Huppke-Brendel Syndrome
Parayil Sankaran Bindu, MD, DM, FRACP, PhD, Shwetha Chiplunkar, MBBS, VP Vandana, PhD, Madhu Nagappa, MD, DM, Periyasamy Govindaraj, PhD, and Arun Taly, MD, DM.
Author Information and AffiliationsInitial Posting: June 13, 2019.
Estimated reading time: 13 minutes
Summary
Clinical characteristics.
Huppke-Brendel syndrome (HBS) is characterized by bilateral congenital cataracts, sensorineural hearing loss, and severe developmental delay. To date, six individuals with HBS have been reported in the literature. All presented in infancy with axial hypotonia; motor delay was apparent in the first few months of life with lack of head control and paucity of limb movement. Seizures have been reported infrequently. In all individuals described to date serum copper and ceruloplasmin levels were very low or undetectable. Brain MRI examination showed hypomyelination, cerebellar hypoplasia mainly affecting the vermis, and wide subarachnoid spaces. None of the individuals reported to date were able to sit or walk independently. All affected individuals died between age ten months and six years.
Diagnosis/testing.
The diagnosis of HBS is established in a proband with characteristic features (bilateral congenital cataracts, sensorineural hearing loss, severe developmental delay, very low serum copper and ceruloplasmin levels) and biallelic (compound heterozygous or homozygous) pathogenic variants in SLC33A1 identified by molecular genetic testing.
Management.
Treatment of manifestations: Cataract extraction is indicated in the first few months of life; early feeding tube placement to manage difficulties with swallowing, ensure adequate nutrition, and reduce the risk of aspiration; developmental intervention; physiotherapy to maintain muscle function and prevent contractures.
Surveillance: Periodic developmental and neurologic assessment; nutritional and growth evaluation; hearing evaluation; ophthalmologic evaluation; orthopedic evaluation for increased risk of scoliosis and contractures.
Genetic counseling.
HBS is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the SLC33A1 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Formal diagnostic criteria for Huppke-Brendel syndrome (HBS) have not been established.
Suggestive Findings
HBS should be suspected in individuals with the following clinical, radiographic, electrophysiologic, and laboratory findings.
Clinical findings
Bilateral congenital cataract
Nystagmus
Sensorineural hearing loss
Severe developmental delay / intellectual disability
Hypotonia
Seizures, hypopigmented hair, and hypogenitalism (reported infrequently)
Radiographic findings on brain MRI examination
Cerebellar hypoplasia
Hypomyelination
Wide subarachnoid spaces
Electrophysiologic findings. Brain stem auditory evoked potentials show absent wave forms.
Laboratory findings
Note: Identification of low serum copper and ceruloplasmin levels may be problematic in infants younger than age six months given the normally low serum concentration in all children at this age.
Establishing the Diagnosis
The diagnosis of HBS is established in a proband with the above Suggestive Findings and identification of biallelic pathogenic (or likely pathogenic) variants in SLC33A1 by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic SLC33A1 variants of uncertain significance (or of one known SLC33A1 pathogenic variant and one SLC33A1 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of HBS is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of HBS has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of HBS, molecular genetic testing approaches can include single-gene testing or use of a multigene panel.
Single-gene testing. Sequence analysis of SLC33A1 detects missense, nonsense, and splice site variants and small intragenic deletions/insertions; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis first. If only one or no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
A multigene panel that includes
SLC33A1 and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Option 2
When the diagnosis of HBS is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Huppke-Brendel Syndrome
View in own window
| Gene 1 | Method | Proportion of Probands with Pathogenic Variants 2 Detectable by Method |
|---|
|
SLC33A1
| Sequence analysis 3 | 6/6 probands |
| Gene-targeted deletion/duplication analysis 4 | Unknown 5 |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include missense, nonsense, and splice site variants and small intragenic deletions/insertions; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 5.
No data on detection rate of gene-targeted deletion/duplication analysis are available.
Clinical Characteristics
Clinical Description
Huppke-Brendel syndrome (HBS) is characterized by cataract, sensorineural deafness, and severe developmental delay in all reported individuals. To date, six individuals with HBS have been reported in the literature [Horváth et al 2005, Huppke et al 2012, Chiplunkar et al 2016].
Ocular features. Bilateral congenital cataracts were reported in all affected individuals. Affected individuals presented with poor visual fixation and rotary nystagmus. Two individuals underwent cataract extraction in early infancy; there was improvement in visual fixation and nystagmus in one child [Horváth et al 2005] and no improvement in vision in the other [Chiplunkar et al 2016].
Sensorineural hearing loss manifests during infancy. Brain stem auditory evoked potentials in two individuals showed absent waveforms. Otoacoustic emissions were absent bilaterally in one individual.
Neurologic features. Axial hypotonia was present in all infants. Motor delay was apparent in the first few months of life in all reported individuals. Lack of head control and paucity of movements in the limbs were evident. None of the individuals reported to date were able to sit or walk independently. None learned to speak. Developmental progress has been reported only in one individual who received copper histidinate therapy from age five months [Horváth et al 2005]. Follow up at age 13 months in this individual showed good head control, rolling over, reaching out for objects, and improved alertness and communication.
Deep tendon reflexes were normal and symmetric.
Two of the six reported individuals had seizures.
Orthopedic complications. Affected individuals are at increased risk for scoliosis and joint contractures [Author, personal communication].
Other. Hypopigmented hair and hypogenitalism were reported in one individual [Chiplunkar et al 2016]. This child had micropenis with bilaterally descended testes. The hair was uniformly hypopigmented and sparse. Hair analysis under polarized light microscopy showed uniform and finely granulated melanin pigment and no clumps. There was no kinking or abnormal polarization.
Prognosis. All affected individuals died between age ten months and six years. Causes of death included pneumonia, kidney failure, and multiorgan failure. The cause of multiorgan failure was not reported.
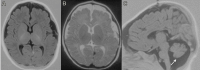
Neuroimaging. Brain MRI showed hypomyelination, cerebellar hypoplasia mainly affecting the vermis, hypoplasia of the temporal lobes, and wide subarachnoid spaces (see ) [Horváth et al 2005, Huppke et al 2012, Chiplunkar et al 2016].
Brain MRI in a child with HBS at age four months A, B. T1-weighted axial view (A) and T2-weighted axial view (B) show presence of myelin only in the posterior limb of the internal capsule, indicating delayed myelination. Widened subarachnoid spaces are (more...)
Histopathology on muscle biopsy
Subsarcolemmal proliferation and vacuolization in few type 1 fibers are reported. No typical ragged red fibers or ragged blue fibers or COX-negative fibers are seen [
Horváth et al 2005,
Chiplunkar et al 2016].
Biochemical measurements of the respiratory chain enzymes showed significantly reduced activity of COX with 30% residual activity in one individual [
Horváth et al 2005] and 35% residual activity in another [
Chiplunkar et al 2016].
Genotype-Phenotype Correlations
The number of individuals with confirmed pathogenic variants in SLC33A1 is too small to make any conclusive genotype-phenotype correlations.
Nomenclature
HBS may also be referred to as congenital cataracts, hearing loss, and neurodegeneration (CCHLND).
Prevalence
Prevalence of HBS is unknown. Only six affected individuals have been reported to date.
Differential Diagnosis
Table 2.
Disorders to Consider in the Differential Diagnosis of Huppke-Brendel Syndrome
View in own window
| Disorder | Gