NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.
Summary
NCBI’s Reference Sequence (RefSeq) database is a collection of taxonomically diverse, non-redundant and richly annotated sequences representing naturally occurring molecules of DNA, RNA, and protein. Included are sequences from plasmids, organelles, viruses, archaea, bacteria, and eukaryotes. Each RefSeq is constructed wholly from sequence data submitted to the International Nucleotide Sequence Database Collaboration (INSDC). Similar to a review article, a RefSeq is a synthesis of information integrated across multiple sources at a given time. RefSeqs provide a foundation for uniting sequence data with genetic and functional information. They are generated to provide reference standards for multiple purposes ranging from genome annotation to reporting locations of sequence variation in medical records. The RefSeq collection is available without restriction and can be retrieved in several different ways, such as by searching or by available links in NCBI resources, including PubMed, Nucleotide, Protein, Gene, and Map Viewer, searching with a sequence via BLAST, and downloading from the RefSeq FTP site.
This chapter describes:
- The database content
- How data are assembled and maintained
- How RefSeqs can be accessed and retrieved
Introduction
NCBI’s Reference Sequence (RefSeq) collection is a freely accessible database of naturally occurring DNA, RNA, and protein sequences. It is a unique resource because it provides a large, multi-species, curated sequence database representing separate but explicitly linked records from genomes to transcripts and translation products, as appropriate. Unlike the sequence redundancy found in the public sequence repositories that comprise the INSDC, (i.e., NCBI’s GenBank, the European Nucleotide Archive [ENA], and the DNA Data Bank of Japan [DDBJ]), the RefSeq collection aims to provide, for each included species, a complete set of non-redundant, extensively cross-linked, and richly annotated nucleic acid and protein records. It is recognized, however, that the coverage and finishing of public sequence data varies from organism to organism so intermediate genomic records are provided in some circumstances.
The non-redundant nature of the RefSeq collection facilitates database inquiries based on genomic location, sequence, or text annotation. Be aware, however, that the RefSeq collection does include alternatively spliced transcripts encoding the same protein or distinct protein isoforms, in addition to orthologs, paralogs, and alternative haplotypes for some organisms, which will affect the outcome of a database query.
RefSeq records are based on sequence records submitted to the INSDC. However, the RefSeq collection is a distinct database. The public archival databases house sequences and annotations supplied by original authors and cannot be altered by others. The RefSeq collection differs from the archival databases in the same way that a review article differs from a related collection of primary research articles on the same subject. Each RefSeq record represents a synthesis, by a person or group, of the primary information that was generated and submitted by others. Other organizing principles or standards of judgment are possible, which is why the work is attributed to the synthesizing "editors". The RefSeq dataset is curated on an ongoing basis by collaborating groups and by NCBI staff. Sequence records are presented in a standard format and subjected to computational validation. The INSDC source of the RefSeq record, the curation status, and attribution to the curation group are also indicated.
The RefSeq collection establishes a useful baseline for integrating diverse data types, including sequence, genetic, expression, and functional information, into one consistent framework with a uniform set of conventions and standards. The RefSeq collection supports the following activities:
- genome annotation
- gene characterization
- comparative genomics
- reporting sequence variation, and
- expression studies
Database Content: Background
The May 2011 RefSeq collection (Release 47) includes sequences from more than 12,000 distinct taxonomic identifiers, ranging from viruses to bacteria to eukaryotes. It represents chromosomes, organelles, plasmids, viruses, transcripts, and more than 12.6 million proteins. Every sequence has a stable accession number, a version number, and an integer identifier (gi) assigned to it. Outdated versions are always available if a sequence is updated. RefSeq records can be distinguished from INSDC records by the inclusion of an underscore (“_”) at the third position of the accession number. The RefSeq accession prefix has an implied meaning in terms of the type of molecule it represents, as outlined in Table 1.
Updates
RefSeq updates are provided daily. These include new records added to the collection, and records updated to reflect sequence or annotation changes, including complete re-annotation of a genome. New and updated records are made available in Entrez and BLAST databases as soon as possible. The RefSeq FTP site also provides daily update information.
Flat File Format and Annotated Features
RefSeq records appear similar in format to GenBank records. Attributes novel to RefSeq records include a unique accession prefix followed by an underscore (Table 1) and a COMMENT field that indicates the RefSeq status and the INSDC source of the sequence information (Figures 1A, 1B, 1C, and 1D). For human RefSeqs, the COMMENT field also indicates whether the RefSeq is a reference standard from the RefSeqGene project. Some RefSeq records may include feature annotations or database cross-references (db_xrefs) that are not seen in the underlying INSDC record. This annotation is provided by computation and by manual curation. For example, nucleotide variation, STS, and tRNA features are computed for a subset of RefSeq entries using the data available in dbSNP (Chapter 5), UniSTS, and through tRNA-scan prediction (Lowe and Eddy, 1997). For human and mouse, exon feature annotation is also calculated for RefSeq transcript and non-transcribed pseudogene records. Db_xrefs provide links to Gene, nomenclature authorities, such as the HUGO Gene Nomenclature Committee (HGNC) for human RefSeq records, and to the Consensus CDS (CCDS) project. RefSeq proteins also report conserved domains computed by NCBI's Conserved Domain Database (Chapter 3). Additional protein features are propagated from the corresponding UniProtKB/Swiss-Prot records for a subset of species. Other nucleotide and protein features, publications, and comments may be added by collaborating groups or NCBI staff.
