Clinical Description
Bohring-Opitz syndrome (BOS) is a rare condition characterized by distinctive facial features and posture, variable but usually severe intellectual disability, growth failure, and variable anomalies. Feeding difficulties have a significant impact on overall health in early childhood; feeding tends to improve with age. This section summarizes clinical data from numerous case reports and case series; see Russell et al [2015] and references therein, Dangiolo et al [2015], and Suggested Reading. Additional references are cited where appropriate.
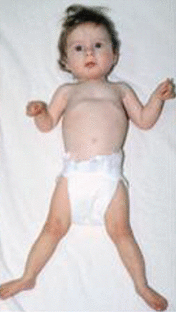
Craniofacial. Individuals with BOS have a characteristic facial appearance (see Suggestive Findings), although significant variability is observed. The most striking and consistent features are a prominent glabellar nevus flammeus (simplex) that fades with age, synophrys that becomes more prominent with age, hypotonic facies with full cheeks, and prominent or proptotic eyes. The majority of affected individuals also have hypertrichosis with rapidly growing hair and nails.
Other features widely, but variably, reported include cleft lip with or without cleft palate, high arched palate, widely spaced eyes (hypertelorism), depressed and wide nasal bridge, anteverted nares, and low-set ears with increased posterior angulation.
Growth. Mild intrauterine growth restriction has been noted, but many infants are products of healthy pregnancies with average or low-average birth weights. Poor growth is typically noted in the first year of life and is often clinically attributed to chronic emesis and feeding intolerance. Adequate nutrition does play a role in early growth, but even those without feeding intolerance typically display poor long-term growth.
Feeding. Most children have had feeding issues beginning in infancy and generally improving or resolving in early childhood. Historically, feeding issues have been presumed to be secondary to severe gastroesophageal reflux, but initial case reports did not produce diagnostic evidence of gastroesophageal reflux or demonstrate improvement on traditional antireflux therapies. Recent publications have suggested a neurogenic etiology, including cyclic vomiting with possible poor gastric motility, as the underlying cause of the chronic emesis [Russell et al 2015]. Given the frequency of emesis, there is a high risk for aspiration and dehydration, typically resulting in multiple hospitalizations.
Development and behavior. All affected individuals reported in the literature have severe-to-profound intellectual disability. Few are able to speak, but many have been able to express basic needs using augmentative and alternative communication (AAC) devices as well as gestures with associated basic vocalizations. Individuals with BOS often have a happy and pleasant demeanor [Russell et al 2015]. Typically, they are able to recognize caregivers and have a social, interactive nature. Most are unable to walk independently, but some have had success using walkers and braces in late childhood. Given the recent increase in rate of diagnosis due to genomic testing, it is expected that affected individuals with a milder phenotype may be reported in the future.
Neurologic. Seizures are common in individuals with BOS and are typically responsive to standard antiepileptic medications. Affected individuals have also been described to have truncal hypotonia with hypertonia of the extremities. A wide range of primary brain anomalies have been reported. Corpus callosum defects are the most common and range from hypoplastic to absent. Dandy-Walker malformation, delayed myelination, and enlarged ventricles have also been described.
Cardiovascular. Idiopathic and transient bradycardia as well as apnea were widely reported in the initial literature that predated the identification of the genetic cause of BOS. Cardiovascular deaths associated with bradycardia and apnea account for four (33%) of the 12 deaths published in the literature (although none of those individuals had a molecular confirmation of BOS). Other minor cardiac anomalies including septal defects and cardiac hypertrophy have also been described in a small number of affected individuals.
Respiratory. In addition to apnea and bradycardia, respiratory infections are common in infancy and account for about 42% of deaths (5/12). When chronic emesis is treated or improves with age, the rate of respiratory infections decreases [Russell et al 2015]. Tracheostomies have been necessary for some (see Management).
Sleep. Obstructive sleep apnea and sleep disturbances, including difficulty falling asleep and staying asleep, are frequently reported. Affected individuals with micrognathia may also exhibit tongue-based airway obstruction.
Ophthalmologic. Myopia, often severe, is common in individuals with BOS. Most affected individuals require corrective lenses in infancy. Retinal and optic nerve abnormalities including colobomas, retinal and optic nerve atrophy, and abnormal coloration of the retinas are also frequently reported.
Immunologic. Recurrent infections may be frequent in early life, although immunodeficiency has not been reported in the literature. The frequency of infections typically declines with age.
Urologic. Urinary retention and recurrent urinary tract infections have been reported in individuals with BOS [Russell et al 2015]. There also appears to be an increased risk for renal stones [Author, personal observation].
Skeletal. The typical BOS posture (see Suggestive Findings) is most notable in early childhood and usually becomes less obvious with age. Congenital contractures, dislocations, and pectus excavatum have also been reported.
Malignancy.
Isolated case reports suggest that children with BOS are at greater risk for Wilms tumor than the general population; large-scale epidemiologic studies have not been conducted due to the limited number of individuals diagnosed with BOS. Of three individuals with a clinical diagnosis of BOS and renal neoplasia, two had documented pathogenic variants in ASXL1 and bilateral Wilms tumor; Wilms tumor was diagnosed in one at age two years and in the other at age six years [Russell et al 2015]. Another individual with BOS had nephroblastomatosis on autopsy at age five months. This infant later underwent molecular genetic testing of ASXL1; no pathogenic variant was identified [Brunner et al 2000, Hoischen et al 2011]. Overall, Wilms tumor appears to affect about 7% of individuals with BOS. This risk estimate is based on a very small number of reported cases and thus is likely to change over time as larger cohorts of children and adults with BOS are investigated.
The only other neoplastic process reported was medulloblastoma in a child age five years with clinical features of BOS in whom a pathogenic ASXL1 variant was not identified [Hastings et al 2010, Hoischen et al 2011].
Other
Annular pancreas has been described in some affected individuals.
Gallstones have been reported in several affected individuals [Author, personal observation].
Historically, this condition had been associated with high infant mortality (27% based on data published before 2015), but the current survival rate is likely to be much better due to advances in pediatric care and more aggressive interventions [
Russell et al 2015].