiers.txt" |
efetch -db pubmed -format docsum
or from a file indicated by an ‑input argument:
efetch -input "file_of_identifiers.txt" -db pubmed -format docsum
As mentioned above, there is no need to use a script to split the identifiers into smaller groups or add a time delay between individual requests, since that functionality is already built into EDirect.
Indexed Fields
The einfo command can report the fields and links that are indexed for each database:
einfo -db protein -fields
This will return a table of field abbreviations and names indexed for proteins:
ACCN Accession
ALL All Fields
ASSM Assembly
AUTH Author
BRD Breed
CULT Cultivar
DIV Division
ECNO EC/RN Number
FILT Filter
FKEY Feature key
...
Qualifying Queries by Indexed Field
Query terms in esearch or efilter can be qualified by entering an indexed field abbreviation in brackets. Boolean operators and parentheses can also be used in the query expression for more complex searches.
Commonly-used fields for PubMed queries include:
[AFFL] Affiliation [LANG] Language
[ALL] All Fields [MAJR] MeSH Major Topic
[AUTH] Author [SUBH] MeSH Subheading
[FAUT] Author - First [MESH] MeSH Terms
[LAUT] Author - Last [PTYP] Publication Type
[CRDT] Date - Create [WORD] Text Word
[PDAT] Date - Publication [TITL] Title
[FILT] Filter [TIAB] Title/Abstract
[JOUR] Journal [UID] UID
and a qualified query looks like:
"Tager HS [AUTH] AND glucagon [TIAB]"
Filters that limit search results to subsets of PubMed include:
humans [MESH]
pharmacokinetics [MESH]
chemically induced [SUBH]
all child [FILT]
english [FILT]
freetext [FILT]
has abstract [FILT]
historical article [FILT]
randomized controlled trial [FILT]
clinical trial, phase ii [PTYP]
review [PTYP]
Sequence databases are indexed with a different set of search fields, including:
[ACCN] Accession [MLWT] Molecular Weight
[ALL] All Fields [ORGN] Organism
[AUTH] Author [PACC] Primary Accession
[GPRJ] BioProject [PROP] Properties
[BIOS] BioSample [PROT] Protein Name
[ECNO] EC/RN Number [SQID] SeqID String
[FKEY] Feature key [SLEN] Sequence Length
[FILT] Filter [SUBS] Substance Name
[GENE] Gene Name [WORD] Text Word
[JOUR] Journal [TITL] Title
[KYWD] Keyword [UID] UID
and a sample query in the protein database is:
"alcohol dehydrogenase [PROT] NOT (bacteria [ORGN] OR fungi [ORGN])"
Additional examples of subset filters in sequence databases are:
mammalia [ORGN]
mammalia [ORGN:noexp]
txid40674 [ORGN]
cds [FKEY]
lacz [GENE]
beta galactosidase [PROT]
protein snp [FILT]
reviewed [FILT]
country united kingdom glasgow [TEXT]
biomol genomic [PROP]
dbxref flybase [PROP]
gbdiv phg [PROP]
phylogenetic study [PROP]
sequence from mitochondrion [PROP]
src cultivar [PROP]
srcdb refseq validated [PROP]
150:200 [SLEN]
(The calculated molecular weight (MLWT) field is only indexed for proteins (and structures), not nucleotides.)
See efilter ‑help for a list of filter shortcuts available for several Entrez databases.
Examining Intermediate Results
EDirect navigation functions produce a custom XML message with the relevant fields (database, web environment, query key, and record count) that can be read by the next command in the pipeline. EDirect may store intermediate results on the Entrez history server or instantiate them in the XML message.
The results of each step in a query can be examined to confirm expected behavior before adding the next step. The Count field in the ENTREZ_DIRECT object contains the number of records returned by the previous step. A good measure of query success is a reasonable (non-zero) count value. For example:
esearch -db protein -query "tryptophan synthase alpha chain [PROT]" |
efilter -query "28000:30000 [MLWT]" |
elink -target structure |
efilter -query "0:2 [RESO]"
produces:
<ENTREZ_DIRECT>
<Db>structure</Db>
<WebEnv> MCID_5fac27e119f45d4eca20b0e6</WebEnv>
<QueryKey>32</QueryKey>
<Count>58</Count>
<Step>4</Step>
</ENTREZ_DIRECT>
with 58 protein structures being within the specified molecular weight range and having the desired (X-ray crystallographic) atomic position resolution.
(The QueryKey value differs from Step because the elink command splits its query into smaller chunks to avoid server truncation limits and timeout errors.)
Combining Independent Queries
Independent esearch, elink, and efilter operations can be performed and then combined at the end by using the history server's "#" convention to indicate query key numbers. (The steps to be combined must be in the same database.) Subsequent esearch commands can take a ‑db argument to override the database piped in from the previous step. (Piping the queries together is necessary for sharing the same history thread.)
Because elink splits a large query into multiple smaller link requests, the new QueryKey value cannot be predicted in advance. The ‑label argument is used to get around this artifact. The label value is prefixed by a "#" symbol and placed in parentheses in the final search. For example, the query:
esearch -db protein -query "amyloid* [PROT]" |
elink -target pubmed -label prot_cit |
esearch -db gene -query "apo* [GENE]" |
elink -target pubmed -label gene_cit |
esearch -query "(#prot_cit) AND (#gene_cit)" |
efetch -format docsum |
xtract -pattern DocumentSummary -element Id Title
uses truncation searching (entering the beginning of a word followed by an asterisk) to return titles of papers with links to amyloid protein sequence and apolipoprotein gene records:
23962925 Genome analysis reveals insights into physiology and ...
23959870 Low levels of copper disrupt brain amyloid-β homeostasis ...
23371554 Genomic diversity and evolution of the head crest in the ...
23251661 Novel genetic loci identified for the pathophysiology of ...
...
Structured Data
Advantages of XML Format
The ability to obtain Entrez records in structured eXtensible Markup Language (XML) format, and to easily extract the underlying data, allows the user to ask novel questions that are not addressed by existing analysis software.
The advantage of XML is that information is in specific locations in a well-defined data hierarchy. Accessing individual units of data that are fielded by name, such as:
<PubDate>2013</PubDate>
<Source>PLoS One</Source>
<Volume>8</Volume>
<Issue>3</Issue>
<Pages>e58144</Pages>
requires matching the same general pattern, differing only by the element name. This is much simpler than parsing the units from a long, complex string:
1. PLoS One. 2013;8(3):e58144 ...
The disadvantage of XML is that data extraction usually requires programming. But EDirect relies on the common pattern of XML value representation to provide a simplified approach to interpreting XML data.
Conversion of XML into Tables
The xtract program uses command-line arguments to direct the selective conversion of data in XML format. It allows record detection, path exploration, element selection, conditional processing, and report formatting to be controlled independently.
The ‑pattern command partitions an XML stream by object name into individual records that are processed separately. Within each record, the ‑element command does an exhaustive, depth-first search to find data content by field name. Explicit paths to objects are not needed.
By default, the ‑pattern argument divides the results into rows, while placement of data into columns is controlled by ‑element, to create a tab-delimited table.
Format Customization
Formatting commands allow extensive customization of the output. The line break between ‑pattern rows is changed with ‑ret, while the tab character between ‑element columns is modified by ‑tab. Multiple instances of the same element are distinguished using ‑sep, which controls their separation independently of the ‑tab command. The following query:
efetch -db pubmed -id 6271474,6092233,16589597 -format docsum |
xtract -pattern DocumentSummary -sep "|" -element Id PubDate Name
returns a tab-delimited table with individual author names separated by vertical bars:
6271474 1981 Casadaban MJ|Chou J|Lemaux P|Tu CP|Cohen SN
6092233 1984 Jul-Aug Calderon IL|Contopoulou CR|Mortimer RK
16589597 1954 Dec Garber ED
The ‑sep value also applies to distinct ‑element arguments that are grouped with commas. This can be used to keep data from multiple related fields in the same column:
-sep " " -element Initials,LastName
Groups of fields are preceded by the ‑pfx value and followed by the ‑sfx value, both of which are initially empty.
The ‑def command sets a default placeholder to be printed when none of the comma-separated fields in an ‑element clause are present:
-def "-" -sep " " -element Year,Month,MedlineDate
Repackaging commands (‑wrp, ‑enc, and ‑pkg) wrap extracted data values with bracketed XML tags given only the object name. For example, "‑wrp Word" issues the following formatting instructions:
-pfx "<Word>" -sep "</Word><Word>" -sfx "</Word>"
and also ensures that data values containing encoded angle brackets, ampersands, quotation marks, or apostrophes remain properly encoded inside the new XML.
Additional commands (‑tag, ‑att, ‑atr, ‑cls, ‑slf, and ‑end) allow generation of XML tags with attributes. Running:
-tag Item -att type journal -cls -element Source -end Item \
-deq "\n" -tag Item -att type journal -atr name Source -slf
will produce regular and self-closing XML objects, respectively:
<Item type="journal">J Bacteriol</Item>
<Item type="journal" name="J Bacteriol" />
Element Variants
Derivatives of ‑element were created to eliminate the inconvenience of having to write post-processing scripts to perform otherwise trivial modifications or analyses on extracted data. They are subdivided into several categories. Substitute for ‑element as needed. A representative selection is shown below:
Positional: -first, -last, -even, -odd, -backward
Numeric: -num, -len, -inc, -dec, -bin, -hex, -bit
Statistics: -sum, -acc, -min, -max, -dev, -med
Averages: -avg, -geo, -hrm, -rms
Logarithms: -lge, -lg2, -log
Character: -encode, -upper, -title, -mirror, -alnum
String: -basic, -plain, -simple, -author, -journal, -prose
Text: -words, -pairs, -letters, -order, -reverse
Citation: -year, -month, -date, -page, -auth
Sequence: -revcomp, -fasta, -ncbi2na, -molwt, -pentamers
Translation: -cds2prot, -gcode, -frame
Coordinate: -0-based, -1-based, -ucsc-based
Variation: -hgvs
Frequency: -histogram
Expression: -reg, -exp, -replace
Substitution: -transform, -translate
Indexing: -aliases, -classify
Miscellaneous: -doi, -wct, -trim, -pad, -accession, -numeric
The original ‑element prefix shortcuts, "#" and "%", are redirected to ‑num and ‑len, respectively.
See xtract ‑help for a brief description of each command.
Exploration Control
Exploration commands provide fine control over the order in which XML record contents are examined, by separately presenting each instance of the chosen subregion. This limits what subsequent commands "see" at any one time, and allows related fields in an object to be kept together.
In contrast to the simpler DocumentSummary format, records retrieved as PubmedArticle XML:
efetch -db pubmed -id 1413997 -format xml |
have authors with separate fields for last name and initials:
<Author>
<LastName>Mortimer</LastName>
<Initials>RK</Initials>
</Author>
Without being given any guidance about context, an ‑element command on initials and last names:
efetch -db pubmed -id 1413997 -format xml |
xtract -pattern PubmedArticle -element Initials LastName
will explore the current record for each argument in turn, printing all initials followed by all last names:
RK CR JS Mortimer Contopoulou King
Inserting a ‑block command adds another exploration layer between ‑pattern and ‑element , and redirects data exploration to present the authors one at a time:
efetch -db pubmed -id 1413997 -format xml |
xtract -pattern PubmedArticle -block Author -element Initials LastName
Each time through the loop, the ‑element command only sees the current author's values. This restores the correct association of initials and last names in the output:
RK Mortimer CR Contopoulou JS King
Grouping the two author subfields with a comma, and adjusting the ‑sep and ‑tab values:
efetch -db pubmed -id 1413997 -format xml |
xtract -pattern PubmedArticle -block Author \
-sep " " -tab ", " -element Initials,LastName
produces a more traditional formatting of author names:
RK Mortimer, CR Contopoulou, JS King
Sequential Exploration
Multiple ‑block statements can be used in a single xtract to explore different areas of the XML. This limits element extraction to the desired subregions, and allows disambiguation of fields with identical names. For example:
efetch -db pubmed -id 6092233,4640931,4296474 -format xml |
xtract -pattern PubmedArticle -element MedlineCitation/PMID \
-block PubDate -sep " " -element Year,Month,MedlineDate \
-block AuthorList -num Author -sep "/" -element LastName |
sort-table -k 3,3n -k 4,4f
generates a table that allows easy parsing of author last names, and sorts the results by author count:
4296474 1968 Apr 1 Friedmann
4640931 1972 Dec 2 Tager/Steiner
6092233 1984 Jul-Aug 3 Calderon/Contopoulou/Mortimer
Like ‑element arguments, the individual ‑block statements are executed sequentially, in order of appearance.
Note that "‑element MedlineCitation/PMID" uses the parent / child construct to prevent the display of additional PMID items that might be present later in CommentsCorrections objects.
Note also that the PubDate object can exist either in a structured form:
<PubDate>
<Year>1968</Year>
<Month>Apr</Month>
<Day>25</Day>
</PubDate>
(with the Day field frequently absent), or in a string form:
<PubDate>
<MedlineDate>1984 Jul-Aug</MedlineDate>
</PubDate>
but would not contain a mixture of both types, so the directive:
-element Year,Month,MedlineDate
will only contribute a single column to the output.
Nested Exploration
Exploration command names (‑group, ‑block, and ‑subset) are assigned to a precedence hierarchy:
-pattern > -group > -block > -subset > -element
and are combined in ranked order to control object iteration at progressively deeper levels in the XML data structure. Each command argument acts as a "nested for-loop" control variable, retaining information about the context, or state of exploration, at its level.
(Hypothetical) census data would need several nested loops to visit each unique address in context:
-pattern State -group City -block Street -subset Number -element Resident
A nucleotide or protein sequence record can have multiple features. Each feature can have multiple qualifiers. And every qualifier has separate name and value nodes. Exploring this natural data hierarchy, with ‑pattern for the sequence, ‑group for the feature, and ‑block for the qualifier:
efetch -db nuccore -id NG_008030.1 -format gbc |
xtract -pattern INSDSeq -element INSDSeq_accession-version \
-group INSDFeature -deq "\n\t" -element INSDFeature_key \
-block INSDQualifier -deq "\n\t\t" \
-element INSDQualifier_name INSDQualifier_value
keeps qualifiers, such as gene and product, associated with their parent features, and keeps qualifier names and values together on the same line:
NG_008030.1
source
organism Homo sapiens
mol_type genomic DNA
db_xref taxon:9606
gene
gene COL5A1
mRNA
gene COL5A1
product collagen type V alpha 1 chain, transcript variant 1
transcript_id NM_000093.4
CDS
gene COL5A1
product collagen alpha-1(V) chain isoform 1 preproprotein
protein_id NP_000084.3
translation MDVHTRWKARSALRPGAPLLPPLLLLLLWAPPPSRAAQP...
