Clinical Description
Mucolipidosis II and mucolipidosis III α/β are distinct clinical disorders with different age of onset and clinical course. Although the experienced clinician observes variation within each phenotype, the phenotypic spectrum between ML II and ML IIIα/β is discontinuous. Knowledge of these two "classic" phenotypes allows recognition of an interesting minority of intermediate clinical types in which physical growth in infancy resembles that in the ML II whereas neuromotor and speech development follow the course of ML IIIα/β.
Mucolipidosis II
Mucolipidosis II (ML II) is slowly progressive with clinical onset at birth and death most often in early childhood [Cathey et al 2010]. The following is a summary of the phenotype by system.
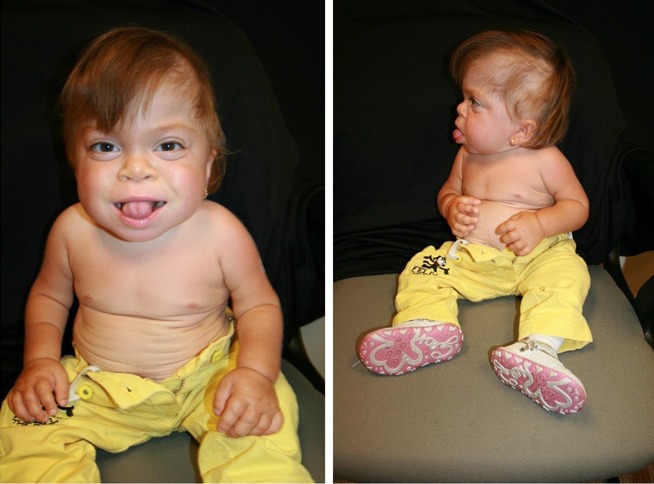
Growth. Birth weight is low to borderline normal. Postnatal growth is limited and ceases during the second year of life. In the event that the diagnosis is not made early, the term "failure to thrive" is often applied. Measured length appears to "decrease" over time as hip and knee contractures worsen (). Head size remains proportional to body size.
Craniofacial. The neonate with ML II has a flat face, depressed nasal bridge, and shallow orbits. The mouth is prominent. Coarsening of facial features is apparent from early infancy and gradually progresses (). Impressive gingival hypertrophy is apparent soon after birth and causes dental eruption to appear incomplete.
The skin is thickened especially around the earlobes. Additional cutaneous findings include prominent periorbital tortuous veins and telangiectatic capillaries in the subcutis over the cheeks. Hair texture and color may be atypical for families of northern European origin as the hair texture is fine and, in some instances, white to golden in color, even in neonates.
Metopic prominence is observed in some children. Craniosynostosis is regularly suspected but not formally confirmed and, in some instances, has resulted in inappropriate cranial surgery.
Ophthalmologic. The epicanthal folds persist. If corneal haziness is present in the slightly proptotic eyes, it is mild and detectable only by slit lamp examination.
Audiologic. Episodes of otitis media occur frequently in nearly all children with ML II. Even when otitis media is treated promptly and adequately, conductive hearing loss is common; however, significant hearing impairment is rare. Sensorineural hearing loss is uncommon.
Respiratory. The voice is consistently hoarse.
Breathing remains noisy throughout life. The airways are narrow and subject to slowly progressive mucosal thickening and overall stiffening of the connective tissues. These factors also adversely affect the lung parenchyma. The gradual stiffening of the thoracic cage compounds the restrictive respiratory insufficiency.
Severe pulmonary hypertension (PH) has been more formally documented in a longer-surviving individual with ML II [Kovacevic et al 2011]. PH is probably the rule instead of the exception in individuals with ML II who survive into childhood. Excessive egress of lysosomal glycoproteins into the extracellular matrix is likely the main cause of adverse and progressive interstitial lung disease, although storage of glycoprotein may contribute. Poor general health in these individuals often precludes invasive diagnostic procedures. Ultimately, cardiorespiratory failure refractory to treatment is the cause of death in most affected children.
