NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
RefSeq Help [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2011-.
General Information about RefSeq
Where can I find more information about the NCBI RefSeq project?
The sequence of a RefSeq accession is identical to that of a GenBank accession. Will one be removed?
How do I cite the RefSeq project, a species-specific dataset, or individual RefSeq records?
How can I evaluate the support for a RefSeq transcript or protein?
Why aren't RefSeq records made for all organisms or for all of the loci available in Gene?
Why are some splice variants for my favorite gene missing in the RefSeq set?
Why is a transcript RefSeq record made from genomic sequence instead of available transcripts?
FTP Downloads
Why are there gaps in sequentially numbered files in the RefSeq release?
Where can I find a report of suppressed or replaced accessions?
Refseq Curation and Content
How are the immunoglobulin and T-cell receptor loci annotated for the human and mouse genomes?
Why is there a stop codon within the CDS annotated on NM_002084.3?
How is the content of RefSeq and Gene synchronized with the content of model organism databases?
How can I identify matches between RefSeq and Ensembl annotation?
How does RefSeq curation impact the Consensus CDS (CCDS) resource?
RefSeq Updates and Removal
How can I determine what information has changed when a RefSeq record has been updated?
Where can I find information about why an accession was removed?
My favorite RefSeq has been removed! How do I determine if it was replaced by a different RefSeq?
I searched for NM_001136525 but see that it has been replaced by NM_001136248. Why?
What causes the version number of a RefSeq record to change?
General Information about RefSeq
What is a Reference Sequence (RefSeq)?
The NCBI Reference Sequence (RefSeq) project provides sequence records and related information for numerous organisms, and provides a baseline for medical, functional, and comparative studies. Whereas the International Nucleotide Sequence Database Collaboration (INSDC, made up of GenBank, the European Nucleotide Archive, and the DNA Data Bank of Japan) represents an archival repository of all sequences, the RefSeq database is a non-redundant set of reference standards derived from the INSDC databases that includes chromosomes, complete genomic molecules (organelle genomes, viruses, plasmids), intermediate assembled genomic contigs, curated genomic regions, mRNAs, RNAs, and proteins. Please see The Reference Sequence (RefSeq) Project chapter in the NCBI Handbook for more details.
What are the distinguishing features of a RefSeq record?
RefSeq records are distinguished from INSDC records by:
Accession format: The most distinguishing feature of a RefSeq record is the distinct accession number format that begins with two characters followed by an underscore (e.g., NP_). INSDC accession numbers never include an underscore. Please see the description of RefSeq accession prefixes.
Comment: RefSeq records contain a COMMENT section that includes the term REFSEQ and identifies the record status, the source accession(s) used to derive the RefSeq sequence (if applicable), and the collaborating group, if any.
Nomenclature: RefSeq records consistently use official nomenclature for the gene feature, when available. When official nomenclature is not available for the species or for individual loci then names typically originate from the INSDC sequence(s) used to generate the RefSeq record, or from publications. Protein names originate from UniProtKB, from the Enzyme Commission, or from NCBI staff curation. Curation by NCBI staff also applies names and alternate aliases from a combination of sources for some records.
db_xrefs: inclusion of db_xrefs on the gene or other features provides links to other sources of information, such as OMIM, Gene, UniProt, CCDS, CDD, and model-organism databases.
DBSOURCE: protein records indicate REFSEQ as the DBSOURCE
How are RefSeq records provided?
Distinct processes are used to generate RefSeq records depending on the organism. The majority of RefSeq nucleotide records are derived solely from the primary sequence data submitted to the archival International Nucleotide Sequence Database Collaboration (INSDC). Protein sequence records represent conceptual translations of the annotated open reading frame (based on curation, collaboration, annotation provided on the nucleotide records or inferred via analysis). The distinct processes used to create RefSeq records include:
Entrez Genomes: This process flow provides genomic, RNA, and protein RefSeq records derived from assembled and annotated whole genome sequence data submitted to the INSDC. This pipeline provides all of the bacterial, viral, organelle, and plasmid RefSeq records and records for some eukaryotic genomes, including plants and fungi, as data becomes publicly available. Protein and transcript records are instantiated from the submitted genome sequence annotation or are predicted by NCBI’s bacterial or eukaryotic computational annotation process.
The Eukaryotic Genome Annotation pipeline: This process flow is an automated computational method that provides a copy of the submitted genome assembly in order to provide an annotated genome. RefSeq records may include chromosomes, intermediate assembled scaffolds and contigs, and transcripts and proteins. Depending on the species, genome annotation may reflect a mixture of transcript-based RefSeq records (below) and computationally predicted transcripts and proteins with varying levels of support from transcript or protein alignments. Please see The NCBI Eukaryotic Genome Annotation Pipeline for more details.
Curation-supported RefSeq pipeline: NCBI staff scientists provide curation support in several ways. Staff leverage the Protein Clusters database to apply consistent nomenclature to orthologous proteins, work with collaborating groups to better represent data ranging from whole genomes to paralogous genes, and react to feedback from users reporting sequence or name improvements.
NCBI curation staff also work closely with developer staff to provide genomic region, RNA, and protein RefSeq records for a subset of species grouped under the Bilateria node. Transcript and protein records are primarily derived from cDNA records submitted to the INSDC. This process flow is supported by a combination of bioinformatics and a significant level of manual curation.
Collaboration: Some RefSeq records are provided by collaborating groups. Different collaborations provide some fully annotated genomes or records for gene families or individual genes. Collaborations with official nomenclature groups, model organism databases, or other database groups also provide descriptive information, including gene symbols, names, publications, mapping data, feature annotation, database cross-references, and more.
Curated RefSeq records are made available in several status levels and represent a compilation of our current knowledge of a gene and its transcripts and proteins.
Where can I find more information about the NCBI RefSeq project?
Additional information about the RefSeq project is available in the NCBI Handbook (Chapter 18), the RefSeq web site, and publications [PubMed; PubMed Central].