Assembling and Maintaining the RefSeq Collection
Summary
The RefSeq collection is the result of data extraction from INSDC submissions, curation, and computation, combined with extensive collaboration with authoritative groups. Each molecule is annotated as accurately as possible with the organism name, strain (or breed, ecotype, cultivar, or isolate), gene symbol for that organism, and informative protein name. Collaborations with authoritative groups outside of NCBI provide a variety of information, including curated sequence data, nomenclature, feature annotations, and links to external organism-specific resources. When no collaboration has been established, NCBI staff assembles the data from the INSDC submission. Each record has a COMMENT, indicating the level of curation that it has received (Table 2), and attribution of the collaborating group. Thus, a RefSeq record may be an essentially unchanged, validated copy of the original INSDC submission, or include updated or additional information supplied by collaborators or NCBI staff.
If multiple INSDC submissions represent the same molecule for an organism, the "best" sequence is chosen to represent as the RefSeq record. Known mutations, sequencing errors, cloning artifacts and erroneous annotation are avoided. Sequences are validated to confirm that the genomic sequence corresponding to an annotated mRNA feature matches the mRNA sequence record, and that coding region features translate into the corresponding protein sequence.
Working groups using distinct process pipelines compile the RefSeq collection for different organisms (Figure 2). RefSeq records are provided via several distinct approaches including:
- collaboration
- extraction from GenBank
- computational genome annotation pipeline
- curation by NCBI staff
Collaboration
RefSeq welcomes collaborations with authoritative groups outside of NCBI that are willing to provide sequences, nomenclature, annotation, or links to phenotypic or organism-specific resources. The RefSeq feedback form can be used to provide corrections or to initiate collaboration. The extent of collaboration may vary. For some species, the sequences and annotation of the entire RefSeq collection is provided by a collaborating authoritative group (see Table 3 for examples). For others, most notably the human and mouse RefSeq collections, numerous collaborations with individual scientists contribute to the representation of specific genes or complete gene families. Nomenclature for human and mouse is also provided via collaboration with the HUGO Gene Nomenclature Committee (HGNC) and the Mouse Genome Informatics group (MGI), respectively; Table 4 provides additional examples. Other collaborations extend across entire sets of organisms; for example, a board of Viral Genomes Advisors supports curation of the viral RefSeq collection. Thus, RefSeq records may contain information provided by an external authoritative source and/or analyses and curation at NCBI. The collaborating group is identified on the record.
Processing of RefSeq records supplied entirely by an external group is largely automated. The sequence and/or annotation is periodically submitted, validated to detect conflicts in the annotation, and modified slightly to format the submission as a RefSeq record, including addition of db_xrefs to Gene. NCBI staff do not directly curate the annotation or modify the sequence of RefSeq records provided by collaborating groups. Any problems identified by the validation process or by the scientific community are reported to the submitting group, and any update made to the annotation or sequence is reflected in a future RefSeq release.
Extraction from GenBank records
Complete genome data for viruses, organelles, prokaryotes, and some eukaryotes is propagated to RefSeq records from the whole genome sequence data and annotation available in GenBank (also in the ENA and DDBJ public archives). Generally, an initial validation step is performed before the RefSeq record is made public. The resulting RefSeq record is a copy of the GenBank submission but may contain some additional annotations as a result of the validation step. In particular, transcripts are provided as separate RefSeq records for most eukaryotic organisms; the GenBank submission of the genome sequence from which the RefSeq record is propagated instantiates the protein only, not the transcript.
This process flow is supported by the BioProject and Genome databases. The BioProject database tracks the status of whole-genome sequencing projects submitted to GenBank, other types of large-scale projects, and provides an overview of the organism and links to data and other resources. The resulting genomic RefSeq data is represented in the Genome database, which includes bacteria, archaea, eukaryotes, viroids, viruses, plasmids, and organelles. The Genome website provides custom displays, analysis, and tools for prokaryotic and some eukaryotic genomes (see Table 5).
Note that processing of most eukaryotic genomes is more complex, requires more than basic extraction from GenBank, and occurs independently, largely because the volume of data is significantly greater.
Extraction of GenBank whole genome data for processing into RefSeq records falls into four primary categories: chromosomes, microbial genomes, small complete genomes, and targeted loci.
Chromosomes
Complete chromosome sequence assembled from individual clones (that are themselves available from the INSDC) is propagated into a RefSeq record. For some genomes, the RefSeq representation uses a unit of interest to the research community; for example, some of the RefSeq genomic records for Drosophila melanogaster represent chromosome arms rather than complete chromosomes. RefSeq records may also be available for some genomes that are not yet fully sequenced but for which complete sequence is available for individual chromosomes. These complete chromosome RefSeq records may be annotated by the NCBI computational annotation pipeline, or they may be curated by an organism-specific collaborating group and undergo NCBI validation before being released.
Microbial genomes
For microbial species, historically all complete and draft genomes submitted to GenBank were propagated to the RefSeq collection. This is no longer tenable because of the volume of genomic data being generated, so additional RefSeq records are created from new GenBank submissions only to span the taxonomic diversity; this means in general, one genomic RefSeq per species is provided. If significant sequence diversity exists, or if subspecies or subgroups require representation as determined by NCBI staff, more than one RefSeq may exist for a given species.
Small complete genomes
RefSeq records representing organelle, viral, and plasmid genomes are based on single GenBank records. For organelle and viral genomes, if more than one GenBank submission is available for a species, typically only one is chosen to propagate to the RefSeq collection. Various factors, including the level of annotation, strain information, and community input are considered when deciding which GenBank submission to represent. There is no plasmid taxonomy; a GenBank submission is propagated to the RefSeq collection if it is part of a larger registered genome sequencing project, or if it exhibits significant sequence divergence when compared to other plasmids.
