

Bookshelf ID: NBK343452PMID: 26844334
NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
PDQ Cancer Information Summaries [Internet]. Bethesda (MD): National Cancer Institute (US); 2002-.

PDQ Cancer Information Summaries [Internet].
Show detailsThis PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood vascular tumors. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
General Information About Childhood Vascular Tumors
The quality of evidence regarding childhood vascular tumors is limited by retrospective data collection, small sample size, cohort selection and participation bias, and heterogeneity of the disorders.
Vascular anomalies are a spectrum of rare diseases classified as vascular tumors or malformations. An updated classification system was adopted at the General Assembly of the International Society for the Study of Vascular Anomalies (ISSVA, April 2014) and recently published.[1] Generally, vascular tumors are proliferative, while malformations enlarge through expansion of a developmental anomaly without underlying proliferation. Growth and/or expansion of vascular anomalies can cause clinical problems such as disfigurement, chronic pain, recurrent infections, coagulopathies (thrombotic and hemorrhagic), organ dysfunction, and death. Individuals often experience progressive clinical symptoms with worsening quality of life. Limited treatment options are available; their efficacy has not been validated in prospective clinical trials. Historically, therapies have been mostly interventional and surgical to palliate symptoms.
Vascular tumors in children are rare. The classification of these tumors has been difficult, especially in the pediatric population, because of their rarity, unusual morphologic appearance, diverse clinical behavior, and the lack of independent stratification for pediatric tumors. In 2013, The World Health Organization (WHO) updated the classification of soft tissue vascular tumors. Pediatric tumors were not independently stratified and the terminology was mostly left unchanged, but the intermediate category of tumors was divided into locally aggressive and rarely metastasizing. The ISSVA classification of tumors is based on the WHO classification (refer to Tables 1 and 2) but the ISSVA classification uses more precise terminology and phenotypes that have been agreed upon by the members of ISSVA.
Table 1. 2013 World Health Organization Classification of Vascular Tumors
| Category | Vascular Tumor Type |
|---|---|
| Benign | Hemangioma |
| Epithelioid hemangioma | |
| Angiomatosis | |
| Lymphangioma | |
| Intermediate (locally aggressive) | Kaposiform hemangioendothelioma |
| Intermediate (rarely metastasizing) | Retiform hemangioendothelioma |
| Papillary intralymphatic angioendothelioma | |
| Composite hemangioendothelioma | |
| Kaposi sarcoma | |
| Malignant | Epithelioid hemangioendothelioma |
| Angiosarcoma of soft tissue |
aAdapted from Fletcher et al.[2]
Table 2. 2014 International Society for the Study of Vascular Anomalies (ISSVA) Classification of Vascular Tumorsa
| Category | Vascular Tumor Type |
|---|---|
| Benign | Infantile hemangioma/hemangioma of infancy |
| Congenital hemangioma | |
| —Rapidly involuting (RICH) | |
| —Non-involuting (NICH) | |
| —Involuting (PICH) | |
| Tufted angioma | |
| Spindle cell hemangioma | |
| Pyogenic granuloma (also known as lobular capillary hemangioma) | |
| Others | |
| Locally aggressive or borderline | Kaposiform hemangioendothelioma |
| Retiform hemangioendothelioma | |
| Papillary intralymphatic angioendothelioma (PILA), Dabska tumor | |
| Composite hemangioendothelioma | |
| Kaposi sarcoma | |
| Others | |
| Malignant | Epithelioid hemangioendothelioma |
| Angiosarcoma | |
| Others |
aAdapted from ISSVA Classification of Vascular Anomalies. ©2014 International Society for the Study of Vascular Anomalies. Available at "issva
.org/classification." Accessed January 2016.[3]
References
- Wassef M, Blei F, Adams D, et al.: Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 136 (1): e203-14, 2015. [PubMed: 26055853]
- Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon, France: IARC Press, 2013.
- International Society for the Study of Vascular Anomalies: ISSVA Classification for Vascular Anomalies. Melbourne, Australia: International Society for the Study of Vascular Anomalies, 2014. Available online. Last accessed August 11, 2016.
Benign Tumors
Infantile Hemangioma
Incidence and epidemiology
Infantile hemangiomas are the most common benign vascular tumor of infancy, occurring in 3% to 10% of infants. They are not usually present at birth and are diagnosed most commonly at age 3 to 6 weeks.[1-3] The lesion proliferates for an average of 5 months, stabilizes, and then involutes over several years. Hemangiomas are more common in females, white non-Hispanic patients, and premature infants and multiple gestations. Hemangiomas are associated with advanced maternal age and placental complications.[1]
Biology
Most hemangiomas occur sporadically. However, they may rarely be caused by an abnormality of chromosome 5 and present in an autosomal dominant pattern.[4] Hemangioma endothelial cells have proven to be clonal in nature.[5] Hemangioma proliferation occurs during vasculogenesis (the formation of new blood vessels from angioblasts), as opposed to angiogenesis (the formation of new blood vessels from existing blood vessels). During proliferation, provasculogenic factors are expressed, such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), CD34, CD31, CD133, LYVE-1, and insulin-like growth factor 2.[6-10] In involution, hemangiomas express increased apoptosis.[11] During this phase, there are also increased mast cells and levels of metalloproteinase, as well as upregulation of interferon and decreased basic FGF (bFGF).[12-21]
Clinical presentation
Most hemangiomas are not present at birth but precursor lesions such as telangiectasia or faint discoloration of the skin or hypopigmentation can be seen. The lesion can be mistaken as a bruise from birth trauma or as a capillary malformation (port wine stain).[22,23] Hemangiomas can be superficial in the dermis, deep in the subcutaneous tissue, combined, or in the viscera. Combined lesions are common. They are most common in the head and neck but can be anywhere on the body. They can be localized, segmental, or multiple in nature. The cutaneous appearance is usually red-blue, firm, and warm in the proliferative phase. The lesion then lightens centrally and becomes less warm and softer; it then flattens and loses its color. The process of involution can take several years and once involution has occurred, regrowth is uncommon. In two patients treated with growth hormone, regrowth after involution was noted.[24] On further investigation, growth hormone receptors were found on the hemangioma cells. Although preliminary, this may advance the research into the etiology of hemangioma growth.
Diagnostic and staging evaluation
Hemangiomas are usually diagnosed by the history and clinical appearance. Biopsy is rarely needed and performed only if there is an atypical appearance and/or atypical history and presentation. Imaging is not usually necessary, but if there is a deeper lesion without a cutaneous component, ultrasound imaging is beneficial for diagnosis because it reveals a high flow lesion with a typical Doppler wave characteristic.[25]
Syndromes associated with infantile hemangioma
Syndromes associated with infantile hemangioma include the following:
- PHACE syndrome: PHACE syndrome represents a spectrum of diseases and is defined by the presence of a large segmental hemangioma, usually on the face or head, in association with one or more congenital malformations.[26-31] Many abnormalities are included in the syndrome.Consensus criteria for definite and possible PHACE syndrome were developed at an expert panel meeting, as follows:
PHACE
- -
Posterior fossa abnormalities. Anomalies include posterior fossa malformations, including Dandy-Walker complex, cerebellar hypoplasia, atrophy, and dysgenesis/agenesis of the vermis.
- -
Hemangioma.
- -
Arterial abnormalities. Cerebrovascular anomalies can include carotid artery abnormalities, absence of cerebral vessels dilation, or narrowing.
- -
Cardiac abnormalities. Cardiac anomalies are most commonly coarctation of the aorta, complex aortic arch anomalies, and ventricular and atrial septal defects.
- -
Eye abnormalities. Ophthalmologic anomalies can include microphthalmos, retinal vascular abnormalities, and persistent fetal retinal vessels.
Diagnosis of PHACE requires clinical examination, cardiac evaluation with echocardiogram, ophthalmologic evaluation, and magnetic resonance imaging (MRI)/magnetic resonance angiogram of the head, neck, and mediastinum. Patients need to be monitored for developmental disorders, progressive arterial occlusion, stroke and neurologic complications, and endocrine issues; some patients report hearing loss. In addition, migraine headaches may be a long-term complication. - LUMBAR/PELVIS/SACRAL syndrome: Hemangiomas located over the lumbar or sacral spine may be associated with genitourinary, anorectal anomalies, or neurological issues such as tethered cord.[8,32,33] The following criteria have been used to describe segmental hemangioma syndrome in the lumbar, pelvic, and sacral areas. This syndrome has been described in the literature using several acronyms.
LUMBAR
- -
Lower-body hemangioma and other cutaneous defects.
- -
Urogenital anomalies or ulceration.
- -
Myelopathy.
- -
Bony deformities.
- -
Anorectal malformations or arterial anomalies.
- -
Renal anomalies.
PELVIS
- -
Perineal hemangioma.
- -
External genital malformations.
- -
Lipomyelomeningocele.
- -
Vesicorenal abnormalities.
- -
Imperforate anus.
- -
Skin tag.
SACRAL
- -
Spinal dysraphism.
- -
Anogenital.
- -
Cutaneous.
- -
Renal and urologic anomalies Associated with an angioma of Lumbosacral localization.
Segmental lesions over the gluteal cleft and lumbar spine need to be evaluated with either ultrasound or MRI.
Infants with more than five hemangiomas need to be evaluated for visceral hemangiomas. The most common site of involvement is the liver, in which multiple or diffuse lesions can be noted.[6,34] Often these lesions are asymptomatic, but in a minority of cases, symptoms such as heart failure secondary to large vessel shunts, compartment syndrome, or profound hypothyroidism can occur. Diffuse liver hemangiomas can occur in the absence of skin lesions. (Refer to the Benign Vascular Tumors of the Liver section of this summary for more information.) Other rare potential complications of visceral hemangiomas, dependent on specific organ involvement, include gastrointestinal hemorrhage, obstructive jaundice, and central nervous system (CNS) sequelae, caused by mass effects.