...
Saving Data in Variables
A value can be recorded in a variable and used wherever needed. Variables are created by a hyphen followed by a name consisting of a string of capital letters or digits (e.g., ‑KEY). Variable values are retrieved by placing an ampersand before the variable name (e.g., "&KEY") in an ‑element statement:
efetch -db nuccore -id NG_008030.1 -format gbc |
xtract -pattern INSDSeq -element INSDSeq_accession-version \
-group INSDFeature -KEY INSDFeature_key \
-block INSDQualifier -deq "\n\t" \
-element "&KEY" INSDQualifier_name INSDQualifier_value
This version prints the feature key on each line before the qualifier name and value, even though the feature key is now outside of the visibility scope (which is the current qualifier):
NG_008030.1
source organism Homo sapiens
source mol_type genomic DNA
source db_xref taxon:9606
gene gene COL5A1
mRNA gene COL5A1
mRNA product collagen type V alpha 1 chain, transcript variant 1
mRNA transcript_id NM_000093.4
CDS gene COL5A1
CDS product collagen alpha-1(V) chain isoform 1 preproprotein
CDS protein_id NP_000084.3
CDS translation MDVHTRWKARSALRPGAPLLPPLLLLLLWAPPPSRAAQP...
...
Variables can be (re)initialized with an explicit literal value inside parentheses:
-block Author -sep " " -tab "" -element "&COM" Initials,LastName -COM "(, )"
They can also be used as the first argument in a conditional statement:
-CHR Chromosome -block GenomicInfoType -if "&CHR" -differs-from ChrLoc
Using a double-hyphen (e.g., ‑‑STATS) appends a value to the variable.
In addition, a variable can also save the the modified data resulting from an ‑element variant operation. This allows multiple sequential transitions within a single xtract command:
-END -sum "Start,Length" -MID -avg "Start,&END"
All variables are reset when the next record is processed.
Conditional Execution
Conditional processing commands (‑if, ‑unless, ‑and, ‑or, and ‑else) restrict object exploration by data content. They check to see if the named field is within the scope, and may be used in conjunction with string, numeric, or object constraints to require an additional match by value. For example:
esearch -db pubmed -query "Havran W [AUTH]" |
efetch -format xml |
xtract -pattern PubmedArticle -if "#Author" -lt 14 \
-block Author -if LastName -is-not Havran \
-sep ", " -tab "\n" -element LastName,Initials[1:1] |
sort-uniq-count-rank
selects papers with fewer than 14 authors and prints a table of the most frequent collaborators, using a range to keep only the first initial so that variants like "Berg, CM" and "Berg, C" are combined:
34 Witherden, D
15 Boismenu, R
12 Jameson, J
10 Allison, J
10 Fitch, F
...
Numeric constraints can also compare the integer values of two fields. This can be used to find genes that are encoded on the minus strand of a nucleotide sequence:
-if ChrStart -gt ChrStop
Object constraints will compare the string values of two named fields, and can look for internal inconsistencies between fields whose contents should (in most cases) be identical:
-if Chromosome -differs-from ChrLoc
The ‑position command restricts presentation of objects by relative location or index number:
-block Author -position last -sep ", " -element LastName,Initials
Multiple conditions are specified with ‑and and ‑or commands:
-if @score -equals 1 -or @score -starts-with 0.9
The ‑else command can supply alternative ‑element or ‑lbl instructions to be run if the condition is not satisfied:
-if MapLocation -element MapLocation -else -lbl "\-"
but setting a default value with ‑def may be more convenient in simple cases.
Parallel ‑if and ‑unless statements can be used to provide a more complex response to alternative conditions that include nested explorations.
Post-processing Functions
Elink ‑cited can perform a reverse citation lookup, thanks to a data service provided by the NIH Open Citation Collection. The extracted author names can be processed by piping to a chain of Unix utilities:
esearch -db pubmed -query "Beadle GW [AUTH]" |
elink -cited |
efetch -format docsum |
xtract -pattern Author -element Name |
sort -f | uniq -i -c
which produces an alphabetized count of authors who cited the original papers:
1 Abellan-Schneyder I
1 Abramowitz M
1 ABREU LA
1 ABREU RR
1 Abril JF
1 Abächerli E
1 Achetib N
1 Adams CM
2 ADELBERG EA
1 Adrian AB
...
Rather than always having to retype a series of common post-processing instructions, frequently-used combinations of Unix commands can be placed in a function, stored in an alias file (e.g., the user's .bash_profile), and executed by name. For example:
SortUniqCountRank() {
grep '.' |
sort -f |
uniq -i -c |
awk '{ n=$1; sub(/[ \t]*[0-9]+[ \t]/, ""); print n "\t" $0 }' |
sort -t "$(printf '\t')" -k 1,1nr -k 2f
}
alias sort-uniq-count-rank='SortUniqCountRank'(An enhanced version of sort-uniq-count-rank that accepts customization arguments is now included with EDirect as a stand-alone script.)
The raw author names can be passed directly to the sort-uniq-count-rank script:
esearch -db pubmed -query "Beadle GW [AUTH]" |
elink -cited |
efetch -format docsum |
xtract -pattern Author -element Name |
sort-uniq-count-rank
to produce a tab-delimited ranked list of authors who most often cited the original papers:
17 Hawley RS
13 Beadle GW
13 PERKINS DD
11 Glass NL
11 Vécsei L
10 Toldi J
9 TATUM EL
8 Ephrussi B
8 LEDERBERG J
...
Similarly, elink ‑cites uses NIH OCC data to return an article's reference list.
Other scripts for tab-delimited files include sort-table, reorder-columns, and align-columns. Unix parameter expansion requires filter-columns and print-columns arguments to be in single quotes.
Note that EDirect commands can also be used inside Unix functions or scripts.
Viewing an XML Hierarchy
Piping a PubmedArticle XML object to xtract ‑outline will give an indented overview of the XML hierarchy:
PubmedArticle
MedlineCitation
PMID
DateCompleted
Year
Month
Day
...
Article
Journal
...
Title
ISOAbbreviation
ArticleTitle
...
Abstract
AbstractText
AuthorList
Author
LastName
ForeName
Initials
AffiliationInfo
Affiliation
Author
...
Using xtract ‑synopsis or ‑contour will show the full paths to all nodes or just the terminal (leaf) nodes, respectively. Piping those results to "sort-uniq-count" will produce a table of unique paths.
Code Nesting Comparison
Sketching with indented pseudo code can clarify relative nesting levels. The extraction command:
xtract -pattern PubmedArticle \
-block Author -element Initials,LastName \
-block MeshHeading \
-if QualifierName \
-element DescriptorName \
-subset QualifierName -element QualifierName
where the rank of the argument name controls the nesting depth, could be represented as a computer program in pseudo code by:
for pat = each PubmedArticle {
for blk = each pat.Author {
print blk.Initials blk.LastName
}
for blk = each pat.MeSHTerm {
if blk.Qual is present {
print blk.MeshName
for sbs = each blk.Qual {
print sbs.QualName
}
}
}
}where the brace indentation count controls the nesting depth.
Extra arguments are held in reserve to provide additional levels of organization, should the need arise in the future for processing complex, deeply-nested XML data. The exploration commands below ‑pattern, in order of rank, are:
-path
-division
-group
-branch
-block
-section
-subset
-unit
Starting xtract exploration with ‑block, and expanding with ‑group and ‑subset, leaves additional level names that can be used wherever needed without having to redesign the entire command.
Complex Objects
Author Exploration
What's in a name? That which we call an author by any other name may be a consortium, investigator, or editor:
<PubmedArticle>
<MedlineCitation>
<PMID>99999999</PMID>
<Article>
<AuthorList>
<Author>
<LastName>Tinker</LastName>
</Author>
<Author>
<LastName>Evers</LastName>
</Author>
<Author>
<LastName>Chance</LastName>
</Author>
<Author>
<CollectiveName>FlyBase Consortium</CollectiveName>
</Author>
</AuthorList>
</Article>
<InvestigatorList>
<Investigator>
<LastName>Alpher</LastName>
</Investigator>
<Investigator>
<LastName>Bethe</LastName>
</Investigator>
<Investigator>
<LastName>Gamow</LastName>
</Investigator>
</InvestigatorList>
</MedlineCitation>
</PubmedArticle>
Within the record, ‑element exploration on last name:
xtract -pattern PubmedArticle -element LastName
prints each last name, but does not match the consortium:
Tinker Evers Chance Alpher Bethe Gamow
Limiting to the author list:
xtract -pattern PubmedArticle -block AuthorList -element LastName
excludes the investigators:
Tinker Evers Chance
Using ‑num on each type of object:
xtract -pattern PubmedArticle -num Author Investigator LastName CollectiveName
displays the various object counts:
4 3 6 1
Date Selection
Dates come in all shapes and sizes:
<PubmedArticle>
<MedlineCitation>
<PMID>99999999</PMID>
<DateCompleted>
<Year>2011</Year>
</DateCompleted>
<DateRevised>
<Year>2012</Year>
</DateRevised>
<Article>
<Journal>
<JournalIssue>
<PubDate>
<Year>2013</Year>
</PubDate>
</JournalIssue>
</Journal>
<ArticleDate>
<Year>2014</Year>
</ArticleDate>
</Article>
</MedlineCitation>
<PubmedData>
<History>
<PubMedPubDate PubStatus="received">
<Year>2015</Year>
</PubMedPubDate>
<PubMedPubDate PubStatus="accepted">
<Year>2016</Year>
</PubMedPubDate>
<PubMedPubDate PubStatus="entrez">
<Year>2017</Year>
</PubMedPubDate>
<PubMedPubDate PubStatus="pubmed">
<Year>2018</Year>
</PubMedPubDate>
<PubMedPubDate PubStatus="medline">
<Year>2019</Year>
</PubMedPubDate>
</History>
</PubmedData>
</PubmedArticle>
Within the record, ‑element exploration on the year:
xtract -pattern PubmedArticle -element Year
finds and prints all nine instances:
2011 2012 2013 2014 2015 2016 2017 2018 2019
Using ‑block to limit the scope:
xtract -pattern PubmedArticle -block History -element Year
prints only the five years within the History object:
2015 2016 2017 2018 2019
Inserting a conditional statement to limit element selection to a date with a specific attribute:
xtract -pattern PubmedArticle -block History \
-if @PubStatus -equals "pubmed" -element Year
surprisingly still prints all five years within History:
2015 2016 2017 2018 2019
This is because the ‑if command uses the same exploration logic as ‑element, but is designed to declare success if it finds a match anywhere within the current scope. There is indeed a "pubmed" attribute within History, in one of the five PubMedPubDate child objects, so the test succeeds. Thus, ‑element is given free rein to do its own exploration in History, and prints all five years.
The solution is to explore the individual PubMedPubDate objects:
xtract -pattern PubmedArticle -block PubMedPubDate \
-if @PubStatus -equals "pubmed" -element Year
This visits each PubMedPubDate separately, with the ‑if test matching only the indicated date type, thus returning only the desired year:
2018
PMID Extraction
Because of the presence of a CommentsCorrections object:
<PubmedArticle>
<MedlineCitation>
<PMID>99999999</PMID>
<CommentsCorrectionsList>
<CommentsCorrections RefType="ErratumFor">
<PMID>88888888</PMID>
</CommentsCorrections>
</CommentsCorrectionsList>
</MedlineCitation>
</PubmedArticle>
attempting to print the record's PubMed Identifier:
xtract -pattern PubmedArticle -element PMID
also returns the PMID of the comment:
99999999 88888888
Using an exploration command cannot exclude the second instance, because it would need a parent node unique to the first element, and the chain of parents to the first PMID:
PubmedArticle/MedlineCitation
is a subset of the chain of parents to the second PMID:
PubmedArticle/MedlineCitation/CommentsCorrectionList/CommentsCorrections
Although ‑first PMID will work in this particular case, the more general solution is to limit by subpath with the parent / child construct:
xtract -pattern PubmedArticle -element MedlineCitation/PMID
That would work even if the order of objects were reversed.
Heterogeneous Data
XML objects can contain a heterogeneous mix of components. For example:
efetch -db pubmed -id 21433338,17247418 -format xml
returns a mixture of book and journal records:
<PubmedArticleSet>
<PubmedBookArticle>
<BookDocument>
...
</PubmedBookData>
</PubmedBookArticle>
<PubmedArticle>
<MedlineCitation>
...
</PubmedData>
</PubmedArticle>
</PubmedArticleSet>
The parent / star construct is used to visit the individual components, even though they may have different names. Piping the output to:
xtract -pattern "PubmedArticleSet/*" -element "*"
separately prints the entirety of each XML component:
<PubmedBookArticle><BookDocument> ... </PubmedBookData></PubmedBookArticle>
<PubmedArticle><MedlineCitation> ... </PubmedData></PubmedArticle>
Use of the parent / child construct can isolate objects of the same name that differ by their location in the XML hierarchy. For example:
efetch -db pubmed -id 21433338,17247418 -format xml |
xtract -pattern "PubmedArticleSet/*" \
-group "BookDocument/AuthorList" -tab "\n" -element LastName \
-group "Book/AuthorList" -tab "\n" -element LastName \
-group "Article/AuthorList" -tab "\n" -element LastName
writes separate lines for book/chapter authors, book editors, and article authors:
Fauci Desrosiers
Coffin Hughes Varmus
Lederberg Cavalli Lederberg
Simply exploring with individual arguments:
-group BookDocument -block AuthorList -element LastName
would visit the editors (at BookDocument/Book/AuthorList) as well as the authors (at BookDocument/AuthorList), and print names in order of appearance in the XML:
Coffin Hughes Varmus Fauci Desrosiers
(In this particular example the book author lists could be distinguished by using ‑if "@Type" ‑equals authors or ‑if "@Type" ‑equals editors, but exploring by parent / child is a general position-based approach.)