Targeted loci
The RefSeq Targeted Loci Project is a collaborative effort to curate and maintain molecular markers of use in the identification and classification of organisms. The initial focus is on ribosomal RNAs, although expansion to other informative sequences is anticipated. From GenBank submissions, the project creates RefSeq records for the small subunit of ribosomal RNA (16S in prokaryotes and 18S in eukaryotes) and the large subunit ribosomal RNA (23S in prokaryotes and 28S in eukaryotes). As of November 2010, there are 3331 16S rDNA RefSeq records from bacteria and archaea and 137 18S rDNA, and 97 28S rDNA RefSeq records from fungi.
Computational Genome Annotation Pipeline
NCBI computes annotation of genomic sequence data for some genomes including some microbes, vertebrates (e.g., human, mouse, rat, cow, and zebrafish, and others) and invertebrates (e.g., honey bee, acorn worm, and pea aphid). The annotation pipeline is automated and yields genomic, transcript, and protein (when appropriate) RefSeq records. Names annotated on the transcript and protein products are based on sequence similarity. Annotation data are refreshed periodically, and records generated from this process flow are not curated or updated between annotation runs (see Chapter 14 for more information on the eukaryotic genome annotation pipeline; information about NCBI’s prokaryotic annotation pipeline is also available). For some species, including human, RefSeq records may be provided by a mixture of methods. In other words, there may be a set of curated transcript and protein records (see the following section) in addition to a set of records generated computationally. RefSeq records that are processed by NCBI's pipelines are displayed in the NCBI Map Viewer (Chapter 20), included in Gene, and are available in NCBI’s sequence databases.
Curation by NCBI Staff
A portion of the RefSeq dataset is curated by NCBI staff. This subset includes viral, mitochondrial, vertebrate, and some invertebrate organisms. Most bacterial, plant, and fungal records are provided either by collaboration or by processing the annotated genome data submitted to the INSDC; however, a small number of bacterial genomes are annotated and curated by NCBI staff.
Curation of Microbial, Viral, and Mitochondrial RefSeqs
Microbial, viral, and metazoan mitochondrial RefSeq records are validated for content propagated from the original GenBank submission, including taxonomy, publications, and annotation, prior to becoming public. This content may be modified, augmented, or deleted by NCBI curation staff.
For microbial genomes, a set of minimal annotation standards (described here) are automatically provided on all legacy and new RefSeq records. These include ribosomal RNAs, transfer RNAs, and protein-coding genes with locus_tags. Ribosomal RNAs are predicted using BLASTn tools against an RNA sequence database and/or using Infernal (Eddy, 2002) and Rfam models (Griffiths-Jones, et al, 2003). Transfer RNAs are predicted using tRNAscan-SE (Lowe and Eddy, 1997). Other annotation above the minimum standards may be added based on an external source or literature review. Annotation associated with the NCBI’s Protein Clusters database is also propagated to the RefSeq records (both proteins and genes) at selected intervals. The Protein Clusters database is a collection of RefSeq proteins from complete genomes broadly organized into the following groups: archeal and bacterial genomes and plasmids, viruses, protists, plants, and chloroplasts and mitochondria, and annotated based on sequence similarity and protein function. This clustering allows the entire group to be curated as a single set, permitting well characterized proteins to seed the annotation of less studied ones within the same cluster. NCBI staff use literature and information from other databases, including UniProtKB/Swiss-Prot, to annotate each cluster with standardized protein names, biochemical descriptions, and other data, which is then transferred to individual proteins within the relevant RefSeq records. A microbial genome RefSeq record typically has a PROVISIONAL review status.
Annotation of viral genomes relies on an established group of Viral RefSeq Genome Advisors, members of the International Committee on the Taxonomy of Viruses, and other experts outside of NCBI. For example, the HIV-1 RefSeq (NC_001802) was curated by NCBI staff in collaboration with the authors of the book Retroviruses, and many of the adenovirus and herpesvirus records have been curated by outside experts. Based on literature review, NCBI curators may modify the CDS and RNA annotation compared to the GenBank submission, as was done for the Measles virus RefSeq record (NC_001498). Additional NCBI resources used during the curation of viral RefSeq records include the Protein Clusters database and PASC, a virus classification tool used to validate the taxonomy of virus RefSeq records across a number of taxonomic families. NCBI also maintains several specialized annotation pipelines for use in the Virus Variation and Influenza Virus resources. Manually curated viral RefSeq records are annotated with a status of REVIEWED or VALIDATED in the RefSeq COMMENT block.
For metazoan mitochondrial RefSeq records, standardized protein, gene, and RNA names are annotated independent of species-specific nomenclature guidelines. Additional curation may include adding common names or missing tRNAs and adjusting the coding region spans based on the Protein Clusters database. Curated metazoan mitochondrial records are annotated with a status of REVIEWED. Non-metazoan and plant chloroplast RefSeq records are not curated, are derived entirely from the original INSDC submission, and have a status of PROVISIONAL.
For targeted loci, vector or primer sequence from the GenBank submission is excluded from the RefSeq record. Any feature annotation may be modified to represent a standard format, and collection identifiers and publications referencing the original GenBank submission may be added.
Curation of Vertebrate and Invertebrate Records
Curation of higher eukaryotic organisms is focused on mammalian genomes, especially human and mouse, but also includes many other species with existing or planned genome assemblies. The RefSeq processing for these organisms provides transcripts and protein records as well as some genomic region records representing gene clusters or pseudogenes; these genomic region records facilitate genome-wide annotation. Because RefSeq uses evidence independent of a genome assembly to represent RNAs and proteins, the dataset can represent sequence not currently part of that genome assembly. RefSeq processing integrates the official nomenclature and other information, including alternate names, Gene Ontology (GO) terms, and literature and GeneRIFs available in Gene. Multiple collaborations support the collection of this descriptive information (Table 4; see also Chapter 19).