Airway hemangiomas are usually associated with segmental hemangiomas in a bearded distribution, which may include all or some of the following: the preauricular skin, mandible, lower lip, chin, or anterior neck although they can be found without skin lesions. It is important for an otolaryngologist to proactively assess lesions in this distribution before signs of stridor occur. The incidence of an airway hemangioma increases with increased area of bearded involvement.[35]
Treatment of infantile hemangioma
Treatment options for infantile hemangioma include the following:
- Pulsed dye laser therapy. Usually used for ulcerated hemangiomas and residual lesions, such as telangiectasia after the proliferative period.[36] Pulsed dye laser therapy helps with pain from ulcerative hemangiomas. The use of pulsed dye laser therapy as upfront treatment for hemangiomas is controversial.
- Excisional surgery. With the advent of new medical treatments, the use of surgery is reserved for ulcerated lesions, residual lesions, and large periocular lesions that interfere with vision.[37]
Propranolol therapy
Propranolol, a nonselective beta-blocker, is the first-line therapy for hemangiomas. Potential mechanisms of action include vasoconstriction, decreased expression of VEGF and bFGF, leading to apoptosis.[40,41] Specific mechanisms of action are under investigation.
The use of propranolol was first noted in two infants treated for cardiac issues in Europe. A change in color, softening, and decrease in hemangioma size was noted. Since that time, the results of a randomized controlled trial have been reported.[7] In 2014, the U.S. Food and Drug Administration approved the drug Hemangeol (propranolol hydrochloride) for the treatment of proliferating infantile hemangioma.
Many propranolol regimens have been reported retrospectively or in small case series.[42-46] Lack of response to treatment is rare. Propranolol therapy is usually used during the proliferative phase but has been effective in patients older than 12 months with hemangiomas.[47]
Evidence (propranolol therapy):
- In a large industry-sponsored randomized trial, 456 infants aged 5 weeks to 5 months with a proliferating hemangioma of at least 1.5 cm received either a placebo or propranolol (1 mg/kg per day or 3 mg/kg per day) for 3 or 6 months. After interim analysis of the first 188 patients who completed 24 weeks of trial treatment, the regimen of 3 mg/kg per day for 6 months was selected for the final efficacy analysis.[7]
- Of patients who received the selected regimen, 88% showed improvement by week 5, compared with 5% of patients who received the placebo.
- Adverse events occurred infrequently.
- In 635 infants with infantile hemangioma, the overall response rate was 91% after 2 mg/kg per day, with most patients showing regression and only 2% with side effects, none of which were severe.[46][Level of evidence: 3iiiDiv]
Based on expert consensus panel recommendations, considerations for the administration of propranolol therapy include the following:[9]
- Initiation of treatment: Treatment should be undertaken in consultation with a pediatric vascular anomaly specialist with expertise in the diagnosis and treatment of pediatric vascular tumors and in the use of propranolol in children. In accord with an expert consensus panel, it is suggested that hospitalization for initiation of oral propranolol be considered in the following circumstances:[9]
- -
Infant aged 8 weeks or younger (corrected for gestational age).
- -
Infant of any age with inadequate social support.
- -
Infant of any age with comorbid conditions affecting the cardiovascular or respiratory system, including symptomatic airway hemangiomas.
- -
Infant of any age with conditions affecting blood glucose maintenance.
The pretreatment evaluation (inpatient or outpatient) includes the following:- -
History, with focus on cardiovascular and respiratory abnormalities (e.g., poor feeding, dyspnea, tachypnea, diaphoresis, wheezing, heart murmur) and family history of heart block or arrhythmia.
- -
Physical examination including cardiac and pulmonary assessment and measurement of heart rate and blood pressure.
- -
Consideration of an electrocardiogram, especially in children with heart rate lower than normal for age and history of arrhythmia or arrhythmia detected during examination.
- -
Family history of congenital heart disease or maternal history of connective tissue disease.
- Dosing: The dosing used is generally 1 mg/kg per day to 3 mg/kg per day divided into two or three doses. Patients are initially started at a dose of 0.5 mg/kg per day to 1 mg/kg per day and increased over time. Initially, dosing of three times per day is recommended for infants younger than 8 weeks and for patients with PHACE syndrome.
- Monitoring: Monitoring varies depending on the institution. However, oral propranolol peaks at 1 to 3 hours after administration and most centers measure heart rate, blood pressure, and glucose 2 hours after each dose with initiation and then when the dose is increased. Parents need to be aware of when to hold the dosing and signs of hypoglycemia. Parents also need to be aware of when to call their physician with any illness that may interfere with oral intake or lead to dehydration or respiratory issues.
- Contraindications: Propranolol treatment is contraindicated in infants and children with the following:
- -
Sinus bradycardia.
- -
Hypotension.
- -
Heart block greater than first degree.
- -
Heart failure.
- -
Asthma.
- -
Hypersensitivity.
- -
PHACE syndrome. PHACE Syndrome with CNS arterial disease and/or coarctation of the aorta may be a relative contraindication. A decision to treat should be made in consultation with neurology and cardiology.
- Adverse effects: Adverse effects of propranolol include the following:[48]
- -
Hypoglycemia.
- -
Hypotension.
- -
Bradycardia.
- -
Sleep disturbance.
- -
Diarrhea/constipation.
- -
Cold extremities.
These complications have been reported in several studies, and severe complications have been rare.[48] The risk of these complications is increased in patients with comorbidities and concomitant diseases. The need for close monitoring and possible periods of drug discontinuation should be considered during periods of illness.
Other selective beta-blocker therapy
Because of the nonselective nature and side effects of propranolol, other beta-blockers are being used for the treatment of hemangiomas with similar results. In one small comparison study, there was no difference in efficacy between propranolol and atenolol.[49] In a retrospective study using nadolol, similar results were seen.[50]
Corticosteroid therapy
Before propranolol, corticosteroids were the first line of treatment for infantile hemangiomas. They were first used in the late 1950s but were never approved by the U.S. Food and Drug Administration. Corticosteroid therapy has become less popular secondary to the acute and long-term side effects of steroids (gastrointestinal irritability, immunosuppression, adrenocortical suppression, cushingoid features, and growth failure).
Corticosteroids (prednisone or methylprednisolone) are used at times when there is a contraindication to beta-blocker therapy or as initial treatment while a patient is started on beta-blocker therapy.[51] No current studies are investigating the effectiveness of using combination therapy with these two agents.
Current Clinical Trials
Check the list of NCI-supported cancer clinical trials that are now accepting patients with infantile hemangioma. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria.
General information about clinical trials is also available from the NCI website.
Congenital Hemangiomas
Congenital hemangiomas are benign vascular tumors that proliferate in utero. Development of these lesions is complete at birth. Pathologically, these lesions are GLUT1 negative, unlike infantile hemangiomas. They are usually cutaneous, but can be in the viscera. Complications include hemorrhage, transient heart failure, and transient coagulopathy.[52]
Congenital hemangiomas are divided into the following three forms:
- Rapidly Involuting Congenital Hemangiomas (RICH). These lesions are large high-flow lesions that are completely formed at birth but gradually start to involute over 12 to 15 months. They can ulcerate and bleed and can cause transient heart failure and mild coagulopathy. After involution, usually some residual changes in the skin are present.[53-56]

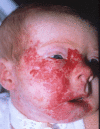
Figure
Figure 2. Typical appearance of a cutaneous congenital hemangioma at birth. Note the pedunculated mass. This RICH lesion involuted over time but some residual skin changes remained. Credit: Denise Adams, M.D.
- Partial Involuting Congenital Hemangiomas (PICH). These lesions are completely formed at birth and involute only partially.[57]
Benign Vascular Tumors of the Liver
In the literature, vascular liver tumors are usually classified as liver hemangioendotheliomas, a broad classification no longer in use. These tumors are classified according to their clinical characteristics and radiologic assessment.
Lesions are usually divided into the following three categories:[34]
- Focal lesions.
- Multifocal lesions.
- Diffuse lesions.
On MRI, vascular liver tumors are hyperintense on T2 imaging and hypointense on T1 imaging, with postcontrast imaging demonstrating early peripheral enhancement with eventual diffuse enhancement.[34]
Treatment of benign vascular tumors of the liver
Focal lesions are usually congenital hemangiomas (RICH or NICH). RICH can present with symptoms of heart failure and mild to moderate coagulopathy. No medication has proven to be an effective treatment, and infants need to be supported during this initial period until involution begins.[34] These lesions can be diagnosed prenatally. In rare situations, maternal treatment with medications such as steroids appears to have been effective.[60] Embolization for shunting has been utilized but needs to be performed by interventional radiologists with expertise in this procedure.[61]
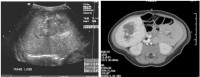
Figure
Figure 3. Single liver lesion (intrahepatic congenital hemangioma). Note the high flow on ultrasound evaluation (left) and the typical hyperintense image with early peripheral enhancement (right). Credit: Denise Adams, M.D.
Multifocal lesions and diffuse lesions are usually infantile hemangiomas. Multifocal lesions may not need to be treated if the patient is asymptomatic and typically follow the same proliferative and involution course as a cutaneous hemangioma.[34]
Diffuse liver lesions can be very serious. Complications include hypothyroidism, congestive heart failure, and compartment syndrome.[6,34,62,63]

Figure
Figure 4. Diffuse liver lesions with classical imaging on MRI. Note the peripheral enhancement in early contrast phase. Credit: Denise Adams, M.D.