Recursive Definitions
Certain XML objects returned by efetch are recursively defined, including Taxon in ‑db taxonomy and Gene-commentary in ‑db gene. Thus, they can contain nested objects with the same XML tag.
Retrieving a set of taxonomy records:
efetch -db taxonomy -id 9606,7227 -format xml
produces XML with nested Taxon objects (marked below with line references) for each rank in the taxonomic lineage:
<TaxaSet>
1 <Taxon>
<TaxId>9606</TaxId>
<ScientificName>Homo sapiens</ScientificName>
...
<LineageEx>
2 <Taxon>
<TaxId>131567</TaxId>
<ScientificName>cellular organisms</ScientificName>
<Rank>no rank</Rank>
3 </Taxon>
4 <Taxon>
<TaxId>2759</TaxId>
<ScientificName>Eukaryota</ScientificName>
<Rank>superkingdom</Rank>
5 </Taxon>
...
</LineageEx>
...
6 </Taxon>
7 <Taxon>
<TaxId>7227</TaxId>
<ScientificName>Drosophila melanogaster</ScientificName>
...
8 </Taxon>
</TaxaSet>
Xtract tracks XML object nesting to determine that the <Taxon> start tag on line 1 is closed by the </Taxon> stop tag on line 6, and not by the first </Taxon> encountered on line 3.
When a recursive object (e.g., Taxon) is given to an exploration command:
efetch -db taxonomy -id 9606,7227,10090 -format xml |
xtract -pattern Taxon \
-element TaxId ScientificName GenbankCommonName Division
subsequent ‑element commands are blocked from descending into the internal objects, and return information only for the main entries:
9606 Homo sapiens human Primates
7227 Drosophila melanogaster fruit fly Invertebrates
10090 Mus musculus house mouse Rodents
The star / child construct will skip past the outer start tag:
efetch -db taxonomy -id 9606,7227,10090 -format xml |
xtract -pattern Taxon -block "*/Taxon" \
-tab "\n" -element TaxId,ScientificName
to visit the next level of nested objects individually:
131567 cellular organisms
2759 Eukaryota
33154 Opisthokonta
...
Recursive objects can be fully explored with a double star / child construct:
esearch -db gene -query "DMD [GENE] AND human [ORGN]" |
efetch -format xml |
xtract -pattern Entrezgene -block "**/Gene-commentary" \
-tab "\n" -element Gene-commentary_type@value,Gene-commentary_accession
which visits every child object regardless of nesting depth:
genomic NC_000023
mRNA XM_006724469
peptide XP_006724532
mRNA XM_011545467
peptide XP_011543769
...
Additional Elink Options
Elink has several additional modes that can be specified with the ‑cmd argument. When not using the default "neighbor_history" command, elink will return an eLinkResult XML object, with the links for each UID presented in separate blocks. For example, the "neighbor" command:
esearch -db pubmed -query "Hoffmann PC [AUTH] AND dopamine [MAJR]" |
elink -related -cmd neighbor |
xtract -pattern LinkSetDb -element Id
will show the original PMID in the first column and related article PMIDs in subsequent columns:
1504781 11754494 3815119 1684029 14614914 12128255 ...
1684029 3815119 1504781 8097798 17161385 14755628 ...
2572612 2903614 6152036 2905789 9483560 1352865 ...
...
The "acheck" command returns all available link names for each record:
esearch -db pubmed -query "Federhen S [AUTH]" |
elink -cmd acheck |
xtract -pattern LinkSet -tab "\n" -element IdLinkSet/Id \
-block LinkInfo -tab "\n" -element LinkName
printing each on its own line:
25510495
pubmed_images
pubmed_pmc
pubmed_pmc_local
pubmed_pmc_refs
pubmed_pubmed
pubmed_pubmed_citedin
...
The "prlinks" command can obtain the URL reference to the publisher web page for an article. The Unix "xargs" command calls elink separately for each identifier:
epost -db pubmed -id 22966225,19880848 |
efetch -format uid |
xargs -n 1 elink -db pubmed -cmd prlinks -id |
xtract -pattern LinkSet -first Id -element ObjUrl/Url
Repackaging XML Results
Splitting abstract paragraphs into individual words, while using XML reformatting commands:
efetch -db pubmed -id 2539356 -format xml |
xtract -stops -rec Rec -pattern PubmedArticle \
-enc Paragraph -wrp Word -words AbstractText
generates:
...
<Paragraph>
<Word>the</Word>
<Word>tn3</Word>
<Word>transposon</Word>
<Word>inserts</Word>
...
<Word>was</Word>
<Word>necessary</Word>
<Word>for</Word>
<Word>immunity</Word>
</Paragraph>
...
with the words from each abstract instance encased in a separate parent object. Word counts for each paragraph could then be calculated by piping to:
xtract -pattern Rec -block Paragraph -num Word
Multi-Step Transformations
Although xtract provides ‑element variants to do simple data manipulation, more complex tasks are sometimes best handled by being broken up into a series of simpler transformations. These are also known as structured data "processing chains".
Document summaries for two bacterial chromosomes:
efetch -db nuccore -id U00096,CP002956 -format docsum |
contain several individual fields and a complex series of self-closing Stat objects:
<DocumentSummary>
<Id>545778205</Id>
<Caption>U00096</Caption>
<Title>Escherichia coli str. K-12 substr. MG1655, complete genome</Title>
<CreateDate>1998/10/13</CreateDate>
<UpdateDate>2020/09/23</UpdateDate>
<TaxId>511145</TaxId>
<Slen>4641652</Slen>
<Biomol>genomic</Biomol>
<MolType>dna</MolType>
<Topology>circular</Topology>
<Genome>chromosome</Genome>
<Completeness>complete</Completeness>
<GeneticCode>11</GeneticCode>
<Organism>Escherichia coli str. K-12 substr. MG1655</Organism>
<Strain>K-12</Strain>
<BioSample>SAMN02604091</BioSample>
<Statistics>
<Stat type="Length" count="4641652"/>
<Stat type="all" count="9198"/>
<Stat type="cdregion" count="4302"/>
<Stat type="cdregion" subtype="CDS" count="4285"/>
<Stat type="cdregion" subtype="CDS/pseudo" count="17"/>
<Stat type="gene" count="4609"/>
<Stat type="gene" subtype="Gene" count="4464"/>
<Stat type="gene" subtype="Gene/pseudo" count="145"/>
<Stat type="rna" count="187"/>
<Stat type="rna" subtype="ncRNA" count="79"/>
<Stat type="rna" subtype="rRNA" count="22"/>
<Stat type="rna" subtype="tRNA" count="86"/>
<Stat source="all" type="Length" count="4641652"/>
<Stat source="all" type="all" count="13500"/>
<Stat source="all" type="cdregion" count="4302"/>
<Stat source="all" type="gene" count="4609"/>
<Stat source="all" type="prot" count="4302"/>
<Stat source="all" type="rna" count="187"/>
</Statistics>
<AccessionVersion>U00096.3</AccessionVersion>
</DocumentSummary>
<DocumentSummary>
<Id>342852136</Id>
<Caption>CP002956</Caption>
<Title>Yersinia pestis A1122, complete genome</Title>
...
which make extracting the single "best" value for gene count a non-trivial exercise.
In addition to repackaging commands that surround extracted values with XML tags, the ‑element "*" construct prints the entirety of the current scope, including its XML wrapper. Piping the document summaries to:
xtract -set Set -rec Rec -pattern DocumentSummary \
-block DocumentSummary -pkg Common \
-wrp Accession -element AccessionVersion \
-wrp Organism -element Organism \
-wrp Length -element Slen \
-wrp Title -element Title \
-wrp Date -element CreateDate \
-wrp Biomol -element Biomol \
-wrp MolType -element MolType \
-block Stat -if @type -equals gene -pkg Gene -element "*" \
-block Stat -if @type -equals rna -pkg RNA -element "*" \
-block Stat -if @type -equals cdregion -pkg CDS -element "*" |
encloses several fields in a Common block, and packages statistics on gene, RNA, and coding region features into separate sections of a new XML object:
...
<Rec>
<Common>
<Accession>U00096.3</Accession>
<Organism>Escherichia coli str. K-12 substr. MG1655</Organism>
<Length>4641652</Length>
<Title>Escherichia coli str. K-12 substr. MG1655, complete genome</Title>
<Date>1998/10/13</Date>
<Biomol>genomic</Biomol>
<MolType>dna</MolType>
</Common>
<Gene>
<Stat type="gene" count="4609"/>
<Stat type="gene" subtype="Gene" count="4464"/>
<Stat type="gene" subtype="Gene/pseudo" count="145"/>
<Stat source="all" type="gene" count="4609"/>
</Gene>
<RNA>
<Stat type="rna" count="187"/>
<Stat type="rna" subtype="ncRNA" count="79"/>
<Stat type="rna" subtype="rRNA" count="22"/>
<Stat type="rna" subtype="tRNA" count="86"/>
<Stat source="all" type="rna" count="187"/>
</RNA>
<CDS>
<Stat type="cdregion" count="4302"/>
<Stat type="cdregion" subtype="CDS" count="4285"/>
<Stat type="cdregion" subtype="CDS/pseudo" count="17"/>
<Stat source="all" type="cdregion" count="4302"/>
</CDS>
</Rec>
...
With statistics from different types of feature now segregated in their own substructures, total counts for each can be extracted with the ‑first command:
xtract -set Set -rec Rec -pattern Rec \
-block Common -element "*" \
-block Gene -wrp GeneCount -first Stat@count \
-block RNA -wrp RnaCount -first Stat@count \
-block CDS -wrp CDSCount -first Stat@count |
This rewraps the data into a third XML form containing specific feature counts:
...
<Rec>
<Common>
<Accession>U00096.3</Accession>
<Organism>Escherichia coli str. K-12 substr. MG1655</Organism>
<Length>4641652</Length>
<Title>Escherichia coli str. K-12 substr. MG1655, complete genome</Title>
<Date>1998/10/13</Date>
<Biomol>genomic</Biomol>
<MolType>dna</MolType>
</Common>
<GeneCount>4609</GeneCount>
<RnaCount>187</RnaCount>
<CDSCount>4302</CDSCount>
</Rec>
...
without requiring extraction commands for the individual elements in the Common block to be repeated at each step.
Assuming the contents are satisfactory, passing the last structured form to:
xtract \
-head accession organism length gene_count rna_count \
-pattern Rec -def "-" \
-element Accession Organism Length GeneCount RnaCount
produces a tab-delimited table with the desired values:
accession organism length gene_count rna_count
U00096.3 Escherichia coli ... 4641652 4609 187
CP002956.1 Yersinia pestis A1122 4553770 4217 86
If a different order of fields is desired after the final xtract has been run, piping to:
reorder-columns 1 3 5 4
will rearrange the output, including the column headings:
accession length rna_count gene_count
U00096.3 4641652 187 4609
CP002956.1 4553770 86 4217
Sequence Records
NCBI Data Model for Sequence Records
The NCBI data model for sequence records is based on the central dogma of molecular biology. Sequences, including genomic DNA, messenger RNAs, and protein products, are "instantiated" with the actual sequence letters, and are assigned identifiers (e.g., accession numbers) for reference.
Each sequence can have multiple features, which contain information about the biology of a given region, including the transformations involved in gene expression. Each feature can have multiple qualifiers, which store specific details about that feature (e.g., name of the gene, genetic code used for protein translation, accession of the product sequence, cross-references to external databases).
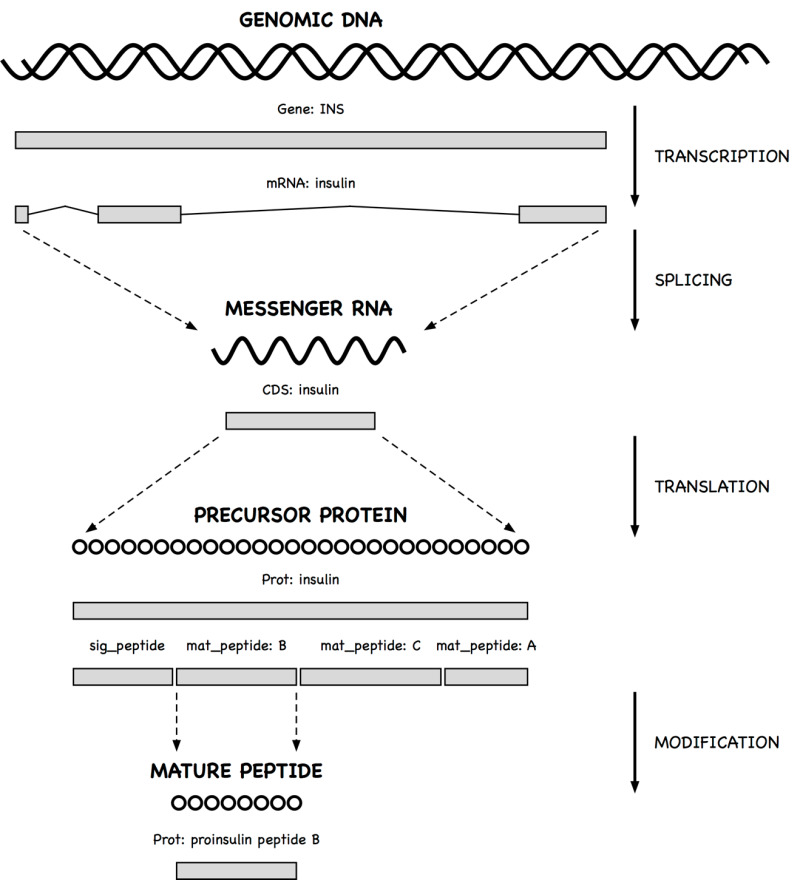
A gene feature indicates the location of a heritable region of nucleic acid that confers a measurable phenotype. An mRNA feature on genomic DNA represents the exonic and untranslated regions of the message that remain after transcription and splicing. A coding region (CDS) feature has a product reference to the translated protein.