Sequences enter RefSeq curation processing by a combination of computational analysis, collaboration, and in-house curation. As illustrated in Figure 2, generation of the initial RefSeq record depends on identifying a representative sequence for a gene. New genes and sequence data are added to the in-house version of the Gene database by RefSeq curators, collaborators, NCBI’s genome annotation pipeline, and NCBI-based mining of UniGene, cDNA alignments, and INSDC submissions. Quality assessment (QA) processes are executed regularly to identify questionable data for review. These assessments include analysis of nomenclature, sequence similarity, genomic placement, and potential cloning errors (e.g., chimeras). The QA steps also leverage data from other NCBI resources, including HomoloGene, Map Viewer, and GenBank related sequences. Data conflicts must be resolved before the INSDC submission is used to generate a RefSeq record.
A sequence record unambiguously associated with a Gene record may be propagated into a RefSeq record. The completeness of the sequence (e.g., complete vs. partial CDS) and the category of the gene (e.g., protein coding, pseudogene) determine whether a RefSeq will be made, and if so, of what type (DNA, RNA, mRNA plus protein). RefSeq records are not made for incomplete proteins, transposable elements, or those loci for which the product type is uncertain (e.g., protein coding or not). It should be noted, however, that the RefSeq collection does include partial transcripts and proteins that are provided by collaborating groups or when the RefSeq is based on an annotated whole genome sequence submitted to the INSDC.
Once a suitable “source” sequence is identified, the RefSeq record is generated using the sequence data from the INSDC submission and the annotation data from the in-house version of the Gene database. Information from Gene includes the GeneID, cross-references to other databases, official nomenclature, aliases, alternate descriptive names, map location, and citations, including those submitted as GeneRIFs. RefSeq records are also subject to programmatic validation to identify annotation format errors and to provide annotation in a more consistent format. Records at this stage have a PROVISIONAL, PREDICTED, or INFERRED status depending on the evidence existing in support of the Gene record.
RefSeq processing for non-protein-coding RNA loci uses the longest defining transcript record associated with the Gene record. For non-transcribed loci (such as non-transcribed pseudogenes), the RefSeq record is typically derived from a region of a larger genomic sequence. Curation of these types of records is minimal because the current focus is on curation of protein-coding loci; however, these records provide an important reagent for the computational annotation pipeline and support annotation of non-protein-coding genes that might otherwise be missed or misrepresented as a predicted protein-coding gene.
Other RefSeq records are provided to represent larger genomic regions, including RefSeqGene sequences, gene clusters, genes requiring rearrangement to express a product (immunoglobulins and T-cell receptors), and haplotypes with known differences in gene content. These genomic region records are annotated by NCBI curation staff, often in collaboration with scientific experts, and are not provided by automatic processing.
RefSeqGene, a partner of the international Locus Reference Genomic (LRG) collaboration, provides stable reference standard genomic, RNA, and protein RefSeqs for medically important genes. These standards support the HGVS expressions used to describe sequence variation in medical records, and thus are constructed to represent standard alleles. The RefSeqGene usually represents a single gene, on the positive strand of the sequence, beginning 5 Kb upstream and extending 2 kb downstream. RefSeqGene records also include alignments of the RefSeq transcripts for the gene. All sequences annotated on the RefSeqGene have a review status of VALIDATED or REVIEWED.
Additional curation of vertebrate and some invertebrate RefSeq records occurs at the request of public users and collaborators, or as indicated by in-house QA analyses. QA analyses focus on, but are not restricted to, HomoloGene-based reporting of inconsistent protein lengths, identification of RefSeqs with repeat elements, questions about gene-to-sequence associations or potentially redundant genes, and reports of genes annotated at one time on a genome but not during subsequent re-annotation of that genome. Additionally, alignment-based tests are conducted for human and mouse that identify RefSeq records with poor quality alignment to the genome, non-consensus splicing, or very short or very long exons. Review of these records by skilled curators results in the most current and complete representation of the nucleotide and protein sequence and feature annotation available at that time. Sequence review may allow removal of vector and linker sequence, extension of the UTRs to define the full-length transcript, modification of the CDS annotation associated with the original INSDC source accession, or the creation of additional RefSeq records to represent the products of alternative splicing. A variety of feature annotations can be added to the RefSeq transcript and protein records. For nucleotide records, these include an indication of the transcript completeness, location of poly(A) signal and site, and sites of sequence variation and RNA editing. Exon annotation is provided for RefSeq transcripts and non-transcribed pseudogenes of human and mouse only; for transcripts, exon annotation is determined from the alignment of the transcript to the reference genome assembly using Splign, and, for non-transcribed pseudogenes, from the Splign alignment of the functional gene to the pseudogene genomic region. For protein records, feature annotations may include alternate or non-AUG initiating codons, Enzyme Commission (EC) numbers, mature peptide products, protein domains, and selenocysteine residues. Finally, literature review is another source of alternate names, aliases, and functional information, the latter which may be used to construct a Reference Sequence Summary on the RefSeq record. A RefSeq record that has undergone the complete review process has a REVIEWED status. Note that for many genes, intermediate levels of manual curation may address issues concerning the RefSeq sequence alone; these records have a review status of VALIDATED pending full review.
The review process may result in updating a RefSeq record, providing new RefSeq records, modifying sequence-to-gene associations, merging Gene records, or discontinuing a RefSeq, GeneID, or both. A RefSeq record is suppressed if it is found to represent a transcribed repeat element, to be derived from the wrong organism (i.e., the INSDC sequence it was based on has incorrect organism annotation), or not to represent a "gene". Records determined to represent an incomplete sequence, such as a partial protein sequence or an incompletely spliced transcript, are temporarily suppressed until more complete sequence data are available. Suppressed records can still be retrieved and will have a disclaimer appearing on the query result document summary (Figure 3a). A suppressed record is not included in BLAST databases, in the calculation of related sequences, in the BLink display (BLink are pre-computed protein BLAST results), or in RefSeq FTP releases. If a RefSeq is found to be redundant with another public RefSeq, then one is retained and the other becomes secondary (Figure 3b). If the sequences were associated with two different Gene records, then the records are merged so that a query of Gene with either of the original GeneIDs will retrieve the remaining single record.