Treatment of multifocal lesions and diffuse liver lesions may include:
- Propranolol: Beta-blockers are the most common treatment for diffuse infantile hemangiomas of the liver but treatment doses of 3mg/kg per day are needed.[7]
- Transplant: If a patient does not respond to propranolol and chemotherapy, rarely, a transplant is indicated.[66]
There have been isolated reports of malignancy in patients with diffuse liver infantile hemangiomas.[67] It is not clear if all cases were transformation of a benign lesion to a malignant phenotype; however, if the lesion does not respond to standard therapy, biopsy should be considered. Further evaluation and consensus is needed to assess whether these patients need to be monitored over a longer period of time with liver ultrasound.
Spindle Cell Hemangioma
Clinical presentation
Spindle cell hemangiomas, initially called spindle cell hemangioendotheliomas, often occur as superficial (skin and subcutis), painful lesions involving distal extremities in children and adults.[10,68] The tumors appear as red-brown or bluish lesions that can begin as a single nodule and develop into multifocal painful lesions over years. The lesions can be seen in Maffucci syndrome (cutaneous spindle cell hemangiomas occurring with cartilaginous tumors, enchondromas) and Klippel-Trenaunay syndrome (capillary/lymphatico/venous malformations), generalized lymphatic anomalies, lymphedema, and organized thrombus.[69,70]
These tumors are well circumscribed, occasionally contain phleboliths, and consist of cavernous blood spaces alternating with areas of nodular spindle cell proliferation. A significant percentage of spindle cell hemangiomas are completely intravascular. The vein containing the tumor is abnormal, as are blood vessels apart from the tumor mass.[69,70]
Epithelioid Hemangioma
Clinical presentation
Epithelioid hemangiomas are benign lesions that usually occur in the skin and subcutis but can occur in other areas, such as the bone.[69,71] Epithelioid hemangiomas may be a reactive process, as they can be associated with local trauma and can develop in pregnancy. Patients usually present with local swelling and pain at the involved site. In the bone, they present as well-defined lytic lesions that involve the metaphysis and diaphysis of long bones.[68] They can have a mixed lytic and sclerotic pattern of bone destruction.
On pathologic evaluation, they have small caliber capillaries with eosinophilic, vacuolated cytoplasm and large oval, grooved, and lobulated nuclei. The endothelial cells are plump and are mature, well-formed vessels surrounded by multiple epithelioid endothelial cells within abundant cytoplasm. They lack cellular atypia and mitotic activity.[72]
Pyogenic Granuloma (Lobular Capillary Hemangioma)
Clinical presentation
Pyogenic granuloma, known as lobular capillary hemangioma, is a benign reactive lesion that can present at any age, including infancy, although it is most common in older children and young adults.[11,73-75] These lesions can arise spontaneously, in sites of trauma, or within capillary malformations. Pyogenic granulomas have also been associated with medications including oral contraceptives and retinoids. Most occur as solitary growths, but multiple (grouped) or rarely disseminated lesions have been described. These lesions appear as small or large, smooth or lobulated vascular nodules that can grow rapidly, sometimes over weeks to months and have a tendency to bleed profusely.
Histologically, these lesions are composed of capillaries and venules with plump endothelial cells separated into lobules by fibromyxoid stroma. Some untreated lesions eventually atrophy, become fibromatous, and slowly regress.
Treatment of pyogenic granuloma
Treatment often consists of full-thickness excision, curettage, or laser photocoagulation, but recurrence is common.[76]
Angiofibroma
Clinical presentation
Angiofibromas are rare, benign neoplasms in the pediatric population. Typically, they are cutaneous lesions associated with tuberous sclerosis, appearing as red papules on the face.
Juvenile Nasopharyngeal Angiofibroma
Clinical presentation
Juvenile nasopharyngeal angiofibromas account for 0.5% of all head and neck tumors.[79] Histologically, juvenile nasopharyngeal angiofibromas are benign vascular tumors but they can be locally destructive, spreading from the nasal cavity to the nasopharynx, paranasal sinuses, and orbit skull base, with intracranial extension. Some publications have suggested a hormonal influence on juvenile nasopharyngeal angiofibroma, with emphasis on the molecular mechanisms involved.[80,81]
Treatment of juvenile nasopharyngeal angiofibroma
Surgical excision is the treatment of choice but this can be challenging because of the extent of the lesion. A single-institution retrospective review of juvenile nasopharyngeal angiofibromas identified 37 patients with lateral extension.[82] Anterior lateral extension to the pterygopalatine fossa occurred in 36 patients (97%) and further to the infratemporal fossa in 20 patients (54%). In 16 patients (43%), posterior lateral spread was observed (posterior to the pterygoid process and/or between its plates). The recurrence rate was 29.7% (11 of 37 patients). The recurrence rate in patients with anterior and/or posterior lateral extension was significantly higher than in patients with anterior lateral extension only.
Juvenile nasopharyngeal angiofibromas have also been treated with radiation therapy, chemotherapy, alpha-interferon therapy, and sirolimus.[83-86]
References
- Munden A, Butschek R, Tom WL, et al.: Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol 170 (4): 907-13, 2014. [PMC free article: PMC4410180] [PubMed: 24641194]
- Darrow DH, Greene AK, Mancini AJ, et al.: Diagnosis and Management of Infantile Hemangioma. Pediatrics 136 (4): e1060-104, 2015. [PubMed: 26416931]
- Darrow DH, Greene AK, Mancini AJ, et al.: Diagnosis and Management of Infantile Hemangioma: Executive Summary. Pediatrics 136 (4): 786-91, 2015. [PubMed: 26416928]
- Blei F, Walter J, Orlow SJ, et al.: Familial segregation of hemangiomas and vascular malformations as an autosomal dominant trait. Arch Dermatol 134 (6): 718-22, 1998. [PubMed: 9645641]
- Boye E, Yu Y, Paranya G, et al.: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107 (6): 745-52, 2001. [PMC free article: PMC208946] [PubMed: 11254674]
- Rialon KL, Murillo R, Fevurly RD, et al.: Risk factors for mortality in patients with multifocal and diffuse hepatic hemangiomas. J Pediatr Surg 50 (5): 837-41, 2015. [PubMed: 25783331]
- Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al.: A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med 372 (8): 735-46, 2015. [PubMed: 25693013]
- Stockman A, Boralevi F, Taïeb A, et al.: SACRAL syndrome: spinal dysraphism, anogenital, cutaneous, renal and urologic anomalies, associated with an angioma of lumbosacral localization. Dermatology 214 (1): 40-5, 2007. [PubMed: 17191046]
- Drolet BA, Frommelt PC, Chamlin SL, et al.: Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics 131 (1): 128-40, 2013. [PMC free article: PMC3529954] [PubMed: 23266923]
- Perkins P, Weiss SW: Spindle cell hemangioendothelioma. An analysis of 78 cases with reassessment of its pathogenesis and biologic behavior. Am J Surg Pathol 20 (10): 1196-204, 1996. [PubMed: 8827025]
- Swerlick RA, Cooper PH: Pyogenic granuloma (lobular capillary hemangioma) within port-wine stains. J Am Acad Dermatol 8 (5): 627-30, 1983. [PubMed: 6863618]
- North PE, Waner M, Mizeracki A, et al.: A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137 (5): 559-70, 2001. [PubMed: 11346333]
- Barnés CM, Huang S, Kaipainen A, et al.: Evidence by molecular profiling for a placental origin of infantile hemangioma. Proc Natl Acad Sci U S A 102 (52): 19097-102, 2005. [PMC free article: PMC1323205] [PubMed: 16365311]
- Walter JW, North PE, Waner M, et al.: Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma. Genes Chromosomes Cancer 33 (3): 295-303, 2002. [PubMed: 11807987]
- Khan ZA, Boscolo E, Picard A, et al.: Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest 118 (7): 2592-9, 2008. [PMC free article: PMC2413184] [PubMed: 18535669]
- Ritter MR, Reinisch J, Friedlander SF, et al.: Myeloid cells in infantile hemangioma. Am J Pathol 168 (2): 621-8, 2006. [PMC free article: PMC1606494] [PubMed: 16436675]
- Bielenberg DR, Bucana CD, Sanchez R, et al.: Progressive growth of infantile cutaneous hemangiomas is directly correlated with hyperplasia and angiogenesis of adjacent epidermis and inversely correlated with expression of the endogenous angiogenesis inhibitor, IFN-beta. Int J Oncol 14 (3): 401-8, 1999. [PubMed: 10024670]
- Nguyen VA, Kutzner H, Fürhapter C, et al.: Infantile hemangioma is a proliferation of LYVE-1-negative blood endothelial cells without lymphatic competence. Mod Pathol 19 (2): 291-8, 2006. [PubMed: 16424896]
- Yu Y, Flint AF, Mulliken JB, et al.: Endothelial progenitor cells in infantile hemangioma. Blood 103 (4): 1373-5, 2004. [PubMed: 14576053]
- Ritter MR, Dorrell MI, Edmonds J, et al.: Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc Natl Acad Sci U S A 99 (11): 7455-60, 2002. [PMC free article: PMC124252] [PubMed: 12032304]