Respiratory support is only infrequently required in newborns. Obstructive sleep apnea necessitates nighttime respiratory support in some children; a minority of longer-surviving children require persistent assisted ventilation. In these cases, invariably, respiratory support was initiated during treatment of an acute infection.
Cardiovascular. Cardiac involvement likely occurs in all children. Thickening and insufficiency of the mitral valve and (less frequently) the aortic valve are the most common findings. Right side and general ventricular hypertrophy and pulmonary artery hypertension have been reported but rarely objectively documented [Kovacevic et al 2011]. Slowly progressive valvular changes are common, but valvular deficiency not consistently observed. Rapidly progressive cardiomyopathy is not a common feature in ML II.
Gastrointestinal/feeding. Children with ML II are usually poor eaters. Growth is minimal and ceases in the second year of life. Small size and paucity of physical movements (most never walk independently) contribute to the limited appetite. Although often a parental request following recommendation of dieticians, only a minority of children require gastrostomy tube feeding. The abdomen is protuberant, although hepatomegaly is equivocal and splenomegaly rarely observed. Inguinal hernias are slightly more common than in the average infant and occur equally in males and females. Umbilical hernias, a nearly constant finding, may gradually enlarge, but are not known to cause gastrointestinal complications and do not require surgical correction.
Skeletal/soft connective tissue. Orthopedic abnormalities, often noticed at birth, may include one or more of the following: thoracic deformity, kyphosis, clubfeet, deformed long bones, and/or dislocation of the hip(s).
The range of motion of all major and small joints is significantly limited. Mobility of the shoulders is significantly reduced despite consistent axial and appendicular hypotonia. The wrists gradually lose range of motion, the hands and fingers broaden gradually in the few years after infancy and become progressively stiffer and fixed in volar claw-like flexion and usually deviate from the appendicular axis.
Neuromotor development and intellect. Alertness is limited in some, but close to normal in most affected children. Children with ML II show affection, happiness, and displeasure as would any child [Cathey et al 2010].
Early motor milestones are significantly delayed: sitting upright with support is usually acquired around age one year; unassisted sitting may not be achieved until age two years. In the majority of affected children, unaided walking is never achieved. Onset of expressive language is late and limited to single words. Receptive communication, which is much better than expressive language, is not age appropriate. Cognitive functioning, although obviously below normal for age, enables the child to understand, interact with, and enjoy the immediate environment.
Mucolipidosis IIIα/β
ML IIIα/β is slowly progressive with clinical onset at approximately age three years and death in early-to-middle adulthood [Leroy 2007, Cathey et al 2010]. Comprehensive data on life expectancy are still lacking.
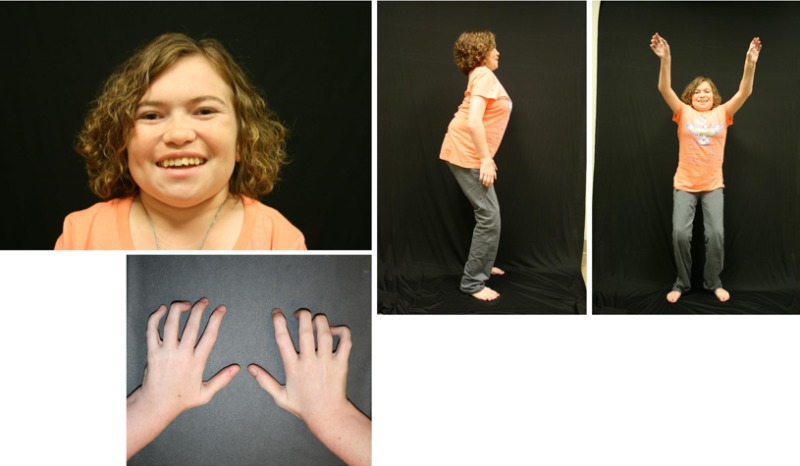
Growth. Weight and length at birth are within normal limits. Gradual slowing of growth rate begins in late infancy to early childhood. Concerns about small stature rarely arise before age three years, when worsening shoulder, hip, and knee contractures adversely affect stature. ML IIIα/β does not cause frank dwarfism as does ML II (); however, stature from early childhood is often below the third centile on standard growth curves. Final stature is well below expected for an individual's average family stature.
Craniofacial. True macrocephaly does not occur. Dysmorphic facial features are absent or minimal in younger children. Coarsening of facial features is gradual and more apparent in profile, including full cheeks, depressed nasal bridge, and prominent mouth. Gingival hypertrophy is mild and does not usually interfere with tooth eruption ().
Ophthalmologic. Epicanthal folds persist longer than normal. Proptosis, often observed in ML II, is rare. The corneas are clear by routine clinical inspection, but corneal clouding, which is visually insignificant, may be appreciated by slit-lamp examination.
Audiologic. Episodes of otitis media occur in individuals with ML IIIα/β more frequently than in the general population. Conductive hearing loss, documented in some affected individuals, has not been studied systematically. Sensorineural hearing loss is not a typical feature of ML III.
Respiratory. Mild hoarseness of the voice is an inconsistent finding. Upper-respiratory infections are more frequent than expected in some (but not all) children. From late childhood onward bronchitis and bronchopneumonia are the most consistent clinical complications.
Adults exhibit restrictive lung disease caused by stiffening of the thoracic cage, slowly progressive sclerosis of bronchi, and hardening and thickening of the interstitial tissue (extracellular matrix) in lung parenchyma.
Cardiovascular. Individuals with ML IIIα/β are at risk for cardiac involvement. Gradual thickening and subsequent insufficiency of the mitral valve and the aortic valve are common from late childhood onward [Steet et al 2005].
Left and/or right ventricular hypertrophy are often documented on echocardiography in older individuals. Pulmonary hypertension may occur in some older individuals, but to date remains insufficiently documented.
Rapid progression of cardiac disease is rarely observed in ML IIIα/β.
Pneumonia may compound mild cardiac insufficiency. Death in early adulthood is often from cardiopulmonary causes, even without complicating factors such as pneumonia.
Gastrointestinal. Prominence of the abdomen especially upon standing upright is caused in part by lumbar hyperlordosis, compensation for hip and knee flexion contractures, and hypotonia of the abdominal wall musculature. Diastasis of the medial recti and small umbilical hernias may also be present. In general, individuals with ML IIIα/β do not have organomegaly.
Skeletal / soft connective tissue. Stiffness of all large and small joints is a cardinal feature. Limited range of motion in the shoulders is frequently the initial evidence of ML IIIα/β and is mainly of soft tissue origin.
Limited range of motion in the hips and knees explains the slow gait and inability of children to run effectively. Flexion contractures in the hips and knees cause the squatting standing posture, most apparent in lateral view ().
Secondary but severe arthritic changes in the hips that can lead to destruction of the proximal femoral epiphyses make walking increasingly difficult and painful. Significant hardening of the surrounding soft tissues contributes to hip dysfunction. Many affected individuals become wheelchair bound before or during early adulthood.
Range of motion is less adversely affected in the wrists and ankles than in the other large joints. Dupuytren-type palmar contractures may appear from late childhood onward and exacerbate the moderate to severe claw-like flexion deformity of the fingers associated with recurrent swelling and progressive stiffness (). Neuropathic carpal tunnel signs can become severe in some individuals.
In ML IIIα/β the hands and fingers are usually of near-normal length in contrast to the severely affected hands in ML II.
Before the appropriate diagnosis is made, many individuals with ML IIIα/β are evaluated for a rheumatologic disorder.
Osteoporosis affects the entire skeleton. Bone pain becomes the most distressing symptom in ML IIIα/β, even in individuals with limited ambulation. Osteolytic bone lesions also are associated with significant bone pain in those who are nonambulatory.