We welcome input from the research community to improve the quality of the RefSeq collection. Interested parties are invited to contact us by sending an email to the NCBI Help Desk (vog.hin.mln.ibcn@ofni) or by using our feedback form.
Access and Retrieval
RefSeq records can be accessed by direct query, BLAST, FTP download, or indirectly through links provided from several NCBI resources, including Gene, Genome, BioProject, and Map Viewer (Table 6). In addition, RefSeq records are included in some computed resources and so links may be found from those pages to individual RefSeq records. Some links from Entrez databases to RefSeq records are based on Gene associations (e.g., links from OMIM; Chapter 7), whereas others are based on sequence similarity or RefSeq annotation content, including links from PubMed. RefSeq records are easy to distinguish in these resources by their unique accession number format (Table 1).
How to access and retrieve RefSeq records is described below.
Entrez Query Access
RefSeq records can be retrieved from the Entrez system (Chapter 15) by querying with an accession number, symbol or locus_tag, name, or by using Entrez Limits and Property terms. All RefSeqs can be found in the Entrez Nucleotide or Protein databases; both RefSeq and INSDC submissions will be included but a filter is provided at the top right hand corner of the results page to allow display of only the RefSeq accessions, if desired. Filters can be configured using the MyNCBI interface. Alternatively, a query can be restricted to retrieve only RefSeq-specific results using the Limits page or by querying with a Property, such as “srcdb_refseq[property]”, or others listed in Table 7. Limits and Properties can also be used to restrict results to molecule type, such as DNA versus mRNA. The Entrez Help document provides additional information about querying.
Gene contains the majority of the RefSeq collection and also supports querying using all the above strategies. RefSeq-to-Gene connections are also provided by direct links; RefSeq records include a link to the Gene report page via the GeneID db_xref link on the gene and CDS features (Figure 1C). Gene reports the RefSeq accession numbers in the RefSeq section of the report, with links to the Nucleotide or Protein records. The Links menu in Gene also provides distinct links to RefSeq RNAs, RefSeq proteins, and RefSeqGene. Gene reports may include a graphical depiction of genome annotation data in the Genomic regions, transcripts, and products section, with links to Nucleotide and Protein displays. When this graphical section is provided, an additional report is available with details about exon and intron boundaries and length. You can change the display format from Full Report to Gene Table to access this report. Note that RefSeq records representing assembled environmental samples (with an NS_ accession prefix) are not included in Gene but can be found in the Genome and Nucleotide databases.
RefSeq records in the Genome or BioProject databases can be retrieved using an accession number for a complete genomic molecule (NC_ accession prefix) or organism name. The BioProject database can also be queried using the property restriction “srcdb_refseq[property]”.
RefSeq records belonging to the RefSeqGene set can be retrieved from the Entrez system using “RefSeqGene[keyword]”.
BLAST
RefSeq transcript records are included in the Nucleotide non-redundant (nr) and the RefSeq mRNA sequences databases. RefSeq protein records are included in the Protein database. Accessions in the results set, either RefSeq or GenBank, that are associated with a Gene record are indicated by a small blue G icon, which is linked to the Gene report. RefSeq genomic records (whole chromosome or scaffold RefSeq records and RefSeqGene records) are provided in the Reference genomic sequences database or via organism-specific genome BLAST databases, which can be accessed via Map Viewer, BioProject reports, or the Genomic Biology webpage. RefSeqGene records are also retrieved from the nr database in BLAST results and in a dedicated RefSeqGene database.
Map Viewer
The NCBI Map Viewer supports queries by RefSeq and RefSeqGene accession numbers if the annotated genome is available in that resource.
FTP
RefSeq data are available in three FTP areas:
- Configured RefSeq BLAST databases are available for download from the BLAST FTP site; separate databases are provided for genomic, transcript, and protein records.
- Organism-specific sequence files are provided in the Genomes FTP site. This area includes RefSeq records that are generated by, or used in, Map Viewer and Genomes processing. NCBI’s annotation of genomic RefSeqs is also available; a file in the latest specification (version 1.20) of Generic Feature Format version 3 (GFF3) is provided in a GFF subdirectory for the latest assembly of many organisms.
- The full RefSeq collection, including the human RefSeqGene set, is available from the RefSeq FTP site, with the exception of the NS_ accession series environmental sample records. The RefSeq collection is provided as comprehensive bi-monthly releases in addition to daily updates for records that are new or updated between RefSeq release cycles. The comprehensive release provides data in multiple file formats, including flat file and FASTA, organized into primary taxonomic groups in addition to the complete dataset. For organisms with more frequent updates to curated records, including human and mouse, subdirectories containing weekly comprehensive releases of transcript and protein RefSeq records are provided also. Information about the RefSeq release is documented on the RefSeq FTP site in the release-notes subdirectory. The availability of new releases is announced on the RefSeq website, on NCBI’s Facebook and Twitter accounts, to subscribers of the refseq-announce email list, and in the NCBI Newsletter.
Related Resources
The Consensus Coding Sequence (CCDS) Project
The CCDS project aims to provide a complete set of high quality annotations of protein-coding genes on the human and mouse genomes. It leverages the computational annotation pipelines of NCBI and Ensembl, and expert curation provided predominantly by the Havana team of the Wellcome Trust Sanger Institute and NCBI’s RefSeq staff, to track identical protein annotations on the reference assemblies of the human and mouse genomes, and to ensure they are consistently and accurately represented in public resources. The CCDS set includes coding regions that are annotated as full-length (with an initiating AUG and valid stop-codon), can be translated from the genome without frameshifts, and use consensus splice-sites. Annotated genes in the CCDS set are associated with a unique identifying number and version. The version number will change with a change to the CDS structure or to the underlying genomic sequence, although any change requires collaborative agreement. See PubMed ID 19498102 for more information.
Related Reading
- Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT., Mouse Genome Database Group. The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucl. Acids Res. 2011;39:D842–8. (PubMed ID ) [PMC free article: PMC3013640] [PubMed: 21051359]
- Coffin JM, Hughes SH, and E Varmus. (1997) Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. [PubMed: 21433340]
- Dwinell MR, Worthey EA, Shimoyama M, Bakir-Gungor B, DePons J, Laulederkind S, Lowry T, Nigram R, Petri V, Smith J, Stoddard A, Twigger SN, Jacob HJ, Team RGD. The Rat Genome Database 2009: variation, ontologies and pathways. Nucl. Acids Res. 2009;37:D744–9. (PubMed ) [PMC free article: PMC2686558] [PubMed: 18996890]
- Eddy SR. A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics. 2002;3:18. (PubMed ID ) [PMC free article: PMC119854] [PubMed: 12095421]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucl. Acids Res. 2003;31:439–441. (PubMed ID ) [PMC free article: PMC165453] [PubMed: 12520045]
- Amberger, J., Bocchini, C. and Hamosh, A. (2011), A new face and new challenges for online mendelian inheritance in man (OMIM®). Human Mutation, 32:n/a. doi: 10.1002/humu.21466. (PubMed ID ).21472891. [PubMed: 21472891] [CrossRef]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl. Acids Res. 1997;25:955–964. (PubMed ID ) [PMC free article: PMC146525] [PubMed: 9023104]
- Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucl. Acids Res. 2011;39:D52–7. (PubMed ID ) [PMC free article: PMC3013746] [PubMed: 21115458]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, Deweese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucl. Acids Res. 2011;39:D225–9. (PubMed ID ) [PMC free article: PMC3013737] [PubMed: 21109532]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2008;19(7):1316–1323. (PubMed ID ) [PMC free article: PMC2704439] [PubMed: 19498102]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI Reference Sequences: current status, policy and new initiatives. Nucl. Acids Res. 2009;37:D32–36. (PubMed ID ) [PMC free article: PMC2686572] [PubMed: 18927115]
- Tatusova TA, Karsch-Mizrachi I, Ostell JA. Complete genomes in WWW Entrez: data representation and analysis. Bioinformatics. 1999;15:536–43. (PubMed ID ) [PubMed: 10487861]
- http://www
.ncbi.nlm.nih .gov/pubmed/18996890Sprague J, Bayraktaroglu L, Clements D, Conlin T, Fashena D, Frazer K, Haendel M, Howe D, Mani P, Ramachandran S, Schaper K, Segerdell E, Song P, Sprunger B, Taylor S, Van Slyke C, and M Westerfield. (2006) The Zebrafish Information Network: the zebrafish model organism database. Nucl. Acids Res. 34:D581-D585 (PubMed ID ).16381936. [PMC free article: PMC1347449] [PubMed: 16381936] - Seal RL, Gordon SM, Lush MJ, Wright MW, Bruford EA. genenames.org: the HGNC resources in 2011. Nucleic Acids Res. 2011;39:D519–9. (PubMed ID ) [PMC free article: PMC3013772] [PubMed: 20929869]
- Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, Zhang Z, and The FlyBase Consortium. (2009) FlyBase: enhancing Drosophila Gene Ontology annotations. Nucl. Acids Res. 37: D555-D559 (PubMed ID ).18948289. [PMC free article: PMC2686450] [PubMed: 18948289]
Figures
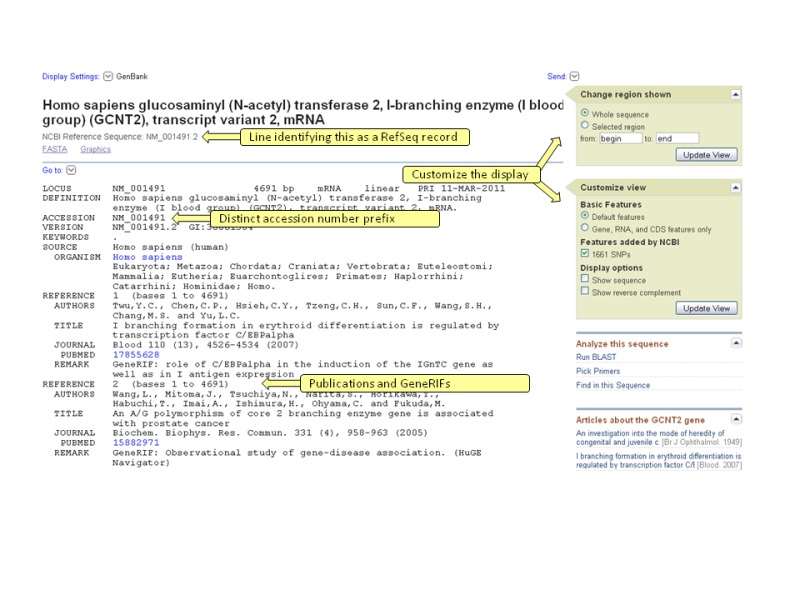
Figure 1A.
Features of a RefSeq record. The beginning of a RefSeq record when displayed in the GenBank flat file format is shown.

Figure 1B.