- Takahashi K, Mulliken JB, Kozakewich HP, et al.: Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest 93 (6): 2357-64, 1994. [PMC free article: PMC294441] [PubMed: 7911127]
- Chang LC, Haggstrom AN, Drolet BA, et al.: Growth characteristics of infantile hemangiomas: implications for management. Pediatrics 122 (2): 360-7, 2008. [PubMed: 18676554]
- Tollefson MM, Frieden IJ: Early growth of infantile hemangiomas: what parents' photographs tell us. Pediatrics 130 (2): e314-20, 2012. [PubMed: 22826568]
- Munabi NC, Tan QK, Garzon MC, et al.: Growth Hormone Induces Recurrence of Infantile Hemangiomas After Apparent Involution: Evidence of Growth Hormone Receptors in Infantile Hemangioma. Pediatr Dermatol 32 (4): 539-43, 2015 Jul-Aug. [PMC free article: PMC5433863] [PubMed: 25690955]
- Dubois J, Patriquin HB, Garel L, et al.: Soft-tissue hemangiomas in infants and children: diagnosis using Doppler sonography. AJR Am J Roentgenol 171 (1): 247-52, 1998. [PubMed: 9648798]
- Metry DW, Garzon MC, Drolet BA, et al.: PHACE syndrome: current knowledge, future directions. Pediatr Dermatol 26 (4): 381-98, 2009 Jul-Aug. [PubMed: 19689512]
- Frieden IJ, Reese V, Cohen D: PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol 132 (3): 307-11, 1996. [PubMed: 8607636]
- Metry D, Heyer G, Hess C, et al.: Consensus Statement on Diagnostic Criteria for PHACE Syndrome. Pediatrics 124 (5): 1447-56, 2009. [PubMed: 19858157]
- Metry DW, Haggstrom AN, Drolet BA, et al.: A prospective study of PHACE syndrome in infantile hemangiomas: demographic features, clinical findings, and complications. Am J Med Genet A 140 (9): 975-86, 2006. [PubMed: 16575892]
- Drolet BA, Dohil M, Golomb MR, et al.: Early stroke and cerebral vasculopathy in children with facial hemangiomas and PHACE association. Pediatrics 117 (3): 959-64, 2006. [PubMed: 16510684]
- Heyer GL, Dowling MM, Licht DJ, et al.: The cerebral vasculopathy of PHACES syndrome. Stroke 39 (2): 308-16, 2008. [PubMed: 18174492]
- Iacobas I, Burrows PE, Frieden IJ, et al.: LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies. J Pediatr 157 (5): 795-801.e1-7, 2010. [PubMed: 20598318]
- Girard C, Bigorre M, Guillot B, et al.: PELVIS Syndrome. Arch Dermatol 142 (7): 884-8, 2006. [PubMed: 16847205]
- Hsi Dickie B, Fishman SJ, Azizkhan RG: Hepatic vascular tumors. Semin Pediatr Surg 23 (4): 168-72, 2014. [PubMed: 25241093]
- Elluru RG, Friess MR, Richter GT, et al.: Multicenter Evaluation of the Effectiveness of Systemic Propranolol in the Treatment of Airway Hemangiomas. Otolaryngol Head Neck Surg 153 (3): 452-60, 2015. [PubMed: 26124263]
- Kessels JP, Hamers ET, Ostertag JU: Superficial hemangioma: pulsed dye laser versus wait-and-see. Dermatol Surg 39 (3 Pt 1): 414-21, 2013. [PubMed: 23279058]
- Keller RG, Patel KG: Evidence-Based Medicine in the Treatment of Infantile Hemangiomas. Facial Plast Surg Clin North Am 23 (3): 373-92, 2015. [PubMed: 26208774]
- Xu DP, Cao RY, Tong S, et al.: Topical timolol maleate for superficial infantile hemangiomas: an observational study. J Oral Maxillofac Surg 73 (6): 1089-94, 2015. [PubMed: 25843815]
- Tawfik AA, Alsharnoubi J: Topical timolol solution versus laser in treatment of infantile hemangioma: a comparative study. Pediatr Dermatol 32 (3): 369-76, 2015 May-Jun. [PubMed: 25740672]
- Sharifpanah F, Saliu F, Bekhite MM, et al.: β-Adrenergic receptor antagonists inhibit vasculogenesis of embryonic stem cells by downregulation of nitric oxide generation and interference with VEGF signalling. Cell Tissue Res 358 (2): 443-52, 2014. [PubMed: 25130141]
- Ma X, Zhao T, Ouyang T, et al.: Propranolol enhanced adipogenesis instead of induction of apoptosis of hemangiomas stem cells. Int J Clin Exp Pathol 7 (7): 3809-17, 2014. [PMC free article: PMC4128992] [PubMed: 25120757]
- Bauman NM: Propanolol effectively treats significant infantile hemangiomas. J Pediatr 167 (1): 210, 2015. [PubMed: 26117643]
- Chang L, Ye X, Qiu Y, et al.: Is Propranolol Safe and Effective for Outpatient Use for Infantile Hemangioma? A Prospective Study of 679 Cases From One Center in China. Ann Plast Surg 76 (5): 559-63, 2016. [PubMed: 26101993]
- Ames JA, Sykes JM: Current trends in medical management of infantile hemangioma. Curr Opin Otolaryngol Head Neck Surg 23 (4): 286-91, 2015. [PubMed: 26101875]
- Lou Y, Peng WJ, Cao Y, et al.: The effectiveness of propranolol in treating infantile haemangiomas: a meta-analysis including 35 studies. Br J Clin Pharmacol 78 (1): 44-57, 2014. [PMC free article: PMC4168379] [PubMed: 24033819]
- Luo Y, Zeng Y, Zhou B, et al.: A retrospective study of propranolol therapy in 635 infants with infantile hemangioma. Pediatr Dermatol 32 (1): 151-2, 2015 Jan-Feb. [PubMed: 24602103]
- Vivas-Colmenares GV, Bernabeu-Wittel J, Alonso-Arroyo V, et al.: Effectiveness of propranolol in the treatment of infantile hemangioma beyond the proliferation phase. Pediatr Dermatol 32 (3): 348-52, 2015 May-Jun. [PubMed: 25721095]
- Prey S, Voisard JJ, Delarue A, et al.: Safety of Propranolol Therapy for Severe Infantile Hemangioma. JAMA 315 (4): 413-5, 2016. [PubMed: 26813215]
- Ábarzúa-Araya A, Navarrete-Dechent CP, Heusser F, et al.: Atenolol versus propranolol for the treatment of infantile hemangiomas: a randomized controlled study. J Am Acad Dermatol 70 (6): 1045-9, 2014. [PubMed: 24656727]
- Randhawa HK, Sibbald C, Garcia Romero MT, et al.: Oral Nadolol for the Treatment of Infantile Hemangiomas: A Single-Institution Retrospective Cohort Study. Pediatr Dermatol 32 (5): 690-5, 2015 Sep-Oct. [PubMed: 26215612]
- Chinnadurai S, Fonnesbeck C, Snyder KM, et al.: Pharmacologic Interventions for Infantile Hemangioma: A Meta-analysis. Pediatrics 137 (2): e20153896, 2016. [PubMed: 26772662]
- Vildy S, Macher J, Abasq-Thomas C, et al.: Life-threatening hemorrhaging in neonatal ulcerated congenital hemangioma: two case reports. JAMA Dermatol 151 (4): 422-5, 2015. [PubMed: 25565634]
- Maguiness S, Uihlein LC, Liang MG, et al.: Rapidly involuting congenital hemangioma with fetal involution. Pediatr Dermatol 32 (3): 321-6, 2015 May-Jun. [PubMed: 25492638]
- Scalise R, Bolton J, Gibbs NF: Rapidly involuting congenital hemangioma (RICH): a brief case report. Dermatol Online J 20 (11): , 2014. [PubMed: 25419759]
- Kumarasamy MT, Castrisios G, Sharma BK: Rapidly involuting congenital haemangioma in a term neonate. BMJ Case Rep 2014: , 2014. [PMC free article: PMC4024566] [PubMed: 24798357]
- Hughes R, McAleer M, Watson R, et al.: Rapidly involuting congenital hemangioma with pustules: two cases. Pediatr Dermatol 31 (3): 398-400, 2014 May-Jun. [PubMed: 24689686]
- Nasseri E, Piram M, McCuaig CC, et al.: Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol 70 (1): 75-9, 2014. [PubMed: 24176519]
- Lee PW, Frieden IJ, Streicher JL, et al.: Characteristics of noninvoluting congenital hemangioma: a retrospective review. J Am Acad Dermatol 70 (5): 899-903, 2014. [PubMed: 24630000]
- Enjolras O, Mulliken JB, Boon LM, et al.: Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg 107 (7): 1647-54, 2001. [PubMed: 11391180]
- Schmitz R, Heinig J, Klockenbusch W, et al.: Antenatal diagnosis of a giant fetal hepatic hemangioma and treatment with maternal corticosteroid. Ultraschall Med 30 (3): 223-6, 2009. [PubMed: 19507116]
- Kayaalp C, Sabuncuoglu MZ: Embolization of Liver Hemangiomas. Hepat Mon 15 (8): e30334, 2015. [PMC free article: PMC4546810] [PubMed: 26322113]
- Rialon KL, Murillo R, Fevurly RD, et al.: Impact of Screening for Hepatic Hemangiomas in Patients with Multiple Cutaneous Infantile Hemangiomas. Pediatr Dermatol 32 (6): 808-12, 2015 Nov-Dec. [PubMed: 26223454]
- Yeh I, Bruckner AL, Sanchez R, et al.: Diffuse infantile hepatic hemangiomas: a report of four cases successfully managed with medical therapy. Pediatr Dermatol 28 (3): 267-75, 2011 May-Jun. [PubMed: 21517953]
- Wasserman JD, Mahant S, Carcao M, et al.: Vincristine for successful treatment of steroid-dependent infantile hemangiomas. Pediatrics 135 (6): e1501-5, 2015. [PubMed: 25986022]
- Vlahovic A, Simic R, Djokic D, et al.: Diffuse neonatal hemangiomatosis treatment with cyclophosphamide: a case report. J Pediatr Hematol Oncol 31 (11): 858-60, 2009. [PubMed: 19829152]
- Sundar Alagusundaramoorthy S, Vilchez V, Zanni A, et al.: Role of transplantation in the treatment of benign solid tumors of the liver: a review of the United Network of Organ Sharing data set. JAMA Surg 150 (4): 337-42, 2015. [PubMed: 25714928]
- Jeng MR, Fuh B, Blatt J, et al.: Malignant transformation of infantile hemangioma to angiosarcoma: response to chemotherapy with bevacizumab. Pediatr Blood Cancer 61 (11): 2115-7, 2014. [PubMed: 24740626]
- Fletcher CD, Beham A, Schmid C: Spindle cell haemangioendothelioma: a clinicopathological and immunohistochemical study indicative of a non-neoplastic lesion. Histopathology 18 (4): 291-301, 1991. [PubMed: 2071088]
- Enjolras O, Mulliken JB, Kozakewich HPW: Vascular tumors and tumor-like lesions. In: Mulliken JB, Burrows PE, Fishman SJ, eds.: Mulliken & Young's Vascular Anomalies: Hemangiomas and Malformations. 2nd ed. New York, NY: Oxford University Press, 2013, pp 259-324.