How do I access RefSeq records?
RefSeq records can be retrieved by querying with an accession number, symbol or locus_tag, name, or by using Entrez Limits and Property terms. RefSeq records can be accessed through several NCBI resources including BLAST, Entrez (Nucleotide, Protein, Gene, Protein Clusters, BioSystems), Genome Data Viewer, and FTP as follows:
BLAST: Transcript, protein, and 'genomic region' (NG_ accession prefix) records are included in the nucleotide and protein non-redundant (nr) databases. RefSeq records only are also available in the Reference mRNA sequences or Reference proteins databases. BLAST against RefSeq genomic records is provided in the Reference genomic sequences database or via organism-specific BLAST pages.
Entrez: Entrez Nucleotide and Protein text query results (e.g., by gene symbol) may include both INSDC and RefSeq records. A filter link (located in the upper right area of the results page) is provided to restrict the display to the RefSeq subset. Alternatively, a query can be formatted to return only RefSeq records using the Limits page, or by querying with a Property such as “srcdb_refseq[property]”. Gene also supports queries with this RefSeq property as well as queries by RefSeq accessions, and Gene reports include information about annotated and current RefSeq records and a display option to view the exon coordinates of RefSeq transcripts that are annotated on a genomic record (see the Gene Table display). Entrez Genomes provides links to RefSeq chromosomes or large linkage groups. Refer to the Entrez Help for general information on constructing queries.
Genome Data Viewer: The NCBI Genome Data Viewer supports queries by RefSeq accession numbers and includes links to RefSeq records in other databases, when the annotated genome is available.
FTP: The complete RefSeq collection is distributed for FTP in bi-monthly releases. Intermediate daily updates are provided between release cycles. Additional weekly reports are available for some species, including human and mouse for which the RefSeq transcript and protein collections are updated more frequently. RefSeq nucleotide and protein records are available from the /refseq ftp directory. See the README file for a description of the directory contents and file formats. NCBI’s annotation of genomes represented in the RefSeq collection is also available as individual GFF3 files from the Genomes FTP site. More information concerning FTP access to the RefSeq collection is detailed below.
What is the difference between XM_ and NM_ accessions?
Accession numbers that begin with the prefix XM_ (mRNA), XR_ (non-coding RNA), and XP_ (protein) are model RefSeqs produced either by NCBI’s genome annotation pipeline or copied from computationally annotated submissions to the INSDC. These RefSeq records are derived from the genome sequence and have varying levels of transcript or protein homology support. They represent the predicted transcripts and proteins annotated on the NCBI RefSeq contigs and may differ from INSDC mRNA submissions or from the subsequently curated RefSeq records (with NM_, NR_, or NP_ accession prefixes). These differences may reflect real sequence variation (polymorphism), or errors or gaps in the available genome sequence. The support for model RefSeq records should be further evaluated by comparing them to other sequence information available in Gene, Related Sequences, and BLAST reports.
The genome annotation pipelines are automated and their predicted products may or may not be subject to manual curation, but the data may be refreshed periodically.
A complete description of RefSeq accessions prefixes can be found here.
What is an NG_ accession and why are they made?
Accession numbers that begin with the prefix NG_ represent genomic sequence records that are provided for some organisms to represent non-transcribed pseudogenes or genomic regions.
Pseudogene records: genomic records representing non-transcribed pseudogenes are provided for organisms that are in scope for NCBI’s eukaryotic genome annotation pipeline or that have an official nomenclature group providing nomenclature for pseudogenes. These records are defined by curation or collaboration with nomenclature groups and Pseudogene.org.
Genomic region records: genomic records are provided to represent RefSeqGene loci, haplotypes with differing gene content, standards that support phylogenetic classification, or genomic regions that are difficult to accurately annotate via NCBI’s eukaryotic genome annotation pipeline, such as nearly-identical paralogs, T-cell receptor loci, and immunoglobulin loci.
What sequence is used to define a RefSeq?
NCBI creates and updates RefSeq records from sequence data available through the INSDC. The COMMENT field on the RefSeq flat file record displays the INSDC accession number(s) used as the source sequence; however, for some organisms the information cited is the annotated genomic record or a locus_tag identifier from the annotated genomic record. Note that INSDC genomic records may include mRNA annotations but, unlike protein annotations, they are not instantiated as an accessioned record. In contrast, these transcripts are explicitly represented as accessioned RefSeq records.
For organisms of the Bilateria node that are part of the NCBI curation-supported pipeline, including human, mouse, rat, cow, and zebrafish, the source INSDC accession selected initially must be annotated with a complete coding sequence. Given more than one accession to choose from, the accession with longer UTR sequence is selected typically.
Reference sequence records are not intended to represent the historical 'first sequenced' record of a gene, although for genes with very limited available sequence data that is frequently true. PROVISIONAL records may be updated automatically to use a longer INSDC source nucleotide sequence that becomes available before the RefSeq record is fully reviewed. While PROVISIONAL RefSeq records do represent a single INSDC source nucleotide sequence, curated RefSeq records (with a status of VALIDATED or REVIEWED) are intended to represent the current state of knowledge as provided by the whole research community rather than by any one laboratory and may be constructed from multiple INSDC sequences to do so. Consequently, RefSeq records which have been manually curated or generated by NCBI automated pipelines may or may not match transcript and protein records in INSDC.
All INSDC submissions used to construct a RefSeq are listed in the COMMENT field on the flat file record. The PRIMARY block, displayed below the COMMENT field, provides the specific coordinates on the RefSeq record (REFSEQ_SPAN) and the corresponding coordinates of each INSDC submission (PRIMARY_IDENTIFIER and PRIMARY_SPAN). This information is provided for vertebrates and a small number of other species.
What is the difference between RefSeq and GenBank?