Since messenger RNA sequences are not always submitted with a genomic region, CDS features (which model the travel of ribosomes on transcript molecules) are traditionally annotated on the genomic sequence, with locations that encode the exonic intervals.
A qualifier can be dynamically generated from underlying data for the convenience of the user. Thus, the sequence of a mature peptide may be extracted from the mat_peptide feature's location on the precursor protein and displayed in a /peptide qualifier, even if a mature peptide is not instantiated.
Sequence Records in INSDSeq XML
Sequence records can be retrieved in an XML version of the GenBank or GenPept flatfile. The query:
efetch -db protein -id 26418308,26418074 -format gpc
returns a set of INSDSeq objects:
<INSDSet>
<INSDSeq>
<INSDSeq_locus>AAN78128</INSDSeq_locus>
<INSDSeq_length>17</INSDSeq_length>
<INSDSeq_moltype>AA</INSDSeq_moltype>
<INSDSeq_topology>linear</INSDSeq_topology>
<INSDSeq_division>INV</INSDSeq_division>
<INSDSeq_update-date>03-JAN-2003</INSDSeq_update-date>
<INSDSeq_create-date>10-DEC-2002</INSDSeq_create-date>
<INSDSeq_definition>alpha-conotoxin ImI precursor, partial [Conus
imperialis]</INSDSeq_definition>
<INSDSeq_primary-accession>AAN78128</INSDSeq_primary-accession>
<INSDSeq_accession-version>AAN78128.1</INSDSeq_accession-version>
<INSDSeq_other-seqids>
<INSDSeqid>gb|AAN78128.1|</INSDSeqid>
<INSDSeqid>gi|26418308</INSDSeqid>
</INSDSeq_other-seqids>
<INSDSeq_source>Conus imperialis</INSDSeq_source>
<INSDSeq_organism>Conus imperialis</INSDSeq_organism>
<INSDSeq_taxonomy>Eukaryota; Metazoa; Lophotrochozoa; Mollusca;
Gastropoda; Caenogastropoda; Hypsogastropoda; Neogastropoda;
Conoidea; Conidae; Conus</INSDSeq_taxonomy>
<INSDSeq_references>
<INSDReference>
...
Biological features and qualifiers (shown here in GenPept format):
FEATURES Location/Qualifiers
source 1..17
/organism="Conus imperialis"
/db_xref="taxon:35631"
/country="Philippines"
Protein <1..17
/product="alpha-conotoxin ImI precursor"
mat_peptide 5..16
/product="alpha-conotoxin ImI"
/note="the C-terminal glycine of the precursor is post
translationally removed"
/calculated_mol_wt=1357
/peptide="GCCSDPRCAWRC"
CDS 1..17
/coded_by="AY159318.1:<1..54"
/note="nAChR antagonist"
are presented in INSDSeq XML as structured objects:
...
<INSDFeature>
<INSDFeature_key>mat_peptide</INSDFeature_key>
<INSDFeature_location>5..16</INSDFeature_location>
<INSDFeature_intervals>
<INSDInterval>
<INSDInterval_from>5</INSDInterval_from>
<INSDInterval_to>16</INSDInterval_to>
<INSDInterval_accession>AAN78128.1</INSDInterval_accession>
</INSDInterval>
</INSDFeature_intervals>
<INSDFeature_quals>
<INSDQualifier>
<INSDQualifier_name>product</INSDQualifier_name>
<INSDQualifier_value>alpha-conotoxin ImI</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier>
<INSDQualifier_name>note</INSDQualifier_name>
<INSDQualifier_value>the C-terminal glycine of the precursor is
post translationally removed</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier>
<INSDQualifier_name>calculated_mol_wt</INSDQualifier_name>
<INSDQualifier_value>1357</INSDQualifier_value>
</INSDQualifier>
<INSDQualifier>
<INSDQualifier_name>peptide</INSDQualifier_name>
<INSDQualifier_value>GCCSDPRCAWRC</INSDQualifier_value>
</INSDQualifier>
</INSDFeature_quals>
</INSDFeature>
...
The data hierarchy is easily explored using a ‑pattern {sequence} ‑group {feature} ‑block {qualifier} construct. However, feature and qualifier names are indicated in data values, not XML element tags, and require ‑if and ‑equals to select the desired object and content.
Generating Qualifier Extraction Commands
As a convenience for exploring sequence records, the xtract ‑insd helper function generates the appropriate nested extraction commands from feature and qualifier names on the command line. (Two computed qualifiers, sub_sequence and feat_location, are also supported.)
Running xtract ‑insd in an isolated command prints a new xtract statement that can then be copied, edited if necessary, and pasted into other queries. Running the ‑insd command within a multi-step pipe dynamically executes the automatically-constructed query.
Providing an optional (complete/partial) location indication, a feature key, and then one or more qualifier names:
xtract -insd complete mat_peptide product peptide
creates a new xtract statement that will produce a table of qualifier values from mature peptide features with complete locations. The statement starts with instructions to record the accession and find features of the indicated type:
xtract -pattern INSDSeq -ACCN INSDSeq_accession-version -SEQ INSDSeq_sequence \
-group INSDFeature -if INSDFeature_key -equals mat_peptide \
-branch INSDFeature -unless INSDFeature_partial5 -or INSDFeature_partial3 \
-clr -pfx "\n" -element "&ACCN" \
Each qualifier then generates custom extraction code that is appended to the growing query. For example:
-block INSDQualifier \
-if INSDQualifier_name -equals product \
-element INSDQualifier_value
Snail Venom Peptide Sequences
Incorporating the xtract ‑insd command in a search on cone snail venom:
esearch -db pubmed -query "conotoxin" |
elink -target protein |
efilter -query "mat_peptide [FKEY]" |
efetch -format gpc |
xtract -insd complete mat_peptide "%peptide" product mol_wt peptide |
prints the accession number, mature peptide length, product name, calculated molecular weight, and amino acid sequence for a sample of neurotoxic peptides:
AAN78128.1 12 alpha-conotoxin ImI 1357 GCCSDPRCAWRC
ADB65789.1 20 conotoxin Cal 16 2134 LEMQGCVCNANAKFCCGEGR
ADB65788.1 20 conotoxin Cal 16 2134 LEMQGCVCNANAKFCCGEGR
AGO59814.1 32 del13b conotoxin 3462 DCPTSCPTTCANGWECCKGYPCVRQHCSGCNH
AAO33169.1 16 alpha-conotoxin GIC 1615 GCCSHPACAGNNQHIC
AAN78279.1 21 conotoxin Vx-II 2252 WIDPSHYCCCGGGCTDDCVNC
AAF23167.1 31 BeTX toxin 3433 CRAEGTYCENDSQCCLNECCWGGCGHPCRHP
ABW16858.1 15 marmophin 1915 DWEYHAHPKPNSFWT
...
Piping the results to a series of Unix commands and EDirect scripts:
grep -i conotoxin |
filter-columns '10 <= $2 && $2 <= 30' |
sort-table -u -k 5 |
sort-table -k 2,2n |
align-columns -
filters by product name, limits the results to a specified range of peptide lengths, removes redundant sequences, sorts the table by peptide length, and aligns the columns for cleaner printing:
AAN78127.1 12 alpha-conotoxin ImII 1515 ACCSDRRCRWRC
AAN78128.1 12 alpha-conotoxin ImI 1357 GCCSDPRCAWRC
ADB43130.1 15 conotoxin Cal 1a 1750 KCCKRHHGCHPCGRK
ADB43131.1 15 conotoxin Cal 1b 1708 LCCKRHHGCHPCGRT
AAO33169.1 16 alpha-conotoxin GIC 1615 GCCSHPACAGNNQHIC
ADB43128.1 16 conotoxin Cal 5.1 1829 DPAPCCQHPIETCCRR
AAD31913.1 18 alpha A conotoxin Tx2 2010 PECCSHPACNVDHPEICR
ADB43129.1 18 conotoxin Cal 5.2 2008 MIQRSQCCAVKKNCCHVG
ADB65789.1 20 conotoxin Cal 16 2134 LEMQGCVCNANAKFCCGEGR
ADD97803.1 20 conotoxin Cal 1.2 2206 AGCCPTIMYKTGACRTNRCR
AAD31912.1 21 alpha A conotoxin Tx1 2304 PECCSDPRCNSSHPELCGGRR
AAN78279.1 21 conotoxin Vx-II 2252 WIDPSHYCCCGGGCTDDCVNC
ADB43125.1 22 conotoxin Cal 14.2 2157 GCPADCPNTCDSSNKCSPGFPG
ADD97802.1 23 conotoxin Cal 6.4 2514 GCWLCLGPNACCRGSVCHDYCPR
AAD31915.1 24 O-superfamily conotoxin TxO2 2565 CYDSGTSCNTGNQCCSGWCIFVCL
AAD31916.1 24 O-superfamily conotoxin TxO3 2555 CYDGGTSCDSGIQCCSGWCIFVCF
AAD31920.1 24 omega conotoxin SVIA mutant 1 2495 CRPSGSPCGVTSICCGRCYRGKCT
AAD31921.1 24 omega conotoxin SVIA mutant 2 2419 CRPSGSPCGVTSICCGRCSRGKCT
ABE27006.1 25 conotoxin p114a 2917 FPRPRICNLACRAGIGHKYPFCHCR
ABE27007.1 25 conotoxin p114.1 2645 GPGSAICNMACRLGQGHMYPFCNCN
...
The xtract ‑insdx variant:
esearch -db protein -query "conotoxin" |
efilter -query "mat_peptide [FKEY]" |
efetch -format gpc |
xtract -insdx complete mat_peptide "%peptide" product mol_wt peptide |
xtract -pattern Rec -select product -contains conotoxin |
xtract -pattern Rec -sort mol_wt
saves the output table directly as XML, with the XML tag names taken from the original qualifier names:
...
<Rec>
<accession>AAO33169.1</accession>
<feature_key>mat_peptide</feature_key>
<peptide_Len>16</peptide_Len>
<product>alpha-conotoxin GIC</product>
<mol_wt>1615</mol_wt>
<peptide>GCCSHPACAGNNQHIC</peptide>
</Rec>
<Rec>
<accession>AIC77099.1</accession>
<feature_key>mat_peptide</feature_key>
<peptide_Len>16</peptide_Len>
<product>conotoxin Im1.2</product>
<mol_wt>1669</mol_wt>
<peptide>GCCSHPACNVNNPHIC</peptide>
</Rec>
...
Qualifier names with prefix shortcuts "#" and "%" are modified to use "_Num" and "_Len" suffixes, respectively.
Missing Qualifiers
For records where a particular qualifier is missing:
esearch -db protein -query "RAG1 [GENE] AND Mus musculus [ORGN]" |
efetch -format gpc |
xtract -insd source organism strain |
sort-table -u -k 2,3
a dash is inserted as a placeholder:
P15919.2 Mus musculus -
AAO61776.1 Mus musculus 129/Sv
NP_033045.2 Mus musculus C57BL/6
EDL27655.1 Mus musculus mixed
BAD69530.1 Mus musculus castaneus -
BAD69531.1 Mus musculus domesticus BALB/c
BAD69532.1 Mus musculus molossinus MOA
Sequence Coordinates
Gene Positions
An understanding of sequence coordinate conventions is necessary in order to use gene positions to retrieve the corresponding chromosome subregion with efetch or with the UCSC browser.
Sequence records displayed in GenBank or GenPept formats use a "one-based" coordinate system, with sequence position numbers starting at "1":
1 catgccattc gttgagttgg aaacaaactt gccggctagc cgcatacccg cggggctgga
61 gaaccggctg tgtgcggcca cagccaccat cctggacaaa cccgaagacg tgagtgaggg
121 tcggcgagaa cttgtgggct agggtcggac ctcccaatga cccgttccca tccccaggga
181 ccccactccc ctggtaacct ctgaccttcc gtgtcctatc ctcccttcct agatcccttc
...
Under this convention, positions refer to the sequence letters themselves:
C A T G C C A T T C
1 2 3 4 5 6 7 8 9 10
and the position of the last base or residue is equal to the length of the sequence. The ATG initiation codon above is at positions 2 through 4, inclusive.
For computer programs, however, using "zero-based" coordinates can simplify the arithmetic used for calculations on sequence positions. The ATG codon in the 0-based representation is at positions 1 through 3. (The UCSC browser uses a hybrid, half-open representation, where the start position is 0-based and the stop position is 1-based.)
Software at NCBI will typically convert positions to 0-based coordinates upon input, perform whatever calculations are desired, and then convert the results to a 1-based representation for display. These transformations are done by simply subtracting 1 from the 1-based value or adding 1 to the 0-based value.
Coordinate Conversions
Retrieving the docsum for a particular gene:
esearch -db gene -query "BRCA2 [GENE] AND human [ORGN]" |
efetch -format docsum |
returns the chromosomal position of that gene in "zero-based" coordinates:
...
<GenomicInfoType>
<ChrLoc>13</ChrLoc>
<ChrAccVer>NC_000013.11</ChrAccVer>
<ChrStart>32315479</ChrStart>
<ChrStop>32399671</ChrStop>
<ExonCount>27</ExonCount>
</GenomicInfoType>
...
Piping the document summary to an xtract command using ‑element:
xtract -pattern GenomicInfoType -element ChrAccVer ChrStart ChrStop
obtains the accession and 0-based coordinate values:
NC_000013.11 32315479 32399671
Efetch has ‑seq_start and ‑seq_stop arguments to retrieve a gene segment, but these expect the sequence subrange to be in 1-based coordinates.
To address this problem, two additional efetch arguments, ‑chr_start and ‑chr_stop, were created to allow direct use of the 0-based coordinates:
efetch -db nuccore -format gb -id NC_000013.11 \
-chr_start 32315479 -chr_stop 32399671
Xtract now has numeric extraction commands to assist with coordinate conversion. Selecting fields with an ‑inc argument:
xtract -pattern GenomicInfoType -element ChrAccVer -inc ChrStart ChrStop
obtains the accession and 0-based coordinates, then increments the positions to produce 1-based values:
NC_000013.11 32315480 32399672
EDirect knows the policies for sequence positions in all relevant Entrez databases (e.g., gene, snp, dbvar), and provides additional shortcuts for converting these to other conventions. For example:
xtract -pattern GenomicInfoType -element ChrAccVer -1-based ChrStart ChrStop
understands that gene docsum ChrStart and ChrStop fields are 0-based, sees that the desired output is 1-based, and translates the command to convert coordinates internally using the ‑inc logic. Similarly:
-element ChrAccVer -ucsc-based ChrStart ChrStop
leaves the 0-based start value unchanged but increments the original stop value to produce the half-open form that can be passed to the UCSC browser:
NC_000013.11 32315479 32399672
Gene Records
Genes in a Region
To list all genes between two markers flanking the human X chromosome centromere, first retrieve the protein-coding gene records on that chromosome:
esearch -db gene -query "Homo sapiens [ORGN] AND X [CHR]" |
efilter -status alive -type coding | efetch -format docsum |
Gene names and chromosomal positions are extracted by piping the records to:
xtract -pattern DocumentSummary -NAME Name -DESC Description \
-block GenomicInfoType -if ChrLoc -equals X \
-min ChrStart,ChrStop -element "&NAME" "&DESC" |
Exploring each GenomicInfoType is needed because of pseudoautosomal regions at the ends of the X and Y chromosomes:
...
<GenomicInfo>
<GenomicInfoType>
<ChrLoc>X</ChrLoc>
<ChrAccVer>NC_000023.11</ChrAccVer>
<ChrStart>155997630</ChrStart>
<ChrStop>156013016</ChrStop>
<ExonCount>14</ExonCount>
</GenomicInfoType>
<GenomicInfoType>
<ChrLoc>Y</ChrLoc>
<ChrAccVer>NC_000024.10</ChrAccVer>
<ChrStart>57184150</ChrStart>
<ChrStop>57199536</ChrStop>
<ExonCount>14</ExonCount>
</GenomicInfoType>
</GenomicInfo>
...
Without limiting to chromosome X, the copy of IL9R near the "q" telomere of chromosome Y would be erroneously placed with genes that are near the X chromosome centromere, shown here in between SPIN2A and ZXDB:
...
57121860 FAAH2 fatty acid amide hydrolase 2
57133042 SPIN2A spindlin family member 2A
57184150 IL9R interleukin 9 receptor
57592010 ZXDB zinc finger X-linked duplicated B
...
With genes restricted to the X chromosome, results can be sorted by position, and then filtered and partitioned:
sort-table -k 1,1n | cut -f 2- |
grep -v pseudogene | grep -v uncharacterized | grep -v hypothetical |
between-two-genes AMER1 FAAH2
to produce an ordered table of known genes located between the two markers:
FAAH2 fatty acid amide hydrolase 2
SPIN2A spindlin family member 2A
ZXDB zinc finger X-linked duplicated B
NLRP2B NLR family pyrin domain containing 2B
ZXDA zinc finger X-linked duplicated A
SPIN4 spindlin family member 4
ARHGEF9 Cdc42 guanine nucleotide exchange factor 9
AMER1 APC membrane recruitment protein 1
Gene Sequence
Genes encoded on the minus strand of a sequence:
esearch -db gene -query "DDT [GENE] AND mouse [ORGN]" |
efetch -format docsum |
xtract -pattern GenomicInfoType -element ChrAccVer ChrStart ChrStop |
have coordinates ("zero-based" in docsums) where the start position is greater than the stop:
NC_000076.6 75773373 75771232
These values can be read into Unix variables by a "while" loop:
while IFS=$'\t' read acn str stp
do
efetch -db nuccore -format gb \
-id "$acn" -chr_start "$str" -chr_stop "$stp"
done
The variables can then be used to obtain the reverse-complemented subregion in GenBank format:
LOCUS NC_000076 2142 bp DNA linear CON 08-AUG-2019
DEFINITION Mus musculus strain C57BL/6J chromosome 10, GRCm38.p6 C57BL/6J.
ACCESSION NC_000076 REGION: complement(75771233..75773374)
...
gene 1..2142
/gene="Ddt"
mRNA join(1..159,462..637,1869..2142)
/gene="Ddt"
/product="D-dopachrome tautomerase"
/transcript_id="NM_010027.1"
CDS join(52..159,462..637,1869..1941)
/gene="Ddt"
/codon_start=1
/product="D-dopachrome decarboxylase"
/protein_id="NP_034157.1"
/translation="MPFVELETNLPASRIPAGLENRLCAATATILDKPEDRVSVTIRP
GMTLLMNKSTEPCAHLLVSSIGVVGTAEQNRTHSASFFKFLTEELSLDQDRIVIRFFP
...
The reverse complement of a plus-strand sequence range can be selected with efetch ‑revcomp
External Data
Querying External Services
The nquire program uses command-line arguments to obtain data from RESTful, CGI, or FTP servers. Queries are built up from command-line arguments. Paths can be separated into components, which are combined with slashes. Remaining arguments (starting with a dash) are tag/value pairs, with multiple values between tags combined with commas.
For example, a POST request:
nquire -url http://w1.weather.gov/xml/current_obs/KSFO.xml |
xtract -pattern current_observation -tab "\n" \
-element weather temp_f wind_dir wind_mph
returns the current weather report at the San Francisco airport:
A Few Clouds
54.0
Southeast
5.8
and a GET query:
nquire -get http://collections.mnh.si.edu/services/resolver/resolver.php \
-voucher "Birds:321082" |
xtract -pattern Result -tab "\n" -element ScientificName StateProvince Country
returns information on a ruby-throated hummingbird specimen:
Archilochus colubris
Maryland
United States
while an FTP request:
nquire -ftp ftp.ncbi.nlm.nih.gov pub/gdp ideogram_9606_GCF_000001305.14_850_V1 |
grep acen | cut -f 1,2,6,7 | awk '/^X\t/'
returns data with the (estimated) sequence coordinates of the human X chromosome centromere (here showing where the p and q arms meet):
X p 58100001 61000000
X q 61000001 63800000
Nquire can also produce a list of files in an FTP server directory:
nquire -lst ftp://nlmpubs.nlm.nih.gov online/mesh/MESH_FILES/xmlmesh
or a list of FTP file names preceded by a column with the file sizes:
nquire -dir ftp.ncbi.nlm.nih.gov gene/DATA
Finally, nquire can download FTP files to the local disk:
nquire -dwn ftp.nlm.nih.gov online/mesh/MESH_FILES/xmlmesh desc2021.zip
If Aspera Connect is installed, the nquire ‑asp command will provide faster retrieval from NCBI servers:
nquire -asp ftp.ncbi.nlm.nih.gov pubmed baseline pubmed22n0001.xml.gz
Without Aspera Connect, nquire ‑asp defaults to using the ‑dwn logic.
XML Namespaces
Namespace prefixes are followed by a colon, while a leading colon matches any prefix:
nquire -url http://webservice.wikipathways.org getPathway -pwId WP455 |
xtract -pattern "ns1:getPathwayResponse" -decode ":gpml" |
The embedded Graphical Pathway Markup Language object can then be processed:
xtract -pattern Pathway -block Xref \
-if @Database -equals "Entrez Gene" \
-tab "\n" -element @ID
Automatic Xtract Format Conversion
Xtract can now detect and convert input data in JSON, text ASN.1, and GenBank/GenPept flatfile formats. The transmute commands or shortcut scripts, described below, are only needed if you want to inspect the intermediate XML, or to override default conversion settings.
JSON Arrays
Consolidated gene information for human β-globin retrieved from a curated biological database service developed at the Scripps Research Institute:
nquire -get http://mygene.info/v3 gene 3043 |
contains a multi-dimensional array of exon coordinates in JavaScript Object Notation (JSON) format:
"position": [
[
5225463,
5225726
],
[
5226576,
5226799
],
[
5226929,
5227071
]
],
"strand": -1,
This can be converted to XML with transmute ‑j2x (or the json2xml shortcut script):
transmute -j2x |
with the default"‑nest element" argument assigning distinct tag names to each level:
<position>
<position_E>5225463</position_E>
<position_E>5225726</position_E>
</position>
...
JSON Mixtures
A query for the human green-sensitive opsin gene:
nquire -get http://mygene.info/v3/gene/2652 |
transmute -j2x |
returns data containing a heterogeneous mixture of objects in the pathway section:
<pathway>
<reactome>
<id>R-HSA-162582</id>
<name>Signal Transduction</name>
</reactome>
...
<wikipathways>
<id>WP455</id>
<name>GPCRs, Class A Rhodopsin-like</name>
</wikipathways>
</pathway>
The parent / star construct is used to visit the individual components of a parent object without needing to explicitly specify their names. For printing, the name of a child object is indicated by a question mark:
xtract -pattern opt -group "pathway/*" \
-pfc "\n" -element "?,name,id"
This displays a table of pathway database references:
reactome Signal Transduction R-HSA-162582
reactome Disease R-HSA-1643685
...
reactome Diseases of the neuronal system R-HSA-9675143
wikipathways GPCRs, Class A Rhodopsin-like WP455
Xtract ‑path can explore using multi-level object addresses, delimited by periods or slashes:
xtract -pattern opt -path pathway.wikipathways.id -tab "\n" -element id
Conversion of ASN.1
Similarly to ‑j2x, transmute ‑a2x (or asn2xml) will convert Abstract Syntax Notation 1 (ASN.1) text files to XML.
Tables to XML
Tab-delimited files are easily converted to XML with transmute ‑t2x (or tbl2xml):
nquire -ftp ftp.ncbi.nlm.nih.gov gene/DATA gene_info.gz |
gunzip -c | grep -v NEWENTRY | cut -f 2,3 |
transmute -t2x -set Set -rec Rec -skip 1 Code Name
This takes a series of command-line arguments with tag names for wrapping the individual columns, and skips the first line of input, which contains header information, to generate a new XML file:
...
<Rec>
<Code>1246500</Code>
<Name>repA1</Name>
</Rec>
<Rec>
<Code>1246501</Code>
<Name>repA2</Name>
</Rec>
...
The transmute ‑t2x ‑header argument will obtain tag names from the first line of the file:
nquire -ftp ftp.ncbi.nlm.nih.gov gene/DATA gene_info.gz |
gunzip -c | grep -v NEWENTRY | cut -f 2,3 |
transmute -t2x -set Set -rec Rec -header
CSV to XML
Similarly to ‑t2x, transmute ‑c2x (or csv2xml) will convert comma-separated values (CSV) files to XML.
GenBank Download
The entire set of GenBank format release files be downloaded with:
fls=$( nquire -lst ftp.ncbi.nlm.nih.gov genbank )
for div in \
bct con env est gss htc htg inv mam pat \
phg pln pri rod sts syn tsa una vrl vrt
do
echo "$fls" |
grep ".seq.gz" | grep "gb${div}" |
sort -V | skip-if-file-exists |
nquire -asp ftp.ncbi.nlm.nih.gov genbank
done
Unwanted divisions can be removed from the "for" loop to limit retrieval to specific sequencing classes or taxonomic regions.
GenBank to XML
The most recent GenBank virus release file can also be downloaded from NCBI servers:
nquire -lst ftp.ncbi.nlm.nih.gov genbank |
grep "^gbvrl" | grep ".seq.gz" | sort -V |
tail -n 1 | skip-if-file-exists |
nquire -asp ftp.ncbi.nlm.nih.gov genbank
GenBank flatfile records can be selected by organism name or taxon identifier, or by presence or absence of an arbitrary text string, with transmute ‑gbf (or filter-genbank):
gunzip -c *.seq.gz | filter-genbank -taxid 11292 |
Since xtract can now read JSON, ASN.1, and GenBank formats, the filtered flatfiles can be piped to xtract to obtain feature location intervals and underlying sequences of individual coding regions:
xtract -insd CDS gene product feat_location sub_sequence
without the need for an explicit transmute ‑g2x (or gbf2xml) step.
GenPept to XML
The latest GenPept daily incremental update file can be downloaded:
nquire -ftp ftp.ncbi.nlm.nih.gov genbank daily-nc Last.File |
sed "s/flat/gnp/g" |
nquire -ftp ftp.ncbi.nlm.nih.gov genbank daily-nc |
gunzip -c | transmute -g2x |
and the extracted INSDSeq XML can be processed in a similar manner:
xtract -pattern INSDSeq -select INSDQualifier_value -equals "taxon:2697049" |
xtract -insd mat_peptide product sub_sequence
Local PubMed Cache
Fetching data from Entrez works well when a few thousand records are needed, but it does not scale for much larger sets of data, where the time it takes to download becomes a limiting factor.
Recent advances in technology provide an affordable and practical alternative. High-performance NVMe solid-state drives (which eliminate rotational delays for file access and bookkeeping operations) are readily available for purchase. Modern high-capacity file systems, such as APFS (which uses 64-bit inodes) or Ext4 (which can be configured for 100 million inodes), are now ubiquitous on contemporary computers. A judicious arrangement of multi-level nested directories (each containing no more than 100 subfolders or record files) ensures maximally-efficient use of these enhanced capabilities.
This combination of features allows local record storage (populated in advance from the PubMed FTP release files) to be an effective replacement for on-demand network retrieval, while avoiding the need to install and support a legacy database product on your computer.
Random Access Archive
EDirect can now preload over 35 million live PubMed records onto an inexpensive external 500 GB (gigabyte) solid-state drive as individual files for rapid retrieval. For example, PMID 12345678 would be stored at:
/Archive/12/34/56/12345678.xml.gz
using a hierarchy of folders to organize the data for random access to any record.
The local archive is a completely self-contained turnkey system, with no need for the user to download, configure, and maintain complicated third-party database software.
Set an environment variable in your configuration file(s) to reference a section of your external drive:
export EDIRECT_LOCAL_ARCHIVE=/Volumes/external_drive_name/
or set separate environment variables to keep the intermediate steps on the external SSD but leave the resulting archive in a designated area of the computer's internal storage:
export EDIRECT_LOCAL_ARCHIVE=$HOME/internal_directory_name/
export EDIRECT_LOCAL_WORKING=/Volumes/external_drive_name/
In the latter case it will store around 180 GB on the internal drive for the local archive, or up to 250 GB if the local search index (see below) is also built.
Then run archive-pubmed to download the PubMed release files and distribute each record on the drive. This process will take several hours to complete, but subsequent updates are incremental, and should finish in minutes.
Retrieving over 125,000 compressed PubMed records from the local archive:
esearch -db pubmed -query "PNAS [JOUR]" -pub abstract |
efetch -format uid | stream-pubmed | gunzip -c |
takes about 20 seconds. Retrieving those records from NCBI's network service, with efetch ‑format xml, would take around 40 minutes.
Even modest sets of PubMed query results can benefit from using the local cache. A reverse citation lookup on 191 papers:
esearch -db pubmed -query "Cozzarelli NR [AUTH]" | elink -cited |
requires 13 seconds to match 9620 subsequent articles. Retrieving them from the local archive:
efetch -format uid | fetch-pubmed |
takes less than one second. Printing the names of all authors in those records:
xtract -pattern PubmedArticle -block Author \
-sep " " -tab "\n" -element LastName,Initials |
allows creation of a frequency table:
sort-uniq-count-rank
that lists the authors who most often cited the original papers:
145 Cozzarelli NR
108 Maxwell A
86 Wang JC
81 Osheroff N
...
Fetching from the network service would extend the 14 second running time to over 2 minutes.
Local Search Index
A similar divide-and-conquer strategy is used to create a local information retrieval system suitable for large data mining queries. Run archive-pubmed ‑index to populate retrieval index files from records stored in the local archive. The initial indexing will also take a few hours. Since PubMed updates are released once per day, it may be convenient to schedule reindexing to start in the late evening and run during the night.
For PubMed titles and primary abstracts, the indexing process deletes hyphens after specific prefixes, removes accents and diacritical marks, splits words at punctuation characters, corrects encoding artifacts, and spells out Greek letters for easier searching on scientific terms. It then prepares inverted indices with term positions, and uses them to build distributed term lists and postings files.
For example, the term list that includes "cancer" in the title or abstract would be located at:
/Postings/TIAB/c/a/n/c/canc.TIAB.trm
A query on cancer thus only needs to load a very small subset of the total index. The software supports expression evaluation, wildcard truncation, phrase queries, and proximity searches.
The phrase-search script (with an implied ‑db pubmed) provides access to the local search system.
Names of indexed fields, all terms for a given field, and terms plus record counts, are shown by:
phrase-search -fields
phrase-search -terms TITL
phrase-search -totals PROP
Terms are truncated with trailing asterisks, and can be expanded to show individual postings counts:
phrase-search -count "catabolite repress*"
phrase-search -counts "catabolite repress*"
Query evaluation includes Boolean operations and parenthetical expressions:
phrase-search -query "(literacy AND numeracy) NOT (adolescent OR child)"
Adjacent words in the query are treated as a contiguous phrase:
phrase-search -query "selective serotonin reuptake inhibitor"
Each plus sign will replace a single word inside a phrase, and runs of tildes indicate the maximum distance between sequential phrases:
phrase-search -query "vitamin c + + common cold"
phrase-search -query "vitamin c ~ ~ common cold"
An exact substring match, without special processing of Boolean operators or indexed field names, can be obtained with -title (on the article title) or -exact (on the title or abstract), while ranked partial term matching in any field is available with -match:
phrase-search -title "Genetic Control of Biochemical Reactions in Neurospora."
phrase-search -match "tn3 transposition immunity [PAIR]" | just-first-key
MeSH identifier code, MeSH hierarchy key, and year of publication are also indexed, and MESH field queries are supported by internally mapping to the appropriate CODE or TREE entries:
phrase-search -query "C14.907.617.812* [TREE] AND 2015:2019 [YEAR]"
phrase-search -query "Raynaud Disease [MESH]"
The phrase-search ‑filter command allows PMIDs to be generated by an EDirect search and then incorporated as a component in a local query:
Data Analysis and Visualization
All query commands return a list of PMIDs, which can be piped directly to fetch-pubmed to retrieve the uncompressed records. For example:
phrase-search -query "selective serotonin ~ ~ ~ reuptake inhibit*" |
fetch-pubmed |
xtract -pattern PubmedArticle -num AuthorList/Author |
sort-uniq-count -n |
reorder-columns 2 1 |
head -n 25 |
align-columns -g 4 -a lr
performs a proximity search with dynamic wildcard expansion (matching phrases like "selective serotonin and norepinephrine reuptake inhibitors") and fetches 12,966 PubMed records from the local archive. It then counts the number of authors for each paper (a consortium is treated as a single author), printing a frequency table of the number of papers per number of authors:
0 51
1 1382
2 1897
3 1906
...
The phrase-search and fetch-pubmed scripts are front-ends to the rchive program, which is used to build and search the inverted retrieval system. Rchive is multi-threaded for speed, retrieving records from the local archive in parallel, and fetching the positional indices for all terms in parallel before evaluating the title words as a contiguous phrase.
The cumulative size of PubMed can be calculated with a running sum of the annual record counts. Exponential growth over time will appear as a roughly linear curve on a semi-logarithmic graph:
phrase-search -totals YEAR |
print-columns '$2, $1, total += $1' |
print-columns '$1, log($2)/log(10), log($3)/log(10)' |
filter-columns '$1 >= 1800' | sed '$d' |
xy-plot annual-and-cumulative.png
Natural Language Processing
NCBI's Biomedical Text Mining Group performs computational analysis to extract chemical, disease, and gene references from article contents. NLM indexing of PubMed records assigns Gene Reference into Function (GeneRIF) mappings.
Running archive-ncbinlp ‑index periodically (monthly) will automatically refresh any out-of-date support files and then index the connections in CHEM, DISZ, GENE, and several gene subfields (GRIF, GSYN, and PREF):
phrase-search -terms DISZ | grep -i Raynaud
phrase-search -counts "Raynaud* [DISZ]"
phrase-search -query "Raynaud Disease [DISZ]"
Following Citation Links
Running archive-nihocc ‑index will download the latest NIH Open Citation Collection monthly release and build CITED and CITES indices, the local equivalent of elink ‑cited and ‑cites commands.
Citation links are retrieved by piping one or more PMIDs to phrase-search ‑link:
phrase-search -db pubmed -query "Havran W* [AUTH]" |
phrase-search -link CITED |
This returns PMIDs for 6504 articles that cite the original 96 papers. The records are then fetched and analyzed:
fetch-pubmed |
xtract -pattern PubmedArticle -histogram Journal/ISOAbbreviation |
sort-table -nr | head -n 10
to display the most popular journals in which the subsequent articles were published:
921 J Immunol
293 Eur J Immunol
248 J Exp Med
168 Front Immunol
149 Proc Natl Acad Sci U S A
139 Cell Immunol
121 Int Immunol
106 J Invest Dermatol
105 Immunol Rev
99 Immunity
Rapidly Scanning PubMed
If the expand-current script is run, an ad hoc scan can be performed on the nonredundant set of live PubMed records:
cat $EDIRECT_LOCAL_WORKING/pubmed/Scratch/Current/*.xml |
xtract -timer -turbo -pattern PubmedArticle -PMID MedlineCitation/PMID \
-group AuthorList -if "#LastName" -eq 7 -element "&PMID" LastName
finding 1,700,652 articles with seven authors. (This query excludes consortia and additional named investigators. Author count is now indexed in the ANUM field.)
Xtract uses the Boyer-Moore-Horspool algorithm to partition an XML stream into individual records, distributing them among multiple instances of the data exploration and extraction function for concurrent execution. A multi-core computer with a solid-state drive can process all of PubMed in under 4 minutes.
The expand-current script now calls xtract -index to place an XML size object immediately before each PubMed record:
...
</PubmedArticle>
<NEXT_RECORD_SIZE>6374</NEXT_RECORD_SIZE>
<PubmedArticle>
...
The xtract ‑turbo flag reads this precomputed information to approximately double the speed of record partitioning, which is the rate-limiting step when many CPU cores are available. With proper cooling, it should allow up to a dozen cores to contribute to batch data extraction throughput.
User-Specified Term Index
Running custom-index with a PubMed indexer script and the names of the fields it populates:
custom-index $( which idx-grant ) GRNT
integrates user-specified indices into the local search system. The idx-grant script:
xtract -set IdxDocumentSet -rec IdxDocument -pattern PubmedArticle \
-wrp IdxUid -element MedlineCitation/PMID -clr -rst -tab "" \
-group PubmedArticle -pkg IdxSearchFields \
-block PubmedArticle -wrp GRNT -element Grant/GrantID
has reusable boilerplate in its first three lines, and indexes PubMed records by Grant Identifier:
...
<IdxDocument>
<IdxUid>2539356</IdxUid>
<IdxSearchFields>
<GRNT>AI 00468</GRNT>
<GRNT>GM 07197</GRNT>
<GRNT>GM 29067</GRNT>
</IdxSearchFields>
</IdxDocument>
...
Once the final inversion:
...
<InvDocument>
<InvKey>ai 00468</InvKey>
<InvIDs>
<GRNT>2539356</GRNT>
</InvIDs>
</InvDocument>
<InvDocument>
<InvKey>gm 07197</InvKey>
<InvIDs>
<GRNT>2539356</GRNT>
</InvIDs>
</InvDocument>
<InvDocument>
<InvKey>gm 29067</InvKey>
<InvIDs>
<GRNT>2539356</GRNT>
</InvIDs>
</InvDocument>
..
and posting steps are completed, the new fields are ready to be searched.
Processing by XML Subset
A query on articles with abstracts published in a chosen journal, retrieved from the local cache, and followed by a multi-step transformation:
esearch -db pubmed -query "PNAS [JOUR]" -pub abstract |
efetch -format uid | fetch-pubmed |
xtract -stops -rec Rec -pattern PubmedArticle \
-wrp Year -year "PubDate/*" -wrp Abst -words Abstract/AbstractText |
xtract -rec Pub -pattern Rec \
-wrp Year -element Year -wrp Num -num Abst > countsByYear.xml
returns structured data with the year of publication and number of words in the abstract for each record:
<Pub><Year>2018</Year><Num>198</Num></Pub>
<Pub><Year>2018</Year><Num>167</Num></Pub>
<Pub><Year>2018</Year><Num>242</Num></Pub>
(The ">" redirect saves the results to a file.)
The following "for" loop limits the processed query results to one year at a time with xtract ‑select, passing the relevant subset to a second xtract command:
for yr in {1960..2021}
do
cat countsByYear.xml |
xtract -set Raw -pattern Pub -select Year -eq "$yr" |
xtract -pattern Raw -lbl "$yr" -avg Num
done |that applies ‑avg to the word counts in order to compute the average number of abstract words per article for the current year:
1969 122
1970 120
1971 127
...
2018 207
2019 207
2020 208
This result can be saved by redirecting to a file, or it can be piped to:
tee /dev/tty |
xy-plot pnas.png
to print the data to the terminal and then display the results in graphical format. The last step should be:
rm countsByYear.xml
to remove the intermediate file.
Identifier Conversion
The archive-pubmed script also downloads MeSH descriptor information from the NLM FTP server and generates a conversion file:
...
<Rec>
<Code>D064007</Code>
<Name>Ataxia Telangiectasia Mutated Proteins</Name>
...
<Tree>D12.776.157.687.125</Tree>
<Tree>D12.776.660.720.125</Tree>
</Rec>
...
that can be used for mapping MeSH codes to and from chemical or disease names. For example:
cat $EDIRECT_LOCAL_ARCHIVE/pubmed/Data/meshconv.xml |
xtract -pattern Rec \
-if Name -starts-with "ataxia telangiectasia" \
-element Code
will return:
C565779
C576887
D001260
D064007
More information on a MeSH term could be obtained by running:
efetch -db mesh -id D064007 -format docsum
Integration with Entrez
Use phrase-search -filter to combine the UID results of a search (here followed by a link step) with a local query:
phrase-search -query "Berg CM [AUTH]" |
phrase-search -link CITED |
phrase-search -filter "Transposases [MESH]"
Intermediate lists of PMIDs can be saved to a file and piped (with "cat") into a subsequent phrase-search ‑filter query. They can also be uploaded to the Entrez history server by piping to epost:
epost -db pubmed
or piped directly to efetch.
Solid-State Drive Preparation
To initialize a solid-state drive for hosting the local archive on a Mac, log into an admin account, run Disk Utility, choose View -> Show All Devices, select the top-level external drive, and press the Erase icon. Set the Scheme popup to GUID Partition Map, and APFS will appear as a format choice. Set the Format popup to APFS, enter the desired name for the volume, and click the Erase button.
To finish the drive configuration, disable Spotlight indexing on the drive with:
sudo mdutil -i off "${EDIRECT_LOCAL_ARCHIVE}"
sudo mdutil -E "${EDIRECT_LOCAL_ARCHIVE}"and disable FSEvents logging with:
sudo touch "${EDIRECT_LOCAL_ARCHIVE}/.fseventsd/no_log"Also exclude the disk from being backed up by Time Machine or scanned by a virus checker.
Automation
Unix Shell Scripting
A shell script can be used to repeat the same sequence of operations on a number of input values. The Unix shell is a command interpreter that supports user-defined variables, conditional statements, and repetitive execution loops. Scripts are usually saved in a file, and referenced by file name.
Comments start with a pound sign ("#") and are ignored. Quotation marks within quoted strings are entered by "escaping" with a backslash ("\"). Subroutines (functions) can be used to collect common code or simplify the organization of the script.
Combining Data from Adjacent Lines
Given a tab-delimited file of feature keys and values, where each gene is followed by its coding regions:
gene matK
CDS maturase K
gene ATP2B1
CDS ATPase 1 isoform 2
CDS ATPase 1 isoform 7
gene ps2
CDS peptide synthetase
the cat command can pipe the file contents to a shell script that reads the data one line at a time:
#!/bin/bash
gene=""
while IFS=$'\t' read feature product
do
if [ "$feature" = "gene" ]
then
gene="$product"
else
echo "$gene\t$product"
fi
done
The resulting output lines, printed by the echo command, have the gene name and subsequent CDS product names in separate columns on individual rows:
matK maturase K
ATP2B1 ATPase 1 isoform 2
ATP2B1 ATPase 1 isoform 7
ps2 peptide synthetase
Dissecting the script, the first line selects the Bash shell on the user's machine:
#!/bin/bash
The latest gene name is stored in the "gene" variable, which is first initialized to an empty string:
gene=""
The while command sequentially reads each line of the input file, IFS indicates tab-delimited fields, and read saves the first field in the "feature" variable and the remaining text in the "product" variable:
while IFS=$'\t' read feature product
The statements between the do and done commands are executed once for each input line. The if statement retrieves the current value stored in the feature variable (indicated by placing a dollar sign ($) in front of the variable name) and compares it to the word "gene":
if [ "$feature" = "gene" ]
If the feature key was "gene", it runs the then section, which copies the contents of the current line's "product" value into the persistent "gene" variable:
then
gene="$product"
Otherwise the else section prints the saved gene name and the current coding region product name:
else
echo "$gene\t$product"
separated by a tab character. The conditional block is terminated with a fi instruction ("if" in reverse):
fi
In addition to else, the elif command can allow a series of mutually-exclusive conditional tests:
if [ "$feature" = "gene" ]
then
...
elif [ "$feature" = "mRNA" ]
then
...
elif [ "$feature" = "CDS" ]
then
...
else
...
fi
A variable can be set to the result of commands that are enclosed between "$(" and ")" symbols:
mrna=$( echo "$product" | grep 'transcript variant' |
sed 's/^.*transcript \(variant .*\).*$/\1/' )
Entrez Direct Commands Within Scripts
EDirect commands can also be run inside scripts. Saving the following text:
#!/bin/bash
printf "Years"
for disease in "$@"
do
frst=$( echo -e "${disease:0:1}" | tr [a-z] [A-Z] )
printf "\t${frst}${disease:1:3}"
done
printf "\n"
for (( yr = 2020; yr >= 1900; yr -= 10 ))
do
printf "${yr}s"
for disease in "$@"
do
val=$(
esearch -db pubmed -query "$disease [TITL]" |
efilter -mindate "${yr}" -maxdate "$((yr+9))" |
xtract -pattern ENTREZ_DIRECT -element Count
)
printf "\t${val}"
done
printf "\n"
done
to a file named "scan_for_diseases.sh" and executing:
chmod +x scan_for_diseases.sh
allows the script to be called by name. Passing several disease names in command-line arguments:
scan_for_diseases.sh diphtheria pertussis tetanus |
returns the counts of papers on each disease, by decade, for over a century:
Years Diph Pert Teta
2020s 104 281 154
2010s 860 2558 1296
2000s 892 1968 1345
1990s 1150 2662 1617
1980s 780 1747 1488
...
A graph of papers per decade for each disease is generated by piping the table to:
xy-plot diseases.png
Passing the data instead to:
align-columns -h 2 -g 4 -a ln
right-justifies numeric data columns for easier reading or for publication:
Years Diph Pert Teta
2020s 104 281 154
2010s 860 2558 1296
2000s 892 1968 1345
1990s 1150 2662 1617
1980s 780 1747 1488
...
while piping to:
transmute -t2x -set Set -rec Rec -header
produces a custom XML structure for further comparative analysis by xtract.
Time Delay
The shell script command:
sleep 1
adds a one second delay between steps, and can be used to help prevent overuse of servers by advanced scripts.
Xargs/Sh Loop
Writing a script to loop through data can sometimes be avoided by creative use of the Unix xargs and sh commands. Within the "sh ‑c" command string, the last name and initials arguments (passed in pairs by "xargs ‑n 2") are substituted at the "$0" and "$1" variables. All of the commands in the sh string are run separately on each name:
echo "Garber ED Casadaban MJ Mortimer RK" |
xargs -n 2 sh -c 'esearch -db pubmed -query "$0 $1 [AUTH]" |
xtract -pattern ENTREZ_DIRECT -lbl "$1 $0" -element Count'
This produces PubMed article counts for each author:
ED Garber 35
MJ Casadaban 46
RK Mortimer 85
While Loop
A "while" loop can also be used to independently process lines of data. Given a file "organisms.txt" containing genus-species names, the Unix "cat" command:
cat organisms.txt |
writes the contents of the file:
Arabidopsis thaliana
Caenorhabditis elegans
Danio rerio
Drosophila melanogaster
Escherichia coli
Homo sapiens
Mus musculus
Saccharomyces cerevisiae
This can be piped to a loop that reads one line at a time:
while read org
do
esearch -db taxonomy -query "$org [LNGE] AND family [RANK]" < /dev/null |
efetch -format docsum |
xtract -pattern DocumentSummary -lbl "$org" \
-element ScientificName Division
done
looking up the taxonomic family name and BLAST division for each organism:
Arabidopsis thaliana Brassicaceae eudicots
Caenorhabditis elegans Rhabditidae nematodes
Danio rerio Cyprinidae bony fishes
Drosophila melanogaster Drosophilidae flies
Escherichia coli Enterobacteriaceae enterobacteria
Homo sapiens Hominidae primates
Mus musculus Muridae rodents
Saccharomyces cerevisiae Saccharomycetaceae ascomycetes
(The "< /dev/null" input redirection construct prevents esearch from "draining" the remaining lines from stdin.)
For Loop
The same results can be obtained with organism names embedded in a "for" loop:
for org in \
"Arabidopsis thaliana" \
"Caenorhabditis elegans" \
"Danio rerio" \
"Drosophila melanogaster" \
"Escherichia coli" \
"Homo sapiens" \
"Mus musculus" \
"Saccharomyces cerevisiae"
do
esearch -db taxonomy -query "$org [LNGE] AND family [RANK]" |
efetch -format docsum |
xtract -pattern DocumentSummary -lbl "$org" \
-element ScientificName Division
done
File Exploration
A for loop can also be used to explore the computer's file system:
for i in *
do
if [ -f "$i" ]
then
echo $(basename "$i")
fi
done
visiting each file within the current directory. The asterisk ("*") character indicates all files, and can be replaced by any pattern (e.g., "*.txt") to limit the file search. The if statement "‑f" operator can be changed to "‑d" to find directories instead of files, and "‑s" selects files with size greater than zero.
Processing in Groups
EDirect supplies a join-into-groups-of script that combines lines of unique identifiers or sequence accession numbers into comma-separated groups:
#!/bin/sh
xargs -n "$@" echo |
sed 's/ /,/g
The following example demonstrates processing sequence records in groups of 200 accessions at a time:
...
efetch -format acc |
join-into-groups-of 200 |
xargs -n 1 sh -c 'epost -db nuccore -format acc -id "$0" |
elink -target pubmed |
efetch -format abstract'
Programming in Go
A program written in a compiled language is translated into a computer's native machine instruction code, and will run much faster than an interpreted script, at the cost of added complexity during development.
Google's Go language (also known as "golang") is "an open source programming language that makes it easy to build simple, reliable, and efficient software". Go eliminates the need for maintaining complicated "make" files. The build system assumes full responsibility for downloading external library packages. Automated dependency management tracks module release numbers to prevent version skew.
As of 2020, the Go development process has been streamlined to the point that it is now easier to use than some popular scripting languages.
To build Go programs, the latest Go compiler must be installed on your computer. A link to the installation URL is in the Documentation section at the end of this web page.
basecount.go Program
Piping FASTA data to the basecount binary executable (compiled from the basecount.go source code file shown below):
efetch -db nuccore -id J01749,U54469 -format fasta | basecount
will return rows containing an accession number followed by counts for each base:
J01749.1 A 983 C 1210 G 1134 T 1034
U54469.1 A 849 C 699 G 585 T 748
The full (uncommented) source code for basecount.go is shown here, and is discussed below:
package main
import (
"eutils"
"fmt"
"os"
"sort"
)
func main() {
fsta := eutils.FASTAConverter(os.Stdin, false)
countLetters := func(id, seq string) {
counts := make(map[rune]int)
for _, base := range seq {
counts[base]++
}
var keys []rune
for ky := range counts {
keys = append(keys, ky)
}
sort.Slice(keys, func(i, j int) bool { return keys[i] < keys[j] })
fmt.Fprintf(os.Stdout, "%s", id)
for _, base := range keys {
num := counts[base]
fmt.Fprintf(os.Stdout, "\t%c %d", base, num)
}
fmt.Fprintf(os.Stdout, "\n")
}
for fsa := range fsta {
countLetters(fsa.SeqID, fsa.Sequence)
}
}
Performance can be measured with the Unix "time" command:
time basecount < NC_000014.fsa
The program reads and counts the 107,043,718 bases of human chromosome 14, from an existing FASTA file, in under 2.5 seconds:
NC_000014.9 A 26673415 C 18423758 G 18559033 N 16475569 T 26911943
2.287
basecount.go Code Review
Go programs start with package main and then import additional software libraries (many included with Go, others residing in commercial repositories like github.com):
package main
import (
"eutils"
"fmt"
"os"
"sort"
)
Each compiled Go binary has a single main function, which is where program execution begins:
func main() {The fsta variable is assigned to a data channel that streams individual FASTA records one at a time:
fsta := eutils.FASTAConverter(os.Stdin, false)
The countLetters subroutine will be called with the identifier and sequence of each FASTA record:
countLetters := func(id, seq string) {An empty counts map is created for each sequence, and its memory is freed when the subroutine exits:
counts := make(map[rune]int)
A for loop on the range of the sequence string visits each sequence letter. The map keeps a running count for each base or residue, with "++" incrementing the current value of the letter's map entry:
for _, base := range seq {
counts[base]++
}(String iteration by range returns position and letter pairs. Since the code does not use the position, its value is absorbed by an underscore ("_") character.)
Maps are not returned in a defined order, so map keys are loaded to a keys array, which is then sorted:
var keys []rune
for ky := range counts {
keys = append(keys, ky)
}
sort.Slice(keys, func(i, j int) bool { return keys[i] < keys[j] })
(The second argument passed to sort.Slice is an anonymous function literal used to control the sort order. It is also a closure, implicitly inheriting the keys array from the enclosing function.)
The sequence identifier is printed in the first column:
fmt.Fprintf(os.Stdout, "%s", id)
Iterating over the array prints letters and base counts in alphabetical order, with tabs between columns:
for _, base := range keys {
num := counts[base]
fmt.Fprintf(os.Stdout, "\t%c %d", base, num)
}A newline is printed at the end of the row, and then the subroutine exits, clearing the map and array:
fmt.Fprintf(os.Stdout, "\n")
}
The remainder of the main function uses a loop to drain the fsta channel, passing the identifier and sequence string of each successive FASTA record to the countLetters function. The main function then ends with a final closing brace:
for fsa := range fsta {
countLetters(fsa.SeqID, fsa.Sequence)
}
}Note that the sequence of human chromosome 14, processed above, is stored in its entirety as a single contiguous Go string. No special coding considerations are needed for input, access, or memory management, even though it is over 107 million characters long.
Go Dependency Management
EDirect includes source code for the eutils helper library, which consolidates common functions used by xtract, transmute, and rchive, including the FASTA parser/streamer used by the basecount program shown above.
In addition to around two dozen eutils "*.go" files, the distribution contains "go.mod" and "go.sum" module files for the eutils package. They were created by running "./build.sh" in the eutils directory prior to release on the FTP site.
Modules provide a mechanism for automatically managing external dependencies. They record version numbers and checksums for the packages imported by eutils source files during development. Go will then retrieve the same versions of those packages, along with all of their internal support packages, if the eutils library is later incorporated into other software development projects.
Use of modules allows external Go packages to evolve independently, publishing newer versions with incompatible function argument signatures on their own schedules, while ensuring that this natural software development cycle does not break a working library or application build at some inopportune time in the future.
Compiling a Go Project
Each project typically resides in its own directory. The source code can be split into multiple files, and the build process will normally compile all of the "*.go" files together.
Create a new directory named "basecount" with:
cd ~
mkdir basecount
and copy the basecount.go source code file into that directory.
The program can then be compiled by running:
cd basecount
go mod init basecount
echo "replace eutils => $HOME/edirect/eutils" >> go.mod
go get eutils
go mod tidy
go build
but for convenience these commands are usually incorporated into a build script. To do this, save the following script to a file named build.sh in the same directory:
#!/bin/bash
if [ ! -f "go.mod" ]
then
go mod init "$( basename $PWD )"
echo "replace eutils => $HOME/edirect/eutils" >> go.mod
go get eutils
fi
if [ ! -f "go.sum" ]
then
go mod tidy
fi
go build
To compile the executable, enter the basecount directory, set the Unix execution permission bit, and run the build script:
cd basecount
chmod +x build.sh
./build.sh
The build script runs "go mod init" to generate "go.mod", and "go mod tidy" to generate " go.sum", if either module file is not already present.
(The "$( basename $PWD )" construct sets the executable's default name to match the parent directory, without needing to manually customize the "go mod init" line for each project.)
(The "replace eutils => $HOME/edirect/eutils" construct computes the path for finding the local eutils source code directory in the standard EDirect installation location.)
The "go build" instruction compiles the source file(s) for the application and all dependent libraries (caching the compiled object files for faster use later). It will then link these into a binary executable file that can run on the development machine.
You can select specific input files, change the executable program's name, and cross-compile for a different platform, with additional arguments to "go build":
env GOOS=darwin GOARCH=arm64 go build -o basecount.Silicon basecount.go
Separate projects in a single directory could be built by changing the "go build" line to:
for fl in *.go
do
go build -o "${fl%.go}" "$fl"
done
Python Integration
Controlling EDirect from Python scripts is easily done with assistance from the edirect.py library file, which is included in the EDirect archive:
import subprocess
import shlex
def execute(cmmd, data=""):
if isinstance(cmmd, str):
cmmd = shlex.split(cmmd)
res = subprocess.run(cmmd, input=data,
capture_output=True,
encoding='UTF-8')
return res.stdout.strip()
def pipeline(cmmds, data=""):
def flatten(cmmd):
if isinstance(cmmd, str):
return cmmd
else:
return shlex.join(cmmd)
if not isinstance(cmmds, str):
cmmds = ' | '.join(map(flatten, cmmds))
res = subprocess.run(cmmds, input=data, shell=True,
capture_output=True,
encoding='UTF-8')
return res.stdout.strip()
def efetch(*, db, id, format, mode=""):
cmmd = ('efetch', '-db', db, '-id', str(id), '-format', format)
if mode:
cmmd = cmmd + ('-mode', mode)
return execute(cmmd))
At the beginning of your program, import the edirect module with the following commands:
#!/usr/bin/env python3
import sys
import os
import shutil
sys.path.insert(1, os.path.dirname(shutil.which('xtract')))
import edirect
(Note that the import command uses "edirect", without the ".py" extension.)
