Clinical Description
To date, 31 individuals have been identified with a pathogenic variant in FAM111B [Mercier et al 2013, Mercier et al 2015, Seo et al 2016, Takeichi et al 2017, Goussot et al 2017, Kazlouskaya et al 2018, Zhang et al 2019, Chen et al 2019, Chasseuil et al 2019, Dokic et al 2020]. The following description of the phenotypic features associated with this condition is based on these reports.
Individuals with hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) can exhibit few or many of the associated clinical features. The severity of the features (e.g., skin or muscle abnormalities) can vary. Intrafamilial clinical variability has been observed [Khumalo et al 2006, Seo et al 2016].
Skin
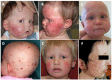
Skin abnormalities are the earliest findings. Of note, skin lesions – particularly facial poikiloderma – improve with time.
Poikiloderma appears during early infancy, typically in the first six months. It is mainly localized to the face (). Transient exacerbations of facial erythema are seen following sun exposure. Mottled pigmentation is also a constituent part of poikiloderma.
Hypohidrosis with heat intolerance is observed in most.
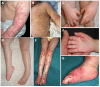
Lymphedema of the lower and/or upper extremities may be present in childhood and is usually mild; it can be complicated by episodes of cellulitis.
Chronic erythematous and scaly skin lesions described as eczema-like, ichthyosis-like, or psoriasis-like lesions are often observed on the extremities.
Sclerosis of the digits and mild palmoplantar keratoderma may also be observed.
Hair/nails. Sparse scalp hair and sparse or absent eyelashes and/or eyebrows of variable severity are found in almost all affected individuals. Four affected individuals had mild nail dysplasia.
Muscle
Muscle contractures are usually seen in childhood and can be present as early as age two years. The most commonly affected muscles are the triceps surae, leading to a shortening of the Achilles tendons and varus deformities of the feet. Contractures of upper limbs (biceps brachii and carpal extensors) are also observed.
Myopathy. The majority of affected individuals develop progressive weakness of the proximal and distal muscles of all four limbs; the first manifestations (observed in lower limbs) are proximal rather than distal. Variability of muscle weakness ranges from loss of ambulation in childhood to absence of symptoms in adulthood [Mercier et al 2013, Mercier et al 2015, Seo et al 2016, Takeichi et al 2017].
Muscle weakness is generally associated with muscle atrophy and sometimes thoracolumbar scoliosis.
Serum creatine kinase is either normal or slightly increased. When performed, electromyography may show a normal or myopathic pattern.
Lung
Recurrent bronchitis can be observed. Abnormal lung function with restrictive pulmonary disease is common.
Some adults develop progressive interstitial pulmonary fibrosis, manifest as progressive breathlessness and dry cough; it can be life threatening within three to four years after the first respiratory symptoms appear.
Gastrointestinal
Pancreatic exocrine insufficiency (common in affected individuals) typically begins in childhood. Manifestations include fatty stools and diarrhea leading to chronic malabsorption of fats and lipid-soluble vitamins if not treated. Diagnosis is confirmed based on fecal elastase deficiency.
Liver impairment (reported in a few affected individuals) manifests initially as mildly elevated transaminases, alkaline phosphatase, gamma-glutamyl transferase, and/or bilirubin. One individual had hepatomegaly and cholestasis [Mercier et al 2013, Mercier et al 2015]. Another individual reported has transaminitis and lymphocytic ductulitis [Dokic et al 2020].
Other Features
Relative short stature and/or poor weight gain associated with delayed puberty have been reported.
Hematologic findings include eosinophilia or mild thrombocytopenia in some. Bone marrow hypocellularity was reported in a multiplex family [Seo et al 2016].
Hypothyroidism was described in a girl age 14 years [Takeichi et al 2017].
Eye. Cataract was reported in a girl age 13 years [Mercier et al 2015].
Cognitive development and function are normal. Of note, one individual had schizophrenia [Mercier et al 2015], which could be an incidental association.
Histopathology
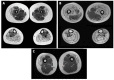
Muscle histology shows extensive fatty infiltration. Residual muscle tissue is composed of fragmented muscle fascicles with either normal fibers or atrophic fibers with central nuclei (). No neuropathic features (i.e., normal ATPase pattern) or mitochondrial network abnormalities are found on histochemistry or immunolabeling. Western blot analysis can show a secondary reduction of calpain.
A-D. Muscle histology: A, B. Fatty tissue, fragmented muscle fascicles next to normal fascicles
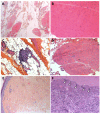
Skin histology shows a characteristic pattern of epidermal atrophy with collagen sclerosis and elastic degeneration in the superficial and deep dermis. Elastic globes in the papillary dermis are associated with a diffuse mild collagen sclerosis ().
Postmortem findings of one affected member of a South African family revealed diffuse fatty infiltration and fibrosis of organs including the lungs, esophagus, and pancreas [Khumalo et al 2006].