The COMMENT and PRIMARY sections. The gene Summary is provided for RefSeqs with a REVIEWED status only. The PRIMARY block, providing the RefSeq assembly details, is displayed for vertebrate records predominantly.

Figure 1C.
The FEATURES section. Only a subset of the available feature annotation is shown.
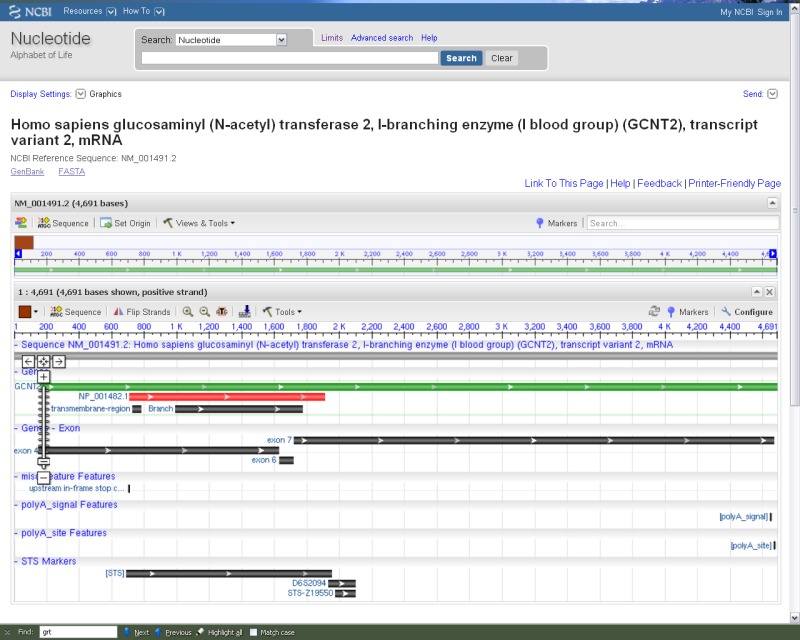
Figure 1D.
NCBI’s Sequence Viewer. The annotated features on a RefSeq record can be displayed in a graphical format (note the link ‘Graphics’ in Figure 1A). The display can be modified by following the ‘Configure’ link. The Help document provides additional information about the display and includes the Graphical View Legend, which provides details on how features are rendered.
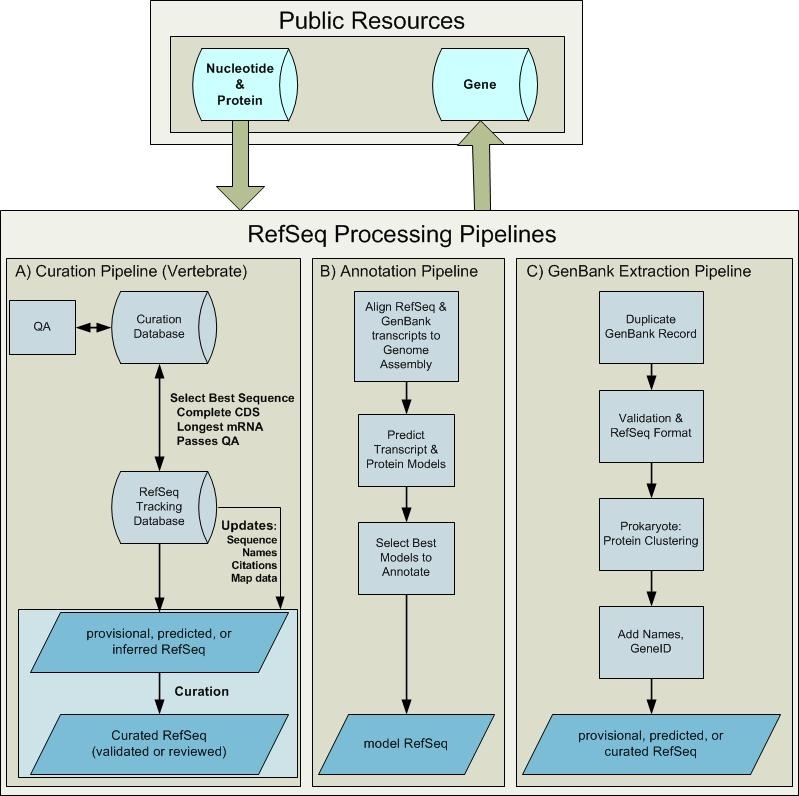
Figure 2.
RefSeq Processing Pipelines. Sequence data deposited in the public archival databases is available for RefSeq processing. Processing pipelines include the vertebrate curation pipeline, the computational genome annotation pipeline, and extraction from GenBank. These pipelines generate new and updated RefSeq records that become publicly available in Entrez Nucleotide, Protein, and Gene databases. (A) Once a gene is defined and associated with sufficient sequence information in an internal curation database, it can be pushed into the RefSeq pipeline. The RefSeq process is initiated by selecting the longest mRNA annotated with a complete coding sequence for each locus. This RefSeq record has a status of PROVISIONAL, PREDICTED, or INFERRED. Subsequent curation may result in a sequence or annotation update and a RefSeq status of VALIDATED or REVIEWED. Records are updated if the underlying INSDC submission is updated or if other associated data are updated, including nomenclature, publications, or map location. (B) Available RefSeq and INSDC data are aligned to an assembled genome, ab initio gene prediction that uses the alignment data is performed, and an analysis program integrates all available data to define the annotation models. New MODEL RefSeq records are generated by this pipeline. (C) When a complete, annotated genome becomes available in the INSDC, a set of corresponding RefSeq records are generated by duplicating the GenBank records, followed by validation and addition of cross-references to Gene (via a db_xref citing the GeneID) and more informative and standardized protein names, when available.