The GenBank archival sequence database includes publicly available DNA sequences submitted from individual laboratories and large-scale sequencing projects. GenBank is part of the International Nucleotide Sequence Database Collaboration (INSDC) along with the European Nucleotide Archive and the DNA Data Bank of Japan (DDBJ). Submitted sequence data is exchanged daily between the three collaborators to achieve comprehensive worldwide coverage. As an archival database, GenBank can be very redundant for some loci. GenBank sequence records are owned by the original submitter and cannot be altered by a third party.
RefSeq sequences are not part of the INSDC but are derived from INSDC sequences to provide non-redundant curated data representing our current knowledge of known genes. Some records include sequence information gathered from more than one INSDC record. Records may include sequence, descriptive information, publications, or feature annotation that is not available from any single INSDC record. RefSeq records are owned by NCBI and therefore can be updated as needed to maintain current annotation or to incorporate additional information. Also see the appendix provided in the NCBI Handbook, GenBank chapter.
Another distinction is that transcripts and proteins annotated on RefSeq genomic records are instantiated as separate records; in contrast, GenBank only instantiates the proteins annotated on genomic sequence records.
The sequence of a RefSeq accession is identical to that of a GenBank accession. Will one be removed?
No, both records will continue to be available. RefSeq and GenBank (a member of the INSDC) are separate databases, and both databases are available at NCBI.
RefSeq records are often quite similar to the source INSDC records they are based upon. Ongoing automatic processing to integrate additional information from external sources, such as nomenclature, together with curation by NCBI staff, may lead to an updated RefSeq record that incorporates more sequence data, biological annotations, and references, at which time the original source INSDC record and the corresponding RefSeq entry can be quite different. Thus, a curated RefSeq record may diverge in either sequence or descriptive information from the INSDC records and may include information that originates from multiple sources.
How can I quickly identify RefSeq records?
A RefSeq record is identified easily by inclusion of an underscore in the accession number.
Querying Entrez Nucleotide or Protein returns results in the default Summary format that includes a Filter option at the top right to restrict the results to only RefSeq records.
The formatted text line of a FASTA file includes the term ‘ref’ preceding the RefSeq accession.version number, where ‘ref’ indicates the database source is RefSeq. e.g.,
>ref|NM_000202.5| Homo sapiens iduronate 2-sulfatase (IDS), transcript variant 1, mRNA
If RefSeq is a non-redundant database why does my BLAST query return hits to more than one RefSeq accession?
It's not surprising to get BLAST hits to more than one RefSeq record. Depending on your query term and BLAST parameters you may get results that include alternative splice variants, paralogs, and orthologs. In addition, alternate strain-specific records or genomic records for particular gene regions are provided for some species.
How do I cite the RefSeq project, a species-specific dataset, or individual RefSeq records?
It is appropriate to cite the RefSeq accession number (with version), the NCBI Handbook, or the most recent RefSeq project article in a Nucleic Acids Research Database issue.
To cite an accession number: Ideally, any accession cited should indicate both the accession and version number. Citing the accession number alone does not provide a specific indication of the sequence if the sequence has been updated over time and the record has a version number greater than ‘1’. Note that the accession number format includes an underscore character (‘_’) and thus citing a RefSeq accession without the underscore is not accurate (i.e., NM 000014.4, or NM000014.4 are invalid citations). A correct citation is RefSeq Accession NM_000014.4.
To cite a species-specific dataset: Please cite the RefSeq FTP release number when you are working with a dataset that has been extracted from a RefSeq release FTP site (/refseq/release/). If your dataset definition does not strictly correspond to a RefSeq release then it is appropriate to indicate the specific method used to define the dataset and the date of the collection.
To cite the project: Please cite the NCBI Handbook as the most current and comprehensive description of the project. If the journal does not allow citation of electronic books then please cite the RefSeq article in the Nucleic Acids Research Database issue.
To cite the whole Handbook: The NCBI handbook [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2002 Oct. Available from http://www.ncbi.nlm.nih.gov/books/NBK21101
To cite the RefSeq chapter (Chapter 18): The NCBI handbook [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2002 Oct. Chapter 18, The Reference Sequence (RefSeq) Project. Available from http://www.ncbi.nlm.nih.gov/books/NBK21091
To cite the RefSeq NAR database issue article: O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover B, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O’Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016 Jan 4;44(D1):D733-45.
How can I evaluate the support for a RefSeq transcript or protein?
Supporting information for a Refseq transcript or protein is best determined by evaluating the results of a BLAST query.
For organisms with an annotated genome displayed in NCBI’s Genome Data Viewer, supporting transcripts can be aligned and visualized in the context of the genome sequence; use the Maps&Options dialog box to add one or more predefined sets of transcripts.
Pre-calculated reports are also available, including
Identical protein accessions reported on the right side of a GenPept protein record.
For human and mouse, a RefSeq transcript record may contain a structured comment indicating explicitly the INSDC transcript(s) that supports its exon combination, when aligned to the reference genome assembly using the alignment program Splign. At most, two supporting transcripts are displayed. The structured comment is found at the bottom of the COMMENT block on the GenBank formatted record.
e.g., NM_053271.2
##Evidence-Data-START##
Transcript_exon_combination :: AB021131.1 [ECO:0000332]
##Evidence-Data-END##
If supporting evidence for the exon combination of the full transcript is not available (for example, a RefSeq transcript with a terminal, untranslated region derived from a partial but otherwise consistently spliced INSDC transcript), supporting evidence for the exon combination of only the coding sequence is reported.
e.g., NM_003181.3
##Evidence-Data-START##
CDS_exon_combination :: AJ001699.1 [ECO:0000331]
##Evidence-Data-END##
The evidence codes cited, ECO:0000331 and ECO:0000332, are derived from the Evidence Code Ontology (ECO) vocabulary.
Why aren't RefSeq records made for all organisms or for all of the loci available in Gene?
A combination of factors influences whether RefSeq records are provided for an organism, including the availability and quality of a genome assembly, annotation for that assembly, the abundance of cDNA sequences in the INSDC, the relevance of the organism to medical and research communities, and input from the research community.
Genomic RefSeq records for nuclear chromosomes, organelles, bacterial and viral genomes, and naturally occurring plasmids are created when the sequence is submitted to the INSDC. If whole genome sequence is submitted without annotation, NCBI may provide annotation from its microbial or eukaryotic genome annotation pipeline.
Provision of RefSeq records for loci represented in Gene depends on the locus type and availability of sufficient sequence data. Not all records in Gene are in scope for the RefSeq project. This includes immunoglobulins, T-cell receptors, most repetitive elements, and records that represent a phenotype only. Lastly, records of an unknown locus type are not provided a curated RefSeq but may be represented with a computationally predicted model.
Why are some splice variants for my favorite gene missing in the RefSeq set?
RefSeq records that represent alternately spliced transcript variants are provided when there is experimental and/or published evidence in support of the full-length nature of the product. When transcript alignments (to the assembled genome) indicate that there is alternate splicing no assumption is made about the naturally-occurring combination(s) of alternate exons in the absence of full-length support. As a consequence, alternately spliced products are underrepresented in the RefSeq collection.
While NM_ and NR_ RefSeq records may be an underrepresented set of variants, model transcripts (XM_ and XR_) generated by NCBI’s eukaryotic genome annotation pipeline may provide other possible variant transcripts. An easy way to view the complete set of annotated RefSeq records for a particular eukaryotic gene is to go to the Gene record and view the customizable genome annotation graphic.
Requests to review additional transcript variants for a gene, and their possible representation in RefSeq, can be made using the Gene and RefSeq Feedback form. We encourage research groups to submit primary sequence data representing alternatively spliced transcripts to the INSDC.
Why is a transcript RefSeq record made from genomic sequence instead of available transcripts?
Transcribed RefSeq records (with NM_ or NR_ accession prefix) may be created in part or entirely from genomic accessions available in the INSDC for several reasons, including 1) to represent a polymorphism that is thought to be the better representative than what is observed among the transcript data, 2) to improve annotation on the RefSeq chromosome and contig accessions of highly related genes, 3) to provide a RefSeq record for a known gene where the transcript data is absent or incomplete but exon structure can be inferred from protein alignments or orthology, and 4) to facilitate RefSeq curation (e.g., using a single genomic range to extend a 3’ UTR rather than multiple, short overlapping ESTs).
What is RefSeqGene?
RefSeqGene, a subset of NCBI's Reference Sequence (RefSeq) project, defines genomic sequences of well-characterized genes to be used as a stable foundation for reporting mutations, for establishing conventions for numbering exons and introns, and for defining the coordinates of other biologically significant variation. RefSeq mRNA and protein sequences already support these functions but have the obvious weakness of not providing explicit coordinates for flanking or intronic sequence. RefSeq chromosome sequences also support these functions, but have awkwardly large coordinate values that may change if the sequence is updated. RefSeqGene sequences counter these drawbacks by providing gene-specific genomic sequence for each gene, as well as including upstream and downstream flanking regions. Sequences in the RefSeqGene set are intended to be well-supported, exist in nature, and, to the extent for which this is possible, represent a prevalent, 'normal' allele.
Search for RefSeqGene records via NCBI's Nucleotide database by adding RefSeqGene[keyword] to your query.
Please see About the NCBI RefSeqGene Project for more information.
What is a readthrough locus and how is it represented?
Please refer to Gene’s FAQ document.
NCBI’s annotation displayed on a genomic RefSeq record may include ‘unclassified transcription discrepancy’ and ‘unclassified translation discrepancy’ exceptions on the mRNA and CDS features, respectively. What do these exceptions mean?
These exceptions refer to the existence of one or more sequence discrepancies (e.g., mismatches, insertions, or deletions) that result in a difference between the transcript or translation product computed from the genomic RefSeq sequence compared to the sequence represented by the corresponding RefSeq mRNA or protein record. When the genomic RefSeq record is viewed using the Graphics display setting, the mRNA and protein features with these exceptions are shaded with a grey background (Figure 1). Add the RefSeq Alignments track using the Configure button of the Graphics display to view the general location of the discrepancies and to access additional information that is revealed by placing your cursor over the RefSeq alignment.
Sequence discrepancies may be the result of errors in the genomic sequence or errors in the mRNA or protein sequence. For human, mouse, and zebrafish, whose genome sequences are maintained by the Genome Reference Consortium (GRC), suspected errors in the genomic sequence may be reported to GRC staff for review. For other organisms, errors may be reported to the original sequencing group that submitted the assembly for consideration in a future update. Suspected errors in mRNA or protein sequences may be corrected, when supported by evidence, by contacting RefSeq staff using the Feedback form available on any Gene record.
FTP Downloads
What data are available for FTP download?
The complete RefSeq collection is distributed for FTP in bi-monthly releases on the RefSeq FTP site (ftp://ftp.ncbi.nlm.nih.gov/refseq/). The RefSeq FTP site provides bulk access to the entire RefSeq database, with data being distributed in several formats and organized by major taxonomic or molecular grouping as well as by type of sequence data (DNA, RNA, protein). The RefSeq release includes reports of the accessions included in the release, accessions removed since the prior release, statistics, files installed, and more. In addition, the RefSeq FTP site displays specific sub-sets and includes content that is not available in the genomes FTP site including: daily updates, RefSeqGene records, viruses, organelles (that are not part of a whole genome submission), the targeted ribosomal RNA project, RefSeq transcript and protein records that are not yet annotated on the corresponding genome, and autonomous non-redundant proteins (WP_ accession prefix) that are not yet directly annotated on a genome. See the README files and the RefSeq release notes for descriptions of directory contents and file formats. Releases are announced to the refseq-announce email list, on the RefSeq web site, and on NCBI’s Facebook and Twitter accounts.
Daily updates are provided between release cycles. We do not provide comprehensive daily FTP updates for information categories that are changing at high frequency, such as citations. Gene is considered the primary source for citation data and many RefSeq records report only a subset of those available.