- Hoeger PH, Colmenero I: Vascular tumours in infants. Part I: benign vascular tumours other than infantile haemangioma. Br J Dermatol 171 (3): 466-73, 2014. [PubMed: 24117053]
- Guo R, Gavino AC: Angiolymphoid hyperplasia with eosinophilia. Arch Pathol Lab Med 139 (5): 683-6, 2015. [PubMed: 25927152]
- O'Connell JX, Nielsen GP, Rosenberg AE: Epithelioid vascular tumors of bone: a review and proposal of a classification scheme. Adv Anat Pathol 8 (2): 74-82, 2001. [PubMed: 11236956]
- Wassef M, Hunt SF, Santa Cruz DJ: Vascular tumors and vascular malformations. In: Barnhi RL, Crowson AN, Magro CM, et al., eds.: Dermatopathology. 3rd ed. New York, NY: McGraw Hill Medical, 2010, pp 802-56.
- Campbell JP, Grekin RC, Ellis CN, et al.: Retinoid therapy is associated with excess granulation tissue responses. J Am Acad Dermatol 9 (5): 708-13, 1983. [PubMed: 6227639]
- Mills SE, Cooper PH, Fechner RE: Lobular capillary hemangioma: the underlying lesion of pyogenic granuloma. A study of 73 cases from the oral and nasal mucous membranes. Am J Surg Pathol 4 (5): 470-9, 1980. [PubMed: 7435775]
- Patrizi A, Gurioli C, Dika E: Pyogenic granulomas in childhood: New treatment modalities. Dermatol Ther 28 (5): 332, 2015 Sep-Oct. [PubMed: 25818498]
- Haemel AK, O'Brian AL, Teng JM: Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Arch Dermatol 146 (7): 715-8, 2010. [PubMed: 20644030]
- Pignatti M, Spaggiari A, Sala P, et al.: Laser treatment of angiofibromas in tuberous sclerosis. Minerva Pediatr 66 (6): 585-6, 2014. [PubMed: 25336102]
- Coutinho-Camillo CM, Brentani MM, Nagai MA: Genetic alterations in juvenile nasopharyngeal angiofibromas. Head Neck 30 (3): 390-400, 2008. [PubMed: 18228521]
- Riggs S, Orlandi RR: Juvenile nasopharyngeal angiofibroma recurrence associated with exogenous testosterone therapy. Head Neck 32 (6): 812-5, 2010. [PubMed: 19626637]
- Liu Z, Wang J, Wang H, et al.: Hormonal receptors and vascular endothelial growth factor in juvenile nasopharyngeal angiofibroma: immunohistochemical and tissue microarray analysis. Acta Otolaryngol 135 (1): 51-7, 2015. [PubMed: 25384380]
- Szymańska A, Szymański M, Czekajska-Chehab E, et al.: Two types of lateral extension in juvenile nasopharyngeal angiofibroma: diagnostic and therapeutic management. Eur Arch Otorhinolaryngol 272 (1): 159-66, 2015. [PMC free article: PMC4282713] [PubMed: 24599598]
- Samanta D: Topical mTOR (mechanistic target of rapamycin) inhibitor therapy in facial angiofibroma. Indian J Dermatol Venereol Leprol 81 (5): 540-1, 2015 Sep-Oct. [PubMed: 26323682]
- Krakowski AC, Nguyen TA: Inhibition of Angiofibromas in a Tuberous Sclerosis Patient Using Topical Timolol 0.5% Gel. Pediatrics 136 (3): e709-13, 2015. [PubMed: 26304829]
- Mallick S, Benson R, Bhasker S, et al.: Long-term treatment outcomes of juvenile nasopharyngeal angiofibroma treated with radiotherapy. Acta Otorhinolaryngol Ital 35 (2): 75-9, 2015. [PMC free article: PMC4443565] [PubMed: 26019389]
- Peters T, Traboulsi D, Tibbles LA, et al.: Sirolimus: a therapeutic advance for dermatologic disease. Skin Therapy Lett 19 (4): 1-4, 2014 Jul-Aug. [PubMed: 25188522]
Intermediate Tumors (Locally Aggressive)
Kaposiform Hemangioendothelioma and Tufted Angioma
Kaposiform hemangioendothelioma and tufted angioma are rare vascular tumors that typically occur during infancy or early childhood but have been reported in adults. Both tumors are thought to be a spectrum of the same disease, because both can be locally aggressive and cause Kasabach-Merritt phenomenon, a serious life-threatening coagulopathy characterized by profound thrombocytopenia and hypofibrinogenemia. They are discussed here as a single entity, kaposiform hemangioendothelioma.
Incidence
The exact incidence of kaposiform hemangioendothelioma is unknown but is estimated to be 0.07 cases per 100,000 children per year.[1-3] The lesions affect both sexes equally, with most developing in the neonatal period, one-half presenting at birth, and others presenting during childhood or adulthood.[4]
Pathology
Kaposiform hemangioendothelioma is characterized by sheets of spindle cells with an infiltrative pattern in the dermis, subcutaneous fat, and muscle. There are often areas of fibrosis, with dilated thin-walled vessels infiltrated around the areas of spindle cells. Mixed with these areas are nests of rounded epithelioid cells of vascular origin and aggregates of capillaries with round or irregularly shaped lumens containing platelet-rich fibrin thrombi. There is usually the presence of abnormal lymphatic spaces, either within or at the periphery of the lesion. The rate of mitosis is variable but usually low. Tufted angioma is characterized by multiple, discrete lobules of tightly packed capillaries (tufts) scattered in the dermis and sometimes in the subcutis, so called cannonball pattern.[5] Mitoses are rare.
The pathogenesis is poorly understood. There is some evidence that kaposiform hemangioendothelioma may be derived from lymphatic endothelium, as the spindle cell expresses the vascular markers CD31 and CD34, the vascular endothelial growth factor receptor-3, a receptor required for lymphangiogenesis, and the lymphatic markers D2-40 and PROX1.[5-7] There is no evidence of association with human herpesvirus 8 infection as is present in Kaposi sarcoma.[7]
Clinical presentation
Kaposiform hemangioendothelioma most frequently involves the extremities and less frequently involves the trunk and head and neck area.[3] Most lesions involve the skin. Deeper lesions (retroperitoneum, thoracic cavity, and muscle) can appear as a bluish-purpuric hue on the skin, whereas superficial lesions can be firm, purpuric or ecchymotic, and painful. Lesions are usually unifocal and growth is contiguous. Local lymph nodes may be involved, but they never metastasize. Rare multifocal presentations have been reported mostly in the bone.[1-3]
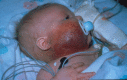
Figure
Figure 5. Kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon. The lesion is indurated, firm, and warm with petechiae and purpura. Credit: Denise Adams, M.D.
Seventy percent of patients with kaposiform hemangioendothelioma develop Kasabach-Merritt phenomenon, which is a life-threatening complication characterized by profound thrombocytopenia (range, 3,000/µL–60,000/µL) and profound hypofibrinogenemia (<1 g/L). D-dimer and fibrin degradation products are elevated. Severe anemia can occur secondary to tumor sequestration. Severe hemorrhage is rare; however, trauma (biopsy, surgical procedure), ulceration, infection, or delay in initiating treatment may induce progression to disseminated intravascular coagulation and serious bleeding and death can occur. Aggressive replacement of blood products, especially platelets, can increase the size of the lesion, causing significant pain and should only be considered with active bleeding and under the direction of a vascular anomaly specialist.[3]
Diagnostic evaluation
The diagnosis is based on the combination of clinical, histologic, and imaging features. Laboratory evaluation is essential for the diagnosis of Kasabach-Merritt phenomenon. Whenever possible, histologic confirmation should be obtained, because prolonged therapy is often needed. However, if clinical and imaging findings are highly suggestive of the diagnosis, deferring biopsy is an option but needs to be planned with an interdisciplinary approach.
Magnetic resonance imaging is the imaging preference. T1-weighted sequences typically show a poorly circumscribed soft tissue mass with soft tissue and dermal thickening and diffuse enhancement with gadolinium. T2-weighted sequences show a diffuse increased signal, with stranding in the subcutaneous fat. Gradient sequences show mildly dilated vessels in and around the soft-tissue mass.[3]
Treatment of kaposiform hemangioendothelioma and tufted angioma
Treatment varies according to severity; there is no evidence-based standard of care. An American and Canadian multidisciplinary expert panel published guidelines for the management of complicated kaposiform hemangioendothelioma.[8] A number of treatment therapies have been reported but none have been uniformly effective.[9,10]
Treatment options for kaposiform hemangioendothelioma include the following:[8-17]
- Steroid therapy.
- Antiplatelet agent (aspirin) therapy.
- Alpha-interferon.