Neuromotor development and intellect are the most variable features in ML IIIα/β ranging from normal to mild developmental delay in reaching motor milestones. Onset and development of receptive and expressive language skills occur at the expected age. Stuttering has not been observed in individuals with ML IIIα/β. Although psychometric tests often reveal an IQ within normal limits, approximately half of affected children require school assistance, often because of their physical limitations.
Other
Genotype-Phenotype Correlations
Genotype-phenotype correlations support the clinical distinction between the phenotypes ML II, ML IIIα/β, and at least the type of intermediate ML described in Phenotypes Intermediate Between ML II and ML IIIα/β.
ML II. Homozygous and compound heterozygous GNPTAB pathogenic variants that result in no functional GlcNAc-1-phosphotransferase (GNPT) activity (typically nonsense or frameshift variants) are consistently associated with the ML II phenotype. This severe phenotype is caused by complete loss of enzyme activity.
ML IIIα/β.
GNPTAB variants in the homozygous or compound heterozygous state in which some retained GNPT enzyme activity (between and 1% and 10% of the normal activity) usually result in the ML IIIα/β phenotype [Paik et al 2005, Steet et al 2005, Tiede et al 2005, Bargal et al 2006, Kudo et al 2006, Otomo et al 2009, Tappino et al 2009, Cathey et al 2010, David-Vizcarra et al 2010]. Some missense and several splice site variants have been associated with ML IIIα/β. Compound heterozygosity for one loss-of-function variant and one reduced function variant are also associated with ML IIIα/β. Hence, a variant that retains some activity protects against the ML II phenotype.
Of note, intellectual disability in one individual with ML IIIα/β homozygous for c.342delCA, whose parents were consanguineous, was significantly below the near-normal range of intellect typically observed in ML IIIα/β; it is unclear to what degree homozygosity for other variants could have affected this aspect of the phenotype.
Intermediate ML. The distinct, consistent intermediate phenotype similar to ML II in physical and radiographic features and to ML IIIα/β in psychomotor development and life expectancy results from compound heterozygosity for the GNPTAB variant c.10A>C (p.Lys4Gln) and a frameshift variant. GNPT enzyme activity is 7%-12% of normal [Leroy et al 2014]. The p.Lys4Gln missense variant located in the N-terminal cytoplasmic tail of the αβ polypeptide impairs its retention in the Golgi complex, but retains in vitro catalytic activity that is nearly normal [van Meel et al 2014].
Other intermediate phenotypes are to date less well defined. Some homozygous variant genotypes appear to be well represented and clinically heterogeneous.
Prevalence
ML II. The few estimates of the prevalence of ML II confirm that it is rare. Estimates include the following:
Ireland. 1.56:100,000 live births in Republic of Ireland (ROI) and Northern Ireland; 114:100, 000 in the Irish Travellers community in ROI. The data suggest a carrier rate for the most common pathogenic variant of 1:512 in the former and of 1:15 in the latter population [
McElligott et al 2011].
If these findings reflect a global prevalence ranging between 2.5x10-6 and 1.10-5, the overall carrier rate ranges between 1:158 and 1:316.
ML II has been reported in nearly all parts of the world.
An unusually high prevalence of ML II in 1:6,184 live births with an estimated carrier rate of 1:39 was found in the northeastern region of the province of Quebec, Canada [Plante et al 2008]. In this region, ML II in several large pedigrees has been attributed to a founder effect as only one GNPTAB pathogenic variant (c.3503_3504delTC) has been detected in all obligate carriers. The variant was introduced into that part of Canada in the 17th century by immigrants from France and Scotland. It is the most common GNPTAB pathogenic variant worldwide.
ML IIIα/β. Estimates of the prevalence of ML IIIα/β based on objective data are not available. It is, however, likely that the prevalence is of the same order of magnitude as that of ML II and hence estimated to range between 2.5x10-6 and 1.10-5. Consequently, the carrier rate is estimated at between 1:158 and 1:316.
The combined prevalence of ML II and ML IIIα/β is 0.22 per 100,000 in the Czech Republic [Poupetová et al 2010].