Additional weekly reports are available for some species, including human and mouse, for which the RefSeq transcript and protein collections are updated at higher frequency. Weekly updates of the human RefSeqGene dataset are also available.
NCBI’s annotation of genomes represented in the RefSeq collection is also available from the Genomes FTP site. The Genomes FTP directories (‘all’, ‘genbank’, and ‘refseq’) provide data based on new or updated prokaryotic or eukaryotic genome assemblies, or updates to whole genome annotations. Over time, this space will include both historical and current assembly and annotation content. This FTP site is oriented on the genome assembly package and corresponds to the content in NCBI’s Assembly resource (http://www.ncbi.nlm.nih.gov/assembly/). The genomes FTP site facilitates access to both GenBank and RefSeq data – including genomic sequence, assembly structure details, accessioned annotated transcripts, and accessioned annotated proteins. RefSeq records that are not part of the genome annotation will not be included here. Please see the genomes FTP FAQ at www.ncbi.nlm.nih.gov/genome/doc/ftpfaq/ for more information.
Why are there gaps in sequentially numbered files in the RefSeq release?
The RefSeq release processing first produces a comprehensive set of ASN.1 files, ordered by tax_id and limited by a size constraint. These initial files (*.bna.gz files) are further processed to export the records by molecule and format type (creating files such as *.genomic.fna.gz, *.protein.faa.gz, etc.). Files with the same numerical increment are related by content. They are all derived from the same initial ASN.1 file (*.bna.gz). If the initial ASN.1 file does not include any records for a given molecule type, such as RNA sequence data, then the corresponding ‘RNA’ FASTA and flatfile records will not be found. This is not an error.
Release file names are not stable over time and cannot be compared between releases. For instance, as genome sequencing projects complete, the data representation in RefSeq may change with a corresponding change in the accession prefix and final release file name. For example, between releases 21 and 22, the draft WGS genome sequence available for Rhodobacter sphaeroides 2.4.1 was completed, resulting in a significant change to the RefSeq records because the accession series NZ_AAAE was retired and a series of accessions with the NC_ prefix was used to represent the finished genome and plasmids.
Note that the set of files provided for a RefSeq release does include a report of files installed. See the README in the RefSeq release catalog directory
Where can I download the human proteome or transcriptome?
A current comprehensive transcript or protein dataset can be downloaded for select species from the RefSeq FTP site. Human, mouse and several other species are included because they are part of the curation-supported, transcript-based Bilateria group, with an expected higher frequency of new or updated records. Data is provided on a weekly basis for transcript and protein records in FASTA and GenBank flat file formats.
Where can I find a report of suppressed or replaced accessions?
The bi-monthly RefSeq release includes a report of accessions that have been suppressed or replaced since the previous release (ftp://ftp.ncbi.nlm.nih.gov/refseq/release/release-catalog/ - see the README file). In addition, reports can be generated for a given list of accessions using the robust functions provided by NCBI’s E-utilities. An ESummary example:
http://eutils.ncbi.nlm.nih.gov/entrez/eutils/esummary.fcgi?db=nucleotide&id=4502272
indicates that NM_000703.1 (gi:4502272) was replaced by NM_152296. The relevant lines are:
<Item Name="Status" Type="String">replaced</Item>
<Item Name="ReplacedBy" Type="String">NM_152296</Item>
For a small subset of taxa, there is also a weekly report of RefSeq accessions that were suppressed or replaced by another:
ftp://ftp.ncbi.nlm.nih.gov/refseq/special_requests
Consult the README file for more information and the file taxid2speciesname for the taxonomic scope of this reporting.
Refseq Curation and Content
How can I tell if a RefSeq record has been curated?
Curated records can be identified by the RefSeq status code of REVIEWED or VALIDATED. This status is displayed in the COMMENT area of the record. Records that have been supplied by a collaborating group are marked as curated by that group, with the group or database identified.
What does a REVIEWED status mean?
Refseq records are made available in several status levels
Reviewed records represent a compilation of our current knowledge of a gene and its transcripts and protein products. These records, reviewed by NCBI staff scientists or collaborating groups, are analogous to a ‘review article’.
Some enhancements to a reviewed record might include the addition or removal of sequence data (to extend UTRs or to remove vector or linker sequence, for example), the addition of relevant publications or nucleotide and protein features, and summary text describing the gene’s function.
When a record is reviewed, sequence data from more than one record may be combined to construct a more complete mRNA record. The review process includes reading of the primary literature, reviewing available sequence data, creating alternative spliced RefSeq records, and providing functional information. Transcript variant records are only made when there is information available on the full-length nature of the product; if multiple, alternate exons are found through the length of the gene, no assumption is made about the combination of alternate exons that exists in vivo. Therefore, the RefSeq collection does under-represent alternatively spliced products.
For the NCBI curation-supported pipeline, the review process includes analysis of all sequences representing the gene, at that time. The list of accessions representing the gene may be expanded, errors in the accession-to-gene association identified and corrected, and problematic accessions, such as chimeric mRNAs, flagged. The curated list of accessions is available in Gene although it is not intended to be a comprehensive list of related sequences; those sequences can be found by a BLAST analysis or by using pre-calculated reports of related sequences provided as links in Entrez Nucleotide and Entrez Protein, or BLink.
Why is the gene symbol or protein name in a RefSeq record different from the symbol or name used in related INSDC records?
RefSeq records use the gene symbols and protein names provided by the original INSDC submission, collaborating or other authoritative groups, including UniProtKB and the Enzyme Commission, or the official nomenclature authority for an organism, if available. For example, the human RefSeq collection uses gene symbols and names supplied by the HUGO Gene Nomenclature Committee. The reference protein name by default is taken from the reviewed UniProtKB (i.e., Swiss-Prot) record associated with the Gene record, if available. RefSeq records may also include alternate symbols and names.
INSDC submissions represent archival sequence data provided by the originators of the data. Submitters maintain editorial control over their records and decide what gene symbol and name to use. Some submitters consult nomenclature authorities to obtain the official gene symbol and name while others may not, or may not update their submissions if the official nomenclature changes. Therefore, it is possible that INSDC records for a given gene may use different gene symbols and names.
What feature annotation is included on RefSeq records?
RefSeq records may include any of the features and qualifiers that are used for GenBank records. Features commonly found on RefSeq records include genes, mRNA or other RNAs, variations (from dbSNP), polyadenylation signals and sites, proteins (CDS), conserved domains (from CDD), and signal and mature peptides, where appropriate. Some feature types are provided for certain species, including propagation of a subset of features from Swiss-Prot to protein records, and exon annotation on human and mouse genomic and transcript records.
How are exons annotated on RefSeqs?
Exon feature annotation is provided on transcript records for human and mouse, and RefSeqGene records. Exon features are determined by aligning RefSeq transcripts for a gene to the assembled genome using the program Splign (Kapustin et al., 2008).
Exon numbering is only reported on RefSeqGene/LRG records. It is based only on the exons represented by the reference standard cDNA(s) for the RefSeqGene/LRG, from 5’ to 3’. Overlapping exons from multiple transcript variants are differentiated by a letter suffix on the RefSeqGene/LRG (e.g., 2a, 2b) but not on the individual transcript records.
The practice of providing exon numbers on all RefSeq transcript records for human and mouse was discontinued in May 2013. These exon numbers were based on ALL the exons known for a gene but were not stable because they were re-calculated as new exons for a gene were discovered. This caused confusion. The RefSeqGene/LRG mechanism will provide stable exon numbers for human genes because reference standard cDNAs change infrequently, and not at all when the LRG is published.
How are PubMed IDs selected for inclusion in RefSeq?
Many RefSeq records simply copy the citation information from the INSDC record that was used to generate the RefSeq. Citations for some species are more actively managed by either a collaborating group or by NCBI staff. For example, citations on mammalian transcript and protein RefSeq records are displayed according to the following: a) if RefSeq curation staff have marked specific citations to include on the RefSeq record, then they are always included; b) otherwise, from the set of publications associated with the GeneID, the RefSeq record displays the five oldest and five newest publications with a comment directing users to access the full bibliography for the gene in the Gene database. Publications are managed at the level of the gene; thus, a set of related RefSeq transcript variants for a gene include the same publications.
How are the immunoglobulin and T-cell receptor loci annotated for the human and mouse genomes?
Immunoglobulin (IG) and T cell receptor (TCR) genes are represented by genomic RefSeqs (NG_ accession prefix); transcript and protein RefSeqs are not made for these genes. In general, each multi-gene locus (e.g., IGH@) is represented by one genomic RefSeq that includes annotation for all the V, D, J and C genes that constitute the locus. Each V, D, J and C gene is annotated with a gene and a CDS feature as well as the appropriate V, D, J or C segment feature. RefSeq uses the defining sequence and nomenclature assigned by the ImMunoGeneTics database. The RefSeq may also include annotation for IG and TCR enhancers, when known, as well as annotation for other genes within the genomic region. The sequence of the genomic RefSeq is based on the components used in the reference assembly. To annotate these loci on the chromosomes of the reference assembly, the manually annotated NG_ record is aligned to the reference assembly and the annotation is lifted over. This process is also used to annotate any alternate assembly; however, the final result is less optimal due to underlying differences in the assemblies.
What is NMD? Why are some transcripts that may be subject to NMD represented in RefSeq, and others not?
Generally, a cDNA with a termination codon more than 50 nucleotides upstream of the last splice junction is believed to be subject to nonsense-mediated mRNA decay (NMD). RefSeq curators review literature, transcript alignments to the genome sequence, protein homology, and genome conservation in the course of deciding whether to represent a protein derived from a transcript that may be subject to NMD. The following curation guidelines are considered:
If a gene is known to be protein-coding, the most supported protein is represented regardless of it being encoded by a transcript that may be subject to NMD. These RefSeq records have the NM_ and NP_ accession prefix for the transcript and protein, respectively.
If a protein can be encoded by a transcript that is unlikely to be subject to NMD, any additional transcript variant that may be subject to NMD is represented as a non-coding RNA (with the NR_ accession prefix).
If review determines that a gene is unlikely to be protein-coding and that all transcripts are candidates for NMD, the gene will be updated to a noncoding RNA gene and be represented by a noncoding RNA (with the NR_ accession prefix). Additional data, as identified, may trigger re-evaluation.
Historically, when curatorial review identified an existing RefSeq as a NMD candidate, the NM_/NP_ records were suppressed. This policy was later changed to replace the existing NMD candidate RefSeq with a non-coding RNA record (with the NR_ accession prefix) and making the NM_ record secondary to it.
This policy for representing NMD transcripts is not yet universally applied across all of the eukaryotic taxa included in RefSeq; it is currently applied to the vertebrates.
Why is there a stop codon within the CDS annotated on NM_002084.3?
The GPX3 gene (GeneID 2878) encodes a protein that includes the amino acid selenocysteine. Selenocysteine is encoded by the codon ‘tga’, which is normally read as a stop codon. NM_002084.3 is explicitly annotated as a selenoprotein; the RefSeq Attribute ‘protein contains selenocysteine’ is displayed in the COMMENT block, and the translation exception qualifier on the CDS feature (transl_except=(pos:434..436, aa:Sec) identifies the location of the stop codon that encodes selenocysteine (which appears as a ‘U’ in the amino acid sequence). In addition, for transcripts and proteins in the Bilateria group, the location of the selenocysteine codon or amino acid residue is annotated as a misc_feature or Site feature, respectively.
Why does the DEFLINE of a predicted model RefSeq include the phrase "PREDICTED: LOW QUALITY PROTEIN"?
