CHILD Syndrome
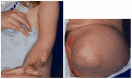
CHILD syndrome is characterized by unilateral ichthyosiform skin lesions typically with sharp midline demarcation with ipsilateral limb defects, onychodystrophy, and periungual hyperkeratosis (see ). Some individuals have scoliosis, joint contractures, central nervous system (CNS) anomalies, and congenital heart defects. CHILD syndrome predominantly affects females and is usually male lethal during gestation. To date, more than 60 individuals have been reported with CHILD syndrome.
Photographs of a female with CHILD syndrome A. Upper left limb. Note the forearm hypoplasia, ectrodactyly, onychodystrophy, and characteristic ichthyosiform skin lesions with yellow scales.
Dermatologic findings
Skeletal features
Limbs. Ipsilateral hypoplasia of the limbs varies from shortening of metacarpals and phalanges and sometimes other long bones (hypomelia) to absence of the entire limb (amelia). Joint contractures are reported. Oligodactyly can be seen; polydactyly and syndactyly have been rarely reported. The skeletal involvement is right-sided in around two thirds of individuals, but can be left-sided or, rarely, bilateral [
König et al 2002,
Hummel et al 2003,
Mi et al 2015,
Rossi et al 2015].
Other musculoskeletal defects (generally evident in infancy) include skeletal asymmetry and scoliosis. Incomplete development (hypoplasia) or absence of vertebrae, pelvis, ribs, mandible, and skull has also been reported [
Bornholdt et al 2005].
Punctate calcifications of cartilaginous structures. Unilateral punctate epiphyseal calcifications (chondrodysplasia punctata) in the pelvis, ribs, vertebrae, and extremities have been reported and are usually seen in the affected limb or body part. These can be visible on radiographs in infancy. In one child, the punctate calcifications were reported to have disappeared completely by age two years [
Happle et al 1980]; it is not known whether stippling always disappears. Ipsilateral stippling has also been observed in the sella turcica and the laryngeal, nasal, and thyroid cartilage [
Happle et al 1980,
Grange et al 2000].
Other structural anomalies
Other findings. Reported additional findings include hearing loss, absence of facial muscles, and unilateral hypoplasia of the thyroid gland, adrenal glands, ovaries, and fallopian tubes [Happle et al 1980, König et al 2002, Bornholdt et al 2005]. Bilateral optic atrophy has been reported in one individual [Knape et al 2010], as have thrombocytosis and congenital bilateral dislocation of the hip [Chander et al 2010]. Ipsilateral vocal cord paralysis and liver lobe and spleen hypertrophy have been reported [Bornholdt et al 2005]. Small intestinal mucosal xanthoma have also been reported [Ryan et al 2013, Tan et al 2022]. Teeth are typically normal.
Prognosis. It is unknown whether life span in females with CHILD syndrome is abnormal. Based on current data, life span is not limited by this condition, as several adults have been reported. CHILD syndrome was reported with phenotypes ranging from mild to severe in three different generations, with the oldest reported individual clinically and molecularly diagnosed at age 82 years [Bittar et al 2006]. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with this condition are underrecognized and underreported.
Hemizygous males. CHILD syndrome-associated NSDHL pathogenic variants are usually lethal to males during gestation. A few males with CHILD syndrome have been reported, and survival in these instances has been attributed to postzygotic somatic mutation in NSDHL. One reported male with the typical skin findings seen in females and normal development was mosaic for NSDHL pathogenic variant c.262C>T (p.Arg88Ter) [Bornholdt et al 2005].
CK Syndrome
CK syndrome is characterized by intellectual disability, behavior issues, seizures, characteristic facial features, thin habitus, and ocular manifestations in males. To date, 25 affected males from three unrelated families have been reported [Garg et al 2021].
Development. Affected males have mild-to-severe intellectual disability. Most cannot speak.
Neurobehavioral/psychiatric manifestations. Most males with CK syndrome manifest aggression, attention-deficit/hyperactivity disorder (ADHD), and irritability. These behaviors appear in infancy and early childhood. According to the Autism Diagnostic Review (ADI-R) and the Autism Diagnostic Observation Schedule (ADOS), affected males do not fulfill the criteria for an autism spectrum disorder.
Neurologic findings. All affected males have developed seizures in infancy. These range from multiple daily episodes of brief unresponsiveness associated with staring and facial and/or limb twitching to prolonged generalized tonic-clonic seizures. These likely arise from cerebral cortical malformations that, by MRI examination, are most consistent with polymicrogyria. Spasticity, tetraparesis, development of contractures, and sensory neuropathy have also been reported [Garg et al 2021].
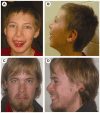
Characteristic craniofacial features (see ). All affected males have microcephaly (greater than 2-3 standard deviations below the mean). Other features include plagiocephaly, a long, thin face, almond-shaped and upslanted palpebral fissures, epicanthal folds, posteriorly rotated ears, prominent nasal bridge, high palate, dental crowding, micrognathia, and malar hypoplasia.
A male age 11 years (A, B) and a male age 22 years (C,D) with CK syndrome. Note the long thin face, epicanthal folds, almond-shaped palpebral fissures, prominent nasal bridge, and micrognathia. The long thin face becomes more apparent with age.
Skeletal manifestations and growth. All males with CK syndrome have a thin habitus, and relatively long, thin limbs, fingers, and toes. Some individuals have scoliosis and kyphosis. Hyperextensibility can be seen as well. Linear growth is normal, and height of affected individuals is average for parental heights.
Ocular findings. Strabismus is common. Optic atrophy is also seen.
Other. Left ventricular concentric hypertrophy has been reported [Garg et al 2021].
Prognosis. It is unknown whether life span in males with CK syndrome is abnormal. One reported individual is alive at age 62 years [du Souich et al 2009], demonstrating that survival into adulthood is possible. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with this condition are underrecognized and underreported.
Heterozygous females. Females heterozygous for a CK syndrome-related NSDHL pathogenic variant have normal physical features, intellect, and brain imaging but may display behavioral problems including irritability and aggression [Herman & Kratz 2012].