The first argument to edirect.execute is the Unix command you wish to run. It can be provided either as a string:
edirect.execute("efetch -db nuccore -id NM_000518.5 -format fasta")or as a sequence of strings:
edirect.execute(('efetch', '-db', 'nuccore', '-id', 'NM_000518.5', '-format', 'fasta'))An optional second argument accepts data to be passed to the Unix command through stdin. Multiple steps are chained together by using the result of the previous command as the data argument in the next command:
seq = edirect.execute("efetch -db nuccore -id NM_000518.5 -format fasta")
sub = edirect.execute("transmute -extract -1-based -loc 51..494", seq)
prt = edirect.execute(('transmute', '-cds2prot', '-every', '-trim'), sub)Alternatively, the edirect.pipeline function can accept a sequence of individual command strings to be piped together for execution:
edirect.pipeline(('efetch -db protein -id NP_000509.1 -format gp',
'xtract -insd Protein mol_wt sub_sequence'))or execute a string containing several piped commands:
edirect.pipeline('''efetch -db nuccore -id J01749 -format fasta |
transmute -replace -offset 1907 -delete GG -insert TC |
transmute -search -circular GGATCC:BamHI GAATTC:EcoRI CTGCAG:PstI |
align-columns -g 4 -a rl''')Data piped to the script itself is relayed by using "sys.stdin.read()" as the second argument.
Hiding details (e.g., isinstance, shlex.join, shlex.split, and subprocess.run) inside a common module means that biologists who are new to coding could control an entire analysis pipeline from their first Python program.
An edirect.efetch shortcut that uses named arguments is also available:
edirect.efetch(db="nuccore", id="NM_000518.5", format="fasta")
To run a custom shell script, make sure the execute permission bit is set, supply the full execution path, and follow it with any command-line arguments:
db = "pubmed"
res = edirect.execute(("./datefields.sh", db), "")
NCBI C++ Toolkit Access
EDirect scripts can be called from the NCBI C++ toolkit using the ncbi::edirect::Execute function:
#include <misc/eutils_client/eutils_client.hpp>
The function signature has separate parameters for the script name and its command-line arguments, followed by an optional string to be passed via stdin:
string Execute (
const string& cmmd,
const vector<string>& args,
const string& data = kEmptyStr
);
Multiple steps are chained together by using the previous result as the data argument in the next command:
string seq = ncbi::edirect::Execute
( "efetch", { "-db", "nuccore", "-id", "NM_000518.5", "-format", "fasta" } );
string sub = ncbi::edirect::Execute
( "transmute", { "-extract", "-1-based", "-loc", "51..494" }, seq );
string prt = ncbi::edirect::Execute
( "transmute", { "-cds2prot", "-every", "-trim" }, sub );
The argument vector can also be generated dynamically, under program control:
vector<string> args;
args.push_back("-db");
args.push_back("pubmed");
args.push_back("-format");
args.push_back("abstract");
args.push_back("-id");
args.push_back(uid);
Citation matching can be performed on a CPub object reference with the -asn argument:
string uid = ncbi::edirect::Execute
( "cit2pmid", { "-asn", FORMAT(MSerial_FlatAsnText << pub) } );
or you can use -title, -author, -journal, -volume, -issue, -pages, and -year arguments.
If a matching PMID is found, it can be retrieved as PubmedArticle XML and transformed into Pubmed-entry ASN.1:
string xml = ncbi::edirect::Execute
( "efetch", { "-db", "pubmed", "-format", "xml" }, uid );
string asn = ncbi::edirect::Execute
( "pma2pme", { "-std" }, xml );
The ASN.1 string can then be read into memory with an object loader for further processing:
#include <objects/pubmed/Pubmed_entry.hpp>
unique_ptr<CObjectIStream> stm;
stm.reset ( CObjectIStream::CreateFromBuffer
( eSerial_AsnText, asn.data(), asn.length() ) );
CRef<CPubmed_entry> pme ( new CPubmed_entry );
stm->Read ( ObjectInfo ( *pme ) );
Additional Examples
EDirect examples demonstrate how to answer ad hoc questions in several Entrez databases. The detailed examples have been moved to a separate document, which can be viewed by clicking on the ADDITIONAL EXAMPLES link.
Appendices
Command-Line Arguments
Each EDirect program has a ‑help command that prints detailed information about available arguments. These include ‑sort values for esearch, ‑format and ‑mode choices for efetch, and ‑cmd options for elink.
Einfo Data
Einfo field data contains status flags for several term list index properties:
<Field>
<Name>ALL</Name>
<FullName>All Fields</FullName>
<Description>All terms from all searchable fields</Description>
<TermCount>280005319</TermCount>
<IsDate>N</IsDate>
<IsNumerical>N</IsNumerical>
<SingleToken>N</SingleToken>
<Hierarchy>N</Hierarchy>
<IsHidden>N</IsHidden>
<IsTruncatable>Y</IsTruncatable>
<IsRangable>N</IsRangable>
</Field>
Unix Utilities
Several useful classes of Unix text processing filters, with selected arguments, are presented below:
Process by Contents:
sort Sorts lines of text
-f Ignore case
-n Numeric comparison
-r Reverse result order
-k Field key (start,stop or first)
-u Unique lines with identical keys
-b Ignore leading blanks
-s Stable sort
-t Specify field separator
uniq Removes repeated lines
-c Count occurrences
-i Ignore case
-f Ignore first n fields
-s Ignore first n characters
-d Only output repeated lines
-u Only output non-repeated lines
grep Matches patterns using regular expressions
-i Ignore case
-v Invert search
-w Search expression as a word
-x Search expression as whole line
-e Specify individual pattern
-c Only count number of matches
-n Print line numbers
-A Number of lines after match
-B Number of lines before match
Regular Expressions:
Characters
. Any single character (except newline)
\w Alphabetic [A-Za-z], numeric [0-9], or underscore (_)
\s Whitespace (space or tab)
\ Escapes special characters
[] Matches any enclosed characters
Positions
^ Beginning of line
$ End of line
\b Word boundary
Repeat Matches
? 0 or 1
* 0 or more
+ 1 or more
{n} Exactly n
Escape Sequences
\n Line break
\t Tab character
Modify Contents:
sed Replaces text strings
-e Specify individual expression
s/// Substitute
/g Global
/I Case-insensitive
/p Print
tr Translates characters
-d Delete character
-s Squeeze runs of characters
rev Reverses characters on line
Format Contents:
column Aligns columns by content width
-s Specify field separator
-t Create table
expand Aligns columns to specified positions
-t Tab positions
fold Wraps lines at a specific width
-w Line width
-s Fold at spaces
Filter by Position:
cut Removes parts of lines
-c Characters to keep
-f Fields to keep
-d Specify field separator
-s Suppress lines with no delimiters
head Prints first lines
-n Number of lines
tail Prints last lines
-n Number of lines
Miscellaneous:
wc Counts words, lines, or characters
-c Characters
-l Lines
-w Words
xargs Constructs arguments
-n Number of words per batch
mktemp Make temporary file
join Join columns in files by common field
paste Merge columns in files by line number
File Compression:
tar Archive files
-c Create archive
-f Name of output file
-z Compress archive with gzip
gzip Compress file
-k Keep original file
-9 Best compression
unzip Decompress .zip archive
-p Pipe to stdout
gzcat Decompress .gz archive and pipe to stdout
Directory and File Navigation:
cd Changes directory
/ Root
~ Home
. Current
.. Parent
- Previous
ls Lists file names
-1 One entry per line
-a Show files beginning with dot (.)
-l List in long format
-R Recursively explore subdirectories
-S Sort files by size
-t Sort by most recently modified
.* Current and parent directory
pwd Prints working directory path
File Redirection:
< Read stdin from file
> Redirect stdout to file
>> Append to file
2> Redirect stderr
2>&1 Merge stderr into stdout
| Pipe between programs
<(cmd) Execute command, read results as file
Shell Script Variables:
$0 Name of script
$n Nth argument
$# Number of arguments
"$*" Argument list as one argument
"$@" Argument list as separate arguments
$? Exit status of previous command
Shell Script Tests:
-d Directory exists
-f File exists
-s File is not empty
-n Length of string is non-zero
-x File is executable
-z Variable is empty or not set
Shell Script Options:
set Set optional behaviors
-e Exit immediately upon error
-u Treat unset variables as error
-x Trace commands and argument
File and Directory Extraction:
BAS=$(printf pubmed%03d $n)
DIR=$(dirname "$0")
FIL=$(basename "$0")
Remove Prefix:
FILE="example.tar.gz"
# ${FILE#.*} -> tar.gz
## ${FILE##.*} -> gz
Remove Suffix:
FILE="example.tar.gz"
TYPE="http://identifiers.org/uniprot_enzymes/"
% ${FILE%.*} -> example.tar
${TYPE%/} -> http://identifiers.org/uniprot_enzymes
%% ${FILE%%.*} -> example
Loop Constructs:
while IFS=$'\t' read ...
for sym in HBB BRCA2 CFTR RAG1
for col in "$@"
for yr in {1960..2020}
for i in $(seq $first $incr $last)
for fl in *.xml.gz
Additional documentation with detailed explanations and examples can be obtained by typing "man" followed by a command name.
Release Notes
EDirect release notes describe the history of incremental development and refactoring, from the original implementation in Perl to the redesign in Go and shell script. The detailed notes have been moved to a separate document, which can be viewed by clicking on the RELEASE NOTES link.
For More Information
Announcement Mailing List
NCBI posts general announcements regarding the E-utilities to the utilities-announce announcement mailing list. This mailing list is an announcement list only; individual subscribers may not send mail to the list. Also, the list of subscribers is private and is not shared or used in any other way except for providing announcements to list members. The list receives about one posting per month. Please subscribe at the above link.
References
The Smithsonian Online Collections Databases are provided by the National Museum of Natural History, Smithsonian Institution, 10th and Constitution Ave. N.W., Washington, DC 20560-0193. https://collections.nmnh.si.edu/.
den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016. https://doi.org/10.1002/humu.22981. (PMID 26931183.)
Holmes JB, Moyer E, Phan L, Maglott D, Kattman B. SPDI: data model for variants and applications at NCBI. Bioinformatics. 2020. https://doi.org/10.1093/bioinformatics/btz856. (PMID 31738401.)
Hutchins BI, Baker KL, Davis MT, Diwersy MA, Haque E, Harriman RM, Hoppe TA, Leicht SA, Meyer P, Santangelo GM. The NIH Open Citation Collection: A public access, broad coverage resource. PLoS Biol. 2019. https://doi.org/10.1371/journal.pbio.3000385. (PMID 31600197.)
Kim S, Thiessen PA, Cheng T, Yu B, Bolton EE. An update on PUG-REST: RESTful interface for programmatic access to PubChem. Nucleic Acids Res. 2018. https://doi.org/10.1093/nar/gky294. (PMID 29718389.)
Mitchell JA, Aronson AR, Mork JG, Folk LC, Humphrey SM, Ward JM. Gene indexing: characterization and analysis of NLM's GeneRIFs. AMIA Annu Symp Proc. 2003:460-4. (PMID 14728215.)
Ostell JM, Wheelan SJ, Kans JA. The NCBI data model. Methods Biochem Anal. 2001. https://doi.org/10.1002/0471223921.ch2. (PMID 11449725.)
Schuler GD, Epstein JA, Ohkawa H, Kans JA. Entrez: molecular biology database and retrieval system. Methods Enzymol. 1996. https://doi.org/10.1016/s0076-6879(96)66012-1. (PMID 8743683.)
Wei C-H, Allot A, Leaman R, Lu Z. PubTator central: automated concept annotation for biomedical full text articles. Nucleic Acids Res. 2019. https://doi.org/10.1093/nar/gkz389. (PMID 31114887.)
Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 2013. https://doi.org/10.1093/nar/gks1114. (PMID 23175613.)
Documentation
EDirect navigation functions call the URL-based Entrez Programming Utilities:
https://www.ncbi.nlm.nih.gov/books/NBK25501
NCBI database resources are described by:
https://www.ncbi.nlm.nih.gov/pubmed/37994677
Information on how to obtain an API Key is described in this NCBI blogpost:
https://ncbiinsights.ncbi.nlm.nih.gov/2017/11/02/new-api-keys-for-the-e-utilities
An introduction to shell scripting for non-programmers is at:
https://missing.csail.mit.edu/2020/shell-tools/
An article on the Go programming language, written by its creators, is at:
https://cacm.acm.org/research/the-go-programming-language-and-environment/
and transcripts of talks on design philosophy and retrospective experience of Go are at:
https://commandcenter.blogspot.com/2012/06/less-is-exponentially-more.html
https://commandcenter.blogspot.com/2024/01/what-we-got-right-what-we-got-wrong.html
Instructions for downloading and installing the Go compiler are at:
https://golang.org/doc/install#download
Additional NCBI website and data usage policy and disclaimer information is located at:
https://www.ncbi.nlm.nih.gov/home/about/policies/
Public Domain Notice
A copy of the NCBI Public Domain Notice, which applies to EDirect, is shown below:
PUBLIC DOMAIN NOTICE
National Center for Biotechnology Information
This software/database is a "United States Government Work" under the
terms of the United States Copyright Act. It was written as part of
the author's official duties as a United States Government employee and
thus cannot be copyrighted. This software/database is freely available
to the public for use. The National Library of Medicine and the U.S.
Government have not placed any restriction on its use or reproduction.
Although all reasonable efforts have been taken to ensure the accuracy
and reliability of the software and data, the NLM and the U.S.
Government do not and cannot warrant the performance or results that
may be obtained by using this software or data. The NLM and the U.S.
Government disclaim all warranties, express or implied, including
warranties of performance, merchantability or fitness for any particular
purpose.
Please cite the author in any work or product based on this material.
Getting Help
Please refer to the PubMed and Entrez help documents for more information about search queries, database indexing, field limitations and database content.
Suggestions, comments, and questions specifically relating to the EUtility programs may be sent to vog.hin.mln.ibcn@seitilitue.