Model RefSeq proteins (with XP_ accession prefixes) which include “PREDICTED: LOW QUALITY PROTEIN” in the DEFLINE are sequences for which the corresponding XM_ has been modified relative to the genome sequence to correct for possible protein-altering mismatches or indels in the genome sequence. These mismatches or indels may arise from errors in the assembled genome sequence and can result in frameshifts and/or nonsense codons in the protein translation. Evidence supporting an XP_ may include the gene type for the species (i.e., protein-coding or otherwise) as determined by NCBI staff or collaborators, as well as data from orthologs and protein alignments. It is important to note that users should critically evaluate “corrected” XP_ models based on supporting evidence. In addition to “PREDICTED: LOW QUALITY PROTEIN” in the DEFLINE, newer models include "corrected model" as a KEYWORD and a structured comment detailing the correction.
An example of a “PREDICTED: LOW QUALITY PROTEIN” XP_ exists for the cat desert hedgehog gene (DHH, GeneID 101095751). When interpreting the combination of transcript and protein alignments, it is clear that there is strong homology to the conserved DHH gene but the homologous full-length protein cannot be derived directly from the genome sequence as the C-terminus of the protein would be frameshifted. By introducing a single 'n' base after position 1225 of XM_003988641.2 the reading frame is restored, yielding a protein (XP_003988690.1) that has strong homology with that of other mammals. Note that since DHH orthologs have their best alignment to the cat DHH locus and because the DHH protein cannot be generated from some other location of the genome, this gene is considered in cat to be protein-coding rather than a pseudogene.
How is the content of RefSeq and Gene synchronized with the content of model organism databases?
Both Gene and RefSeq records are updated daily when new information is received from an external source, including model organism databases. The scope of new information and the timing of its receipt are variable. For some organisms, updates may affect single gene records; changes to individual human and mouse records from the HUGO Gene Nomenclature Committee and Mouse Gene Nomenclature Committee, respectively, are received daily, for example. For other organisms, updates to Gene and RefSeq records coincide with a periodic new data release from a model organism database; updates from FlyBase to Drosophila melanogaster records are one example and may affect a large number of records. There may be a lack of synchronization between Gene and RefSeq compared to the model organism database depending on the frequency of data release.
How can I identify matches between RefSeq and Ensembl annotation?
Matches between NCBI and Ensembl annotations can be found in several ways using data provided in Gene, including: the Reference Sequences section of the Full Report display; by using the Gene "matches Ensembl" index property in a query; and in the gene2ensembl FTP file.
A summary of species’ whose annotations have been compared, including release and assembly information and the date when the comparison was last performed, can be found at:
ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/README_ensembl
Matching NCBI and Ensembl annotations is based on comparison of RNA and CDS features. For a transcript or protein to be identified as a match between RefSeq and Ensembl, there must be at least 80% overlap between the two. Furthermore, splice site matches must meet certain conditions: either 60% or more of the splice sites must match, or there may be at most one splice site mismatch.
How does RefSeq curation impact the Consensus CDS (CCDS) resource?
The consensus CDS (CCDS) project is a collaboration between the RefSeq group at NCBI, WTSI Havana curators, Ensembl, and UCSC that identifies human and mouse proteins that are annotated identically by these organizations on the reference genome assembly and pass quality assessment metrics. The matched annotation dataset represents RefSeq annotation compared to the union of WTSI gene models that are manually curated by the Havana curation group and Ensembl Genebuild models. Once a consensus CDS is identified, it is assigned an identifier and further modifications to the CDS annotation coordinates on the reference genome are done by collaborative agreement between NCBI, WTSI, and UCSC. Thus, for human and mouse RefSeq records that cite a CCDS identifier, updates that change the CDS coordinates, such as using an alternate translation initiation site or alternate exon, have been jointly agreed upon by members of the CCDS collaboration.
RefSeq Updates and Removal
How frequently are RefSeq records updated?
Updates become available on a daily basis. Records are updated, and new records are added, at different frequencies for different species. For some species, including human and mouse, individual records are updated (or added) and released on a daily basis following ongoing staff review or nomenclature changes. For other species, the entire RefSeq dataset is generated when a new annotated genome becomes available in the public archives, or the dataset may be updated if a collaborating group provides an update for the annotated genome.
How can I determine what information has changed when a RefSeq record has been updated?
Use NCBI’s Revision History report to determine how a sequence or its feature annotation has changed over time. From a nucleotide or protein display page, click on “Display Settings” in the top left hand corner. This opens a menu listing that includes 'Revision History', a page providing links to display each update, including minor updates that do not change the sequence. Tools are provided to compare any two versions either by displaying highlighted differences in text details for each update, or via a sequence alignment. Note that the tool support is optimized for shorter sequences rather than whole chromosomes. For example, see the revision history for NM_002020.
A RefSeq for my favorite gene has a problem with the sequence or annotation (or a RefSeq record isn’t available). What should I do?
We welcome feedback, suggestions, and error reports to help maintain the quality of the RefSeq collection and the Gene database. Contact us using the Gene and RefSeq Feedback form.
Because RefSeq records are created from submissions to the International Nucleotide Sequence Database Collaboration (INSDC), any sequence data which contributes to defining the gene, splice variants, and feature annotation is very useful. Submitting sequence data to the INSDC, composed of the DNA DataBank of Japan (DDBJ), the European Molecular Biology Laboratory (EMBL), and GenBank at NCBI, is straightforward. These three organizations exchange data on a daily basis, so submission to one is sufficient.
Sequence data can be submitted to GenBank using one of their submission tools. More information is available at the GenBank submission page.
Why are RefSeq accessions removed?
RefSeq records are removed for a variety of reasons related to curation decisions and updates to genome annotation. A comment that often explains the reason for removal is displayed on the Summary and GenBank flat file display of the removed record. For RefSeqs of Bilateria node organisms that have been removed by curation staff, this comment is also displayed in the Reference Sequence section of the Gene record. Additional information about the removal may be available for some accessions by contacting the NCBI help desk.