Figure 3.
Suppressed or redundant RefSeq records. (A) A standard text statement is included on the Entrez document summary for suppressed RefSeq records. (A) If redundant RefSeq records are merged, then both accession numbers appear on the flat file ACCESSION line (yellow arrow). The first ACCESSION number listed is the primary identifier and all others listed are "secondary" accession numbers.
Tables
Table 1.
RefSeq accession numbers and molecule types.
| Accession prefix | Molecule type | Comment |
|---|---|---|
| AC_ | Genomic | Complete genomic molecule, usually alternate assembly |
| NC_ | Genomic | Complete genomic molecule, usually reference assembly |
| NG_ | Genomic | Incomplete genomic region |
| NT_ | Genomic | Contig or scaffold, clone-based or WGSa |
| NW_ | Genomic | Contig or scaffold, primarily WGSa |
| NZ_b | Genomic | Complete genomes and unfinished WGS data |
| NM_ | mRNA | Protein-coding transcripts (usually curated) |
| NR_ | RNA | Non-protein-coding transcripts |
| XM_c | mRNA | Predicted model protein-coding transcript |
| XR_c | RNA | Predicted model non-protein-coding transcript |
| AP_ | Protein | Annotated on AC_ alternate assembly |
| NP_ | Protein | Associated with an NM_ or NC_ accession |
| YP_c | Protein | Annotated on genomic molecules without an instantiated transcript record |
| XP_c | Protein | Predicted model, associated with an XM_ accession |
| WP_ | Protein | Non-redundant across multiple strains and species |
- a
Whole Genome Shotgun sequence data.
- b
An ordered collection of WGS sequence for a genome.
- c
Computed.
Table 2.
RefSeq status codes.
| Code | Description |
|---|---|
| MODEL | The RefSeq record is provided by the NCBI Genome Annotation pipeline and is not subject to individual review or revision between annotation runs. |
| INFERRED | The RefSeq record has been predicted by genome sequence analysis, but it is not yet supported by experimental evidence. The record may be partially supported by homology data. |
| PREDICTED | The RefSeq record has not yet been subject to individual review, and some aspect of the RefSeq record is predicted. |
| PROVISIONAL | The RefSeq record has not yet been subject to individual review. The initial sequence-to-gene association has been established by outside collaborators or NCBI staff. |
| REVIEWED | The RefSeq record has been reviewed by NCBI staff or by a collaborator. The NCBI review process includes assessing available sequence data and the literature. Some RefSeq records may incorporate expanded sequence and annotation information. |
| VALIDATED | The RefSeq record has undergone an initial review to provide the preferred sequence standard. The record has not yet been subject to final review at which time additional functional information may be provided. |
| WGS | The RefSeq record is provided to represent a collection of whole genome shotgun sequences. These records are not subject to individual review or revisions between genome updates. |
Table 3.
Examples of collaborators who contribute RefSeq records.
| Organism | Collaborator |
|---|---|
| Saccharomyces cerevisiae | Saccharomyces Genome Database (SGD) |
| Arabidopsis thaliana | The Arabidopsis Information Resource (TAIR) |
| Pseudomonas aeruginosa | Pseudomonas aeruginosa Community Annotation Project (PseudoCAP) |
| Drosophila melanogaster | FlyBase |
| multiple invertebrates | VectorBase |
Table 4.
Examples of collaborating groups
| FlyBase |
| HUGO Gene Nomenclature Committee (HGNC) |
| Microbial genomes |
| Mouse Genome Informatics (MGI) |
| Online Mendelian Inheritance in Man (OMIM) |
| Rat Genome Database (RGD) |
| VectorBase |
| Viral Genome Advisors |
| XenBase |
| Zebrafish Information Network (ZFIN) |
Table 5.
Selected Entrez Genome resources.
| Web Page | Web Site |
|---|---|
| Genome homepage |
http://www |
| Eukaryotes |
http://www |
| Prokaryotes |
|
| Viral Genomes | http://www.ncbi.nlm.nih.gov/genomes/GenomesHome.cgi?taxid=10239 |
| Organelles |
http://www |
| Plant Genomes |
http://www |
Table 6.
NCBI resources with links to RefSeq records.
| BioSystems | Gene Expression Omnibus ( Chapter 6 ) |
|---|---|
| BLAST results (Chapter 16) | Genome |
| BLink (pre-computed BLASTp) | BioProject |
| Bookshelf (Chapter 8) | HomoloGene |
| Consensus CDS project | Map Viewer (Chapter 20) |
| dbSNP (Chapter 5) | Probe |
| dbVar | Protein Clusters |
| Entrez (Chapter 15) | PubMed Central (Chapter 9) |
| Epigenomics | UniGene (Chapter 21) |
| Gene (Chapter 19) | UniSTS |
Table 7.
Entrez queries to retrieve sets of RefSeq records.
| Query | Accession prefix | RefSeq status retrieved |
|---|---|---|
|
srcdb | All RefSeq accessions | All |
|
srcdb | NC_, AC_, NG_, NM_, NR_, NP_, AP_ | REVIEWED, PROVISIONAL, PREDICTED, INFERRED, and VALIDATED |
|
srcdb | NC_, AC_,NG_, NM_, NR_, NP_, AP_ | REVIEWED |
|
srcdb | NC_, NM_,NR_,NP_ | VALIDATED |
|
srcdb | NC_, AC_, NG_, NM_, NR_, NP_, AP_ | PROVISIONAL |
|
srcdb | NM_, NR_, NP_ | PREDICTED |
|
srcdb | AC_, AP_, NM_,NR_,NP_ | INFERRED |
|
srcdb | NT_, NW_, XM_, XR_, XP_, ZP_ | Genome annotation models |