- Antifibrinolytic agent therapy.
- Chemotherapy, including vincristine, cyclophosphamide, actinomycin, and methotrexate used alone or in combination.
- Propranolol therapy.
- Surgical excision with or without embolization.
- Sirolimus.
Initial treatment is most commonly steroids followed by vincristine. A retrospective review identified 37 children with kaposiform hemangioendothelioma whose lesions did not respond to steroids.[11][Level of evidence: 3iiiDiv] Twenty-six kaposiform hemangioendothelioma lesions achieved complete remission, with platelet counts reaching normal levels within 7.6 ± 5.2 weeks after vincristine treatment.
Sirolimus as monotherapy has been used. Initially using sirolimus with steroids is emerging as another treatment modality, but evidence of efficacy is very preliminary. In a prospective study that assessed the efficacy and safety of sirolimus for the treatment of complicated vascular anomalies, 13 patients with kaposiform hemangioendothelioma were treated. In patients with kaposiform hemangioendothelioma and Kasabach-Merritt phenomenon, ten of ten patients had a partial response, with normalization of their platelet count and fibrinogen at the end of six and 12 courses. In three of three patients with kaposiform hemangioendothelioma without Kasabach-Merritt phenomenon, one patient with multifocal bony disease had disease progression while the other two patients revealed a partial response at the end of course 12. Side effects were minimal in this group of young patients and no patient with kaposiform hemangioendothelioma required a dose adjustment or was removed from study secondary to toxicity.[18] Further studies are needed to determine the long-term efficacy and safety of sirolimus for the treatment of vascular tumors associated with Kasabach-Merritt phenomenon.
Surgical excision may be possible for lesions that are smaller, have failed medical management, or are life threatening. Embolization may be performed in conjunction with surgery of medical therapy; usually it is a temporizing measure.
Even with therapy, these lesions do not fully regress and can recur. The mortality associated with this tumor is primarily from the extensive coagulopathy associated with Kasabach-Merritt phenomenon.
Long-term effects include chronic pain, lymphedema, heart failure, and orthopedic issues. These lesions prove to be a difficult dilemma for the practitioner because they have a varied clinical spectrum and response to therapy.
References
- Rodriguez V, Lee A, Witman PM, et al.: Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 31 (7): 522-6, 2009. [PubMed: 19564750]
- Ryan C, Price V, John P, et al.: Kasabach-Merritt phenomenon: a single centre experience. Eur J Haematol 84 (2): 97-104, 2010. [PubMed: 19889011]
- Croteau SE, Liang MG, Kozakewich HP, et al.: Kaposiform hemangioendothelioma: atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J Pediatr 162 (1): 142-7, 2013. [PMC free article: PMC3494787] [PubMed: 22871490]
- Lee B, Chiu M, Soriano T, et al.: Adult-onset tufted angioma: a case report and review of the literature. Cutis 78 (5): 341-5, 2006. [PubMed: 17186794]
- Enjolras O, Soupre V, Picard A: Uncommon benign infantile vascular tumors. Adv Dermatol 24: 105-24, 2008. [PubMed: 19263597]
- Zukerberg LR, Nickoloff BJ, Weiss SW: Kaposiform hemangioendothelioma of infancy and childhood. An aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol 17 (4): 321-8, 1993. [PubMed: 8494101]
- Arai E, Kuramochi A, Tsuchida T, et al.: Usefulness of D2-40 immunohistochemistry for differentiation between kaposiform hemangioendothelioma and tufted angioma. J Cutan Pathol 33 (7): 492-7, 2006. [PubMed: 16872472]
- Drolet BA, Trenor CC 3rd, Brandão LR, et al.: Consensus-derived practice standards plan for complicated Kaposiform hemangioendothelioma. J Pediatr 163 (1): 285-91, 2013. [PubMed: 23796341]
- Haisley-Royster C, Enjolras O, Frieden IJ, et al.: Kasabach-merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol 24 (6): 459-62, 2002 Aug-Sep. [PubMed: 12218593]
- Hauer J, Graubner U, Konstantopoulos N, et al.: Effective treatment of kaposiform hemangioendotheliomas associated with Kasabach-Merritt phenomenon using four-drug regimen. Pediatr Blood Cancer 49 (6): 852-4, 2007. [PubMed: 16411198]
- Wang Z, Li K, Yao W, et al.: Steroid-resistant kaposiform hemangioendothelioma: a retrospective study of 37 patients treated with vincristine and long-term follow-up. Pediatr Blood Cancer 62 (4): 577-80, 2015. [PubMed: 25346262]
- Fernandez-Pineda I, Lopez-Gutierrez JC, Ramirez G, et al.: Vincristine-ticlopidine-aspirin: an effective therapy in children with Kasabach-Merritt phenomenon associated with vascular tumors. Pediatr Hematol Oncol 27 (8): 641-5, 2010. [PubMed: 20863161]
- Kai L, Wang Z, Yao W, et al.: Sirolimus, a promising treatment for refractory Kaposiform hemangioendothelioma. J Cancer Res Clin Oncol 140 (3): 471-6, 2014. [PubMed: 24464150]
- Hammill AM, Wentzel M, Gupta A, et al.: Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer 57 (6): 1018-24, 2011. [PubMed: 21445948]
- Blatt J, Stavas J, Moats-Staats B, et al.: Treatment of childhood kaposiform hemangioendothelioma with sirolimus. Pediatr Blood Cancer 55 (7): 1396-8, 2010. [PubMed: 20730884]
- Fernandez-Pineda I, Lopez-Gutierrez JC, Chocarro G, et al.: Long-term outcome of vincristine-aspirin-ticlopidine (VAT) therapy for vascular tumors associated with Kasabach-Merritt phenomenon. Pediatr Blood Cancer 60 (9): 1478-81, 2013. [PubMed: 23609996]
- Chiu YE, Drolet BA, Blei F, et al.: Variable response to propranolol treatment of kaposiform hemangioendothelioma, tufted angioma, and Kasabach-Merritt phenomenon. Pediatr Blood Cancer 59 (5): 934-8, 2012. [PMC free article: PMC3528889] [PubMed: 22648868]
- Adams DM, Trenor CC 3rd, Hammill AM, et al.: Efficacy and Safety of Sirolimus in the Treatment of Complicated Vascular Anomalies. Pediatrics 137 (2): e20153257, 2016. [PMC free article: PMC4732362] [PubMed: 26783326]
Intermediate Tumors (Rarely Metastasizing)
Retiform Hemangioendothelioma
Pathology and clinical presentation
Retiform hemangioendotheliomas are slow growing, exophytic, flat tumors found in young adults and occasionally children.[1] They are usually located in the limbs and trunk. Pathologically, they are located in the dermis and subcutaneous tissue. Vessels exhibit a pattern resembling the rete testis and are lined by protruding endothelial cells. They do not express lymphatic endothelial markers but stain positive for endothelial cells.[2]
Prognostic factors
Local recurrences are common, but distinct metastases are extremely rare.[2]
Papillary Intralymphatic Angioendothelioma
Pathology and clinical presentation
Papillary intralymphatic angioendothelioma, also known as Dabska tumor, can occur in the adult and pediatric population.[7] The lesions occur in the dermis and subcutis on all body parts and there have been some reports of lymph node involvement. They can be large or small raised purplish firm nodules.
Pathologically, they reveal intravascular growth of well-differentiated endothelial cells in a columnar configuration. They have thickened hyaline walls with hobnailed endothelium. Vascular endothelial growth factor receptor type 3, a marker for lymphatic endothelium, is positive in most cases. There is minimal cytologic atypia.[8] Some are associated with vascular malformations.
Treatment of papillary intralymphatic angioendothelioma
Surgical excision is the treatment of choice.[9]
Composite Hemangioendothelioma
Pathology and clinical presentation
Composite hemangioendothelioma is a very rare vascular tumor classified because of the combined benign and malignant vascular components. Usually, combined epithelioid and retiform variants are noted but some tumors have three components (epithelioid, retiform, and spindle cell).[10] Angiosarcoma foci have been noted. Pathology reveals positivity for CD31, factor VIII, and vimentin.[10,11] Rarely, D-240 is positive with a Ki-67 index of approximately 20%.[10]
This tumor usually occurs in the dermis and subdermis of the distal extremities but has been found in other areas such as the head, neck, and mediastinum.[10] They have been reported in all age groups.[10]
Kaposi Sarcoma
Pathology and clinical presentation
Kaposi sarcoma is a rare malignant vascular tumor associated with a viral etiology (human herpesvirus 8).[14] The skin lesions were first described in 1872 by Moritz Kaposi. The incidence has increased worldwide secondary to the HIV-AIDS epidemic. It is an extremely rare diagnosis in children. Epidemic and iatrogenic forms of Kaposi sarcoma in children result from profound acquired T-cell deficiency that results from HIV infection and rare immune disorders.
A retrospective study has investigated the presentation of Kaposi sarcoma in children in endemic areas of Africa. Children usually present with cutaneous lesions, lymphadenopathy, and intrathoracic and oral lesions. Cutaneous lesions initially appear as red, purple, or brown macules, later developing into plaques and then nodules.[15]
Treatment of Kaposi sarcoma
Children with Kaposi sarcoma have responded to treatment with chemotherapy. (Refer to the PDQ summary on Kaposi Sarcoma Treatment for information about the treatment of Kaposi sarcoma in adults.)
References
- El Darouti M, Marzouk SA, Sobhi RM, et al.: Retiform hemangioendothelioma. Int J Dermatol 39 (5): 365-8, 2000. [PubMed: 10849129]
- Colmenero I, Hoeger PH: Vascular tumours in infants. Part II: vascular tumours of intermediate malignancy [corrected] and malignant tumours. Br J Dermatol 171 (3): 474-84, 2014. [PubMed: 24965196]
- Keiler SA, Honda K, Bordeaux JS: Retiform hemangioendothelioma treated with Mohs micrographic surgery. J Am Acad Dermatol 65 (1): 233-5, 2011. [PubMed: 21679834]
- Hirsh AZ, Yan W, Wei L, et al.: Unresectable retiform hemangioendothelioma treated with external beam radiation therapy and chemotherapy: a case report and review of the literature. Sarcoma 2010: , 2010. [PMC free article: PMC2948930] [PubMed: 20936126]
- Enjolras O, Mulliken JB, Kozakewich HPW: Vascular tumors and tumor-like lesions. In: Mulliken JB, Burrows PE, Fishman SJ, eds.: Mulliken & Young's Vascular Anomalies: Hemangiomas and Malformations. 2nd ed. New York, NY: Oxford University Press, 2013, pp 259-324.
- Tamhankar AS, Vaidya A, Pai P: Retiform hemangioendothelioma over forehead: A rare tumor treated with chemoradiation and a review of literature. J Cancer Res Ther 11 (3): 657, 2015 Jul-Sep. [PubMed: 26458658]
- Dabska M: Malignant endovascular papillary angioendothelioma of the skin in childhood. Clinicopathologic study of 6 cases. Cancer 24 (3): 503-10, 1969. [PubMed: 5343389]
- Fanburr-Smith JC: Papillary intralymphatic angioendothelioma. In: Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon, France: IARC Press, 2013, pp 148.
- Neves RI, Stevenson J, Hancey MJ, et al.: Endovascular papillary angioendothelioma (Dabska tumor): underrecognized malignant tumor in childhood. J Pediatr Surg 46 (1): e25-8, 2011. [PubMed: 21238627]
- Shang Leen SL, Fisher C, Thway K: Composite hemangioendothelioma: clinical and histologic features of an enigmatic entity. Adv Anat Pathol 22 (4): 254-9, 2015. [PubMed: 26050262]
- Mahmoudizad R, Samrao A, Bentow JJ, et al.: Composite hemangioendothelioma: An unusual presentation of a rare vascular tumor. Am J Clin Pathol 141 (5): 732-6, 2014. [PubMed: 24713748]
- Tateishi J, Saeki H, Ito K, et al.: Cutaneous composite hemangioendothelioma on the nose treated with electron beam. Int J Dermatol 52 (12): 1618-9, 2013. [PubMed: 22998114]
- Soldado F, Fontecha CG, Haddad S, et al.: Composite vascularized fibular epiphyseo-osteo-periosteal transfer for hip reconstruction after proximal femoral tumoral resection in a 4-year-old child. Microsurgery 32 (6): 489-92, 2012. [PubMed: 22511340]
- Jackson CC, Dickson MA, Sadjadi M, et al.: Kaposi Sarcoma of Childhood: Inborn or Acquired Immunodeficiency to Oncogenic HHV-8. Pediatr Blood Cancer 63 (3): 392-7, 2016. [PMC free article: PMC4984265] [PubMed: 26469702]
- Dow DE, Cunningham CK, Buchanan AM: A Review of Human Herpesvirus 8, the Kaposi's Sarcoma-Associated Herpesvirus, in the Pediatric Population. J Pediatric Infect Dis Soc 3 (1): 66-76, 2014. [PMC free article: PMC3933043] [PubMed: 24567845]
Malignant Tumors
Epithelioid Hemangioendothelioma
Incidence and outcome
This tumor was first described in soft tissue by Weiss and Enzinger in 1982. Epithelioid hemangioendotheliomas can occur at younger ages, but the peak incidence is in the fourth and fifth decades of life. The tumors can have an indolent or very aggressive course, with overall survival of 73% at 5 years. There are case reports of patients with untreated multiple lesions who have a very benign course compared with other patients who have a very aggressive course. Some pathologists have tried to stratify patients to evaluate risks and adjust treatment, but more research is needed.[1-7]
Pathology and biology
A WWTR1-CAMTA1 gene fusion has been found in a large percentage of patients; less commonly, a YAP1-TFE3 gene fusion has been reported.[1] These fusions are not directly targetable with medicines. Monoclonality has been described in multiple liver lesions, suggesting a metastatic process. Histologically, these lesions are characterized as epithelioid lesions arranged in nests, strands, and trabecular patterns, with infrequent vascular spaces. Features that may be associated with aggressive clinical behavior include cellular atypia, one or more mitoses per 10 high-power fields, an increased proportion of spindled cells, focal necrosis, and metaplastic bone formation.[3]
Clinical presentation and diagnostic evaluation
Common sites of involvement are liver alone (21%), liver plus lung (18%), lung alone (12%), and bone alone (14%).[3,8,9] Clinical presentation depends on site of involvement, as follows:
- Liver: Hepatic nodules have central vascularity on ultrasound, contrast-enhancing lesions by computed tomography, and low T1 signal and moderate T2 signal on magnetic resonance imaging.
- Lung: Pulmonary epithelioid hemangioendothelioma may be an asymptomatic finding on chest x-ray or be associated with pleuritic pain, hemoptysis, anemia, and fibrosis.
- Bone: Bone metastasis may be associated with pathologic fracture. On x-rays, they are well-defined osteolytic lesions and can be multiple or solitary.
- Soft tissue: Thirty percent of soft tissue cases are associated with metastases, and when present, can have a very aggressive course, with limited response to chemotherapy.
- Skin: Cutaneous lesions can be raised and nodular or can be warm red-brown plaques.
Treatment of epithelioid hemangioendothelioma
Treatment options for epithelioid hemangioendothelioma include the following:
- Observation.
- Surgery.
- Immunotherapy.
- Targeted therapy.
- Chemotherapy.
For indolent cases, observation is warranted. For more aggressive cases, multiple medications have been used, including interferon, thalidomide, sorafenib, pazopanib, and sirolimus. The most aggressive cases are treated with angiosarcoma-type chemotherapy. Surgery is used when possible. Liver transplantation has been used with aggressive liver lesions, both with and without metastases.[3,10-13]
Current Clinical Trials
Check the list of NCI-supported cancer clinical trials that are now accepting patients with childhood epithelioid hemangioendothelioma. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria.
General information about clinical trials is also available from the NCI website.
Angiosarcoma of the Soft Tissue
Incidence
Angiosarcoma is a rare (accounting for 2% of sarcomas), aggressive, vascular tumor that can arise in any part of the body, but is more common in the soft tissue. Angiosarcoma has an estimated incidence of 2 cases per 1 million; in the United States, it annually affects approximately 600 people who are typically aged 60 to 70 years.[14]
Angiosarcomas are extremely rare in children. However, cases have been reported in neonates and toddlers, with presentation of multiple cutaneous lesions and liver lesions, some of which are GLUT1 positive.[15-18] Most angiosarcomas involve the skin and superficial soft tissue, although the liver, spleen, and lung can be affected; bone is rarely affected.
Risk factors
Established risk factors include vinyl chloride exposure, radiation exposure, and chronic lymphedema from any cause, including Stewart-Treves syndrome.[19]
Pathology and biology
Angiosarcomas are largely aneuploidy tumors. The rare cases of angiosarcoma that arise from benign lesions such as hemangiomas have a distinct pathway that needs to be investigated. MYC amplification is seen in radiation-induced angiosarcoma. KDR-VEGFR2 mutations and FLT4-VEGFR3 amplifications have been seen with a frequency of less than 50%.[19]
Histopathologic diagnosis can be very difficult because there can be areas of varied atypia. The common feature is an irregular network of channels in a dissective pattern along dermal collagen bundles. There is varied cellular shape, size, mitosis, endothelial multilayering, and papillary formation. Epithelioid cells can also be present. Necrosis and hemorrhage are common. Tumors stain for factor VIII, CD31, and CD34. Some liver lesions can mimic infantile hemangiomas and have focal GLUT1 positivity. Nomenclature of these liver lesions has been difficult and confusing with use of terminology from 1971 (e.g., type I hemangioendothelioma: infantile hemangioma; type II hemangioendothelioma: low-grade angiosarcoma; type III hemangioendothelioma: high-grade angiosarcoma).[16]
Treatment of angiosarcoma of the soft tissue
Treatment options for angiosarcoma of the soft tissue include the following:
- Surgery.
- Surgery, chemotherapy, and radiation therapy (metastatic disease).
- Bevacizumab and chemotherapy (angiosarcoma secondary to infantile hemangioma).
Localized disease is cured by aggressive surgery. Complete surgical excision appears to be crucial for angiosarcomas and lymphangiosarcomas despite evidence of tumor shrinkage in some patients who were treated with local or systemic therapy.[17,20-22] A review of 222 patients (median age, 62 years; range, age 15–90 years) showed an overall disease-specific survival (DSS) rate of 38% at 5 years. Five-year DSS was 44% in 138 patients with localized, resected tumors but only 16% in 43 patients with metastases at diagnosis.[22] Data on liver transplantation for localized angiosarcoma are limited.[23][Level of evidence: 3iiA]
Multimodal treatment with surgery, systemic chemotherapy, and radiation therapy is used for metastatic disease, although it is rarely curative.[24] Disease control is the objective in metastatic angiosarcoma, with published progression-free survival rates between 3 months and 7 months [25] and a median overall survival (OS) rate of 14 months to 18 months.[26] In both adults and children, 5-year OS rates between 20% and 35% are reported.[17,18,27]
In a child diagnosed with angiosarcoma secondary to malignant transformation from infantile hemangioma, response to treatment with bevacizumab, a monoclonal antibody against vascular endothelial growth factor, combined with systemic chemotherapy, has been reported.[15]
Biologic agents that inhibit angiogenesis have shown activity in adults with angiosarcoma.[16,27]
Treatment options under clinical evaluation
The following is an example of a national and/or institutional clinical trial that is currently being conducted. Information about ongoing clinical trials is available from the NCI website.
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery [PAZNTIS]): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with angiosarcoma of the soft tissue are eligible for this trial.
Current Clinical Trials
Check the list of NCI-supported cancer clinical trials that are now accepting patients with childhood angiosarcoma. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria.
General information about clinical trials is also available from the NCI website.
References
- Mehrabi A, Kashfi A, Fonouni H, et al.: Primary malignant hepatic epithelioid hemangioendothelioma: a comprehensive review of the literature with emphasis on the surgical therapy. Cancer 107 (9): 2108-21, 2006. [PubMed: 17019735]
- Haro A, Saitoh G, Tamiya S, et al.: Four-year natural clinical course of pulmonary epithelioid hemangioendothelioma without therapy. Thorac Cancer 6 (4): 544-7, 2015. [PMC free article: PMC4511336] [PubMed: 26273413]
- Sardaro A, Bardoscia L, Petruzzelli MF, et al.: Epithelioid hemangioendothelioma: an overview and update on a rare vascular tumor. Oncol Rev 8 (2): 259, 2014. [PMC free article: PMC4419652] [PubMed: 25992243]
- Dong K, Wang XX, Feng JL, et al.: Pathological characteristics of liver biopsies in eight patients with hepatic epithelioid hemangioendothelioma. Int J Clin Exp Pathol 8 (9): 11015-23, 2015. [PMC free article: PMC4637634] [PubMed: 26617819]
- Adams DM, Hammill A: Other vascular tumors. Semin Pediatr Surg 23 (4): 173-7, 2014. [PubMed: 25241094]
- Xiao Y, Wang C, Song Y, et al.: Primary epithelioid hemangioendothelioma of the kidney: the first case report in a child and literature review. Urology 82 (4): 925-7, 2013. [PubMed: 23726166]
- Reich S, Ringe H, Uhlenberg B, et al.: Epithelioid hemangioendothelioma of the lung presenting with pneumonia and heart rhythm disturbances in a teenage girl. J Pediatr Hematol Oncol 32 (4): 274-6, 2010. [PubMed: 20445417]
- Daller JA, Bueno J, Gutierrez J, et al.: Hepatic hemangioendothelioma: clinical experience and management strategy. J Pediatr Surg 34 (1): 98-105; discussion 105-6, 1999. [PubMed: 10022152]
- Ackermann O, Fabre M, Franchi S, et al.: Widening spectrum of liver angiosarcoma in children. J Pediatr Gastroenterol Nutr 53 (6): 615-9, 2011. [PubMed: 21832953]
- Semenisty V, Naroditsky I, Keidar Z, et al.: Pazopanib for metastatic pulmonary epithelioid hemangioendothelioma-a suitable treatment option: case report and review of anti-angiogenic treatment options. BMC Cancer 15: 402, 2015. [PMC free article: PMC4437555] [PubMed: 25967676]
- Raheja A, Suri A, Singh S, et al.: Multimodality management of a giant skull base hemangioendothelioma of the sphenopetroclival region. J Clin Neurosci 22 (9): 1495-8, 2015. [PubMed: 25986183]
- Ahmad N, Adams DM, Wang J, et al.: Hepatic epithelioid hemangioendothelioma in a patient with hemochromatosis. J Natl Compr Canc Netw 12 (9): 1203-7, 2014. [PubMed: 25190690]
- Otte JB, Zimmerman A: The role of liver transplantation for pediatric epithelioid hemangioendothelioma. Pediatr Transplant 14 (3): 295-7, 2010. [PubMed: 20331517]
- Cioffi A, Reichert S, Antonescu CR, et al.: Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am 27 (5): 975-88, 2013. [PubMed: 24093171]
- Jeng MR, Fuh B, Blatt J, et al.: Malignant transformation of infantile hemangioma to angiosarcoma: response to chemotherapy with bevacizumab. Pediatr Blood Cancer 61 (11): 2115-7, 2014. [PubMed: 24740626]
- Dehner LP, Ishak KG: Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol 92 (2): 101-11, 1971. [PubMed: 5559952]
- Ferrari A, Casanova M, Bisogno G, et al.: Malignant vascular tumors in children and adolescents: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Med Pediatr Oncol 39 (2): 109-14, 2002. [PubMed: 12116058]
- Deyrup AT, Miettinen M, North PE, et al.: Pediatric cutaneous angiosarcomas: a clinicopathologic study of 10 cases. Am J Surg Pathol 35 (1): 70-5, 2011. [PubMed: 21164289]
- Elliott P, Kleinschmidt I: Angiosarcoma of the liver in Great Britain in proximity to vinyl chloride sites. Occup Environ Med 54 (1): 14-8, 1997. [PMC free article: PMC1128629] [PubMed: 9072028]
- Lezama-del Valle P, Gerald WL, Tsai J, et al.: Malignant vascular tumors in young patients. Cancer 83 (8): 1634-9, 1998. [PubMed: 9781959]
- Fata F, O'Reilly E, Ilson D, et al.: Paclitaxel in the treatment of patients with angiosarcoma of the scalp or face. Cancer 86 (10): 2034-7, 1999. [PubMed: 10570428]
- Lahat G, Dhuka AR, Hallevi H, et al.: Angiosarcoma: clinical and molecular insights. Ann Surg 251 (6): 1098-106, 2010. [PubMed: 20485141]
- Orlando G, Adam R, Mirza D, et al.: Hepatic hemangiosarcoma: an absolute contraindication to liver transplantation--the European Liver Transplant Registry experience. Transplantation 95 (6): 872-7, 2013. [PubMed: 23354302]
- Dickson MA, D'Adamo DR, Keohan ML, et al.: Phase II Trial of Gemcitabine and Docetaxel with Bevacizumab in Soft Tissue Sarcoma. Sarcoma 2015: 532478, 2015. [PMC free article: PMC4446476] [PubMed: 26074722]
- North PE, Waner M, Mizeracki A, et al.: A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137 (5): 559-70, 2001. [PubMed: 11346333]
- Boye E, Yu Y, Paranya G, et al.: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107 (6): 745-52, 2001. [PMC free article: PMC208946] [PubMed: 11254674]
- Ravi V, Patel S: Vascular sarcomas. Curr Oncol Rep 15 (4): 347-55, 2013. [PubMed: 23852636]
Special Considerations for the Treatment of Children with Cancer
Fortunately, cancer in children and adolescents is rare, although the overall incidence of childhood cancer has been slowly increasing since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the primary care physician, a surgeon experienced in vascular tumors, a pathologist, radiation oncologists, pediatric oncologists, rehabilitation specialists, pediatric nurse specialists, social workers, and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life. (Refer to the PDQ summaries on Supportive and Palliative Care for specific information about supportive care for children and adolescents with cancer.)
Guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[2] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate in these trials is offered to most patients and families. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1] Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.)
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PMC free article: PMC4136455] [PubMed: 24853691]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004. [PubMed: 15173520]
Changes to this Summary (08/11/2016)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Added text about how once a hemangioma has involuted, regrowth is uncommon. Also added text about how two patients were treated with growth hormone, and regrowth after involution was noted; on further investigation, growth hormone receptors were found on the hemangioma cells (cited Munabi et al. as reference 24).
Added corticosteroid therapy as a treatment option for infantile hemangioma.
Added text to state that propranolol therapy is usually used during the proliferative phase but has been effective in patients older than 12 months with hemangiomas (cited Vivas-Colmenares et al. as reference 47).
Added text to state that propranolol dosing of three times per day is recommended for infants younger than 8 weeks and for patients with PHACE syndrome.
Added text to state that parents also need to be aware of when to call their physician with any illness that may interfere with oral intake or lead to dehydration or respiratory issues.
Added PHACE syndrome as a contraindication to propranolol treatment in infants and children. Also added text to state that PHACE Syndrome with central nervous system arterial disease and/or coarctation of the aorta may be a relative contraindication. A decision to treat should be made in consultation with neurology and cardiology.
Added Prey et al. as reference 48.
Added text to state that complications have been reported in several studies, and severe complications have been rare. The risk of these complications is increased in patients with comorbidities and concomitant diseases. The need for close monitoring and possible periods of drug discontinuation should be considered during periods of illness.
Added Corticosteroid therapy as a new subsection.
Special Considerations for the Treatment of Children with Cancer
Added this new section.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood vascular tumors. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Vascular Tumors Treatment are:
- Denise Adams, MD (Children's Hospital Boston)
- Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
- Holcombe Edwin Grier, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (AFLAC Cancer Center and Blood Disorders Service of Children's Healthcare of Atlanta - Egleston Campus)
- Alberto S. Pappo, MD (St. Jude Children's Research Hospital)
- R Beverly Raney, MD (Consultant)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Vascular Tumors Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: http://www.cancer.gov/types/soft-tissue-sarcoma/hp/child-vascular-tumors-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26844334]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
- Childhood Vascular Tumors Treatment (PDQ®) - PDQ Cancer Information SummariesChildhood Vascular Tumors Treatment (PDQ®) - PDQ Cancer Information Summaries
- PhenolphthaleinsPhenolphthaleinsA family of 3,3-bis(p-hydroxyphenyl)phthalides. They are used as CATHARTICS, indicators, and COLORING AGENTS.<br/>MeSH
- ResveratrolResveratrolA stilbene and non-flavonoid polyphenol produced by various plants including grapes and blueberries. It has anti-oxidant, anti-inflammatory, cardioprotective, anti-mutagenic, ...<br/>Year introduced: 2019 (1989)MeSH
Your browsing activity is empty.
Activity recording is turned off.
See more...