RefSeq records may be removed due to:
Replacement: A record may be replaced by another record to remove redundancy. Replacement events are indicated by the explanatory comment and the appearance of the secondary (replaced) accession number on the ACCESSION line, following the primary accession number. For example, XM_001232266.1 was replaced by NM_001080874; NM_001080874 is the primary accession number and XM_001232266 is a secondary identifier.
Suppression by curation staff: A record may be suppressed if a curator determines there is insufficient support for the record, or if the record is determined to be out of scope for the RefSeq project. For example, suppression may occur if a record represents the wrong protein (e.g., was annotated in the wrong frame), is no longer thought to encode a protein (e.g., a protein-coding locus was updated to a pseudogene locus), or is considered out of scope (e.g., a transcribed ALU sequence, a T-cell receptor, or a chimeric sequence). Examples include NM_001013738.1 and NM_198519.1.
Suppression by automatic processing: A record may be suppressed coincident with whole genome annotation updates. This may happen for a variety of reasons, including a) for updates to some whole genome shotgun assemblies that lack robust tracking methods, all previous genomic contig RefSeq accessions are suppressed and new contig accessions are assigned (e.g., NW_633586.1); b) if a previously annotated model is no longer predicted; and c) if an update to a genome assembly results in removal of a redundant contig, along with any associated annotation.
Any replaced or suppressed record will always be retrievable from the public sequence databases. A BLAST analysis in many cases will help identify a replacement accession, if available.
Where can I find information about why an accession was removed?
Removed records are clearly identified by a brief text description included in query results and at the top of the Entrez Nucleotide or Protein displays. The description varies depending on whether the record was removed due to batch processing (e.g., following an update to whole genome annotation) or removed due to a curation decision. For example (see XM_221470.3), the default text provided for suppressions related to a whole genome update is:
Record removed: This record was removed as a result of standard genome annotation processing. See the genome build documentation at http://www.ncbi.nlm.nih.gov/genome/guide/build.html for further information, or contact info@ncbi.nlm.nih.gov.
Additional information may be available for some accessions and can be obtained by contacting the NCBI help desk.
My favorite RefSeq has been removed! How do I determine if it was replaced by a different RefSeq?
A BLAST analysis, particularly one taking advantage of the RefSeq mRNA sequence or RefSeq protein sequence databases and specifying the organism (available as options when choosing the search set) is a rapid, accurate means to identify a new RefSeq accession that may be the replacement for a withdrawn, removed, or suppressed RefSeq record. If necessary, additional information may be available for some accessions and can be obtained by contacting the NCBI help desk.
Also note that suppressed RefSeq records remain retrievable from the INSDC databases when retrieving by accession.version.
I searched for NM_001136525 but see that it has been replaced by NM_001136248. Why?
A record may be replaced by another record to remove redundancy. This is a common occurrence following the merging of two Gene records and is clearly indicated in Entrez Nucleotide and Protein databases by a comment in the query results and the page header of the GenBank display (for example, NM_001136525.1). This information is also indicated on the retained RefSeq record by the appearance of the replaced accession number on the ACCESSION line, following the primary accession number. There may be more than one replaced accession listed. As displayed on the flat file [primary accession, then replaced accession(s)]:
ACCESSION NM_001136248 NM_001136525
What causes the version number of a RefSeq record to change?
A version number change (e.g., NM_111111.1 -> NM_111111.2) occurs to a RefSeq record when there is any update to the sequence of that record. Sequence updates include the alteration, addition, or removal of nucleotides or amino acids from a record. Please note that other alterations such as updates to annotation or associated publications will not trigger a version number change. Also, nucleotide (NM_, XM_) and protein (NP_, XP) records for the same transcript may not have the same version number after an update. For example, an update to the 5’ UTR of a RefSeq nucleotide record would cause a version number change for the updated nucleotide record but not for the corresponding protein record. Conversely, a change in the annotated start site of the coding sequence without a change in the underlying nucleotide sequence would cause a version number update for the NP_ but not for the NM_.
What updates to RefSeq records need a simple version number change and which require a new accession number?
The following cases require a simple version change:
All other cases require the old record to be suppressed rather than updated, and a new record with a new accession number to be created. In addition, RefSeqGene records cannot be updated when the exon definition or protein length is changed.
Figures
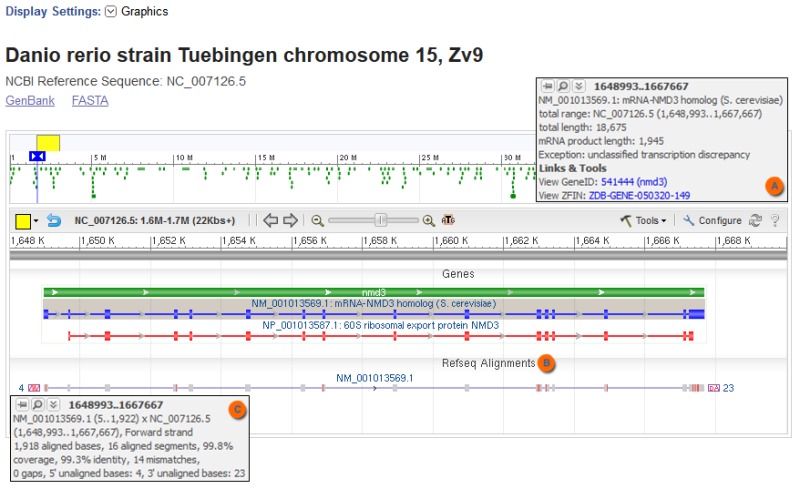
Figure 1.
Compared to the genomic sequence of zebrafish chromosome 15 (NC_007126.5), the RefSeq transcript NM_001013569.1 contains mismatches, as indicated by the grey background and the unclassified transcription discrepancy exception noted in the ‘mouseover’ text (A). Further information about the number and location of mismatches (red vertical lines on the RefSeq transcript) is provided by the RefSeq Alignments track (B), both graphically and by additional ‘mouseover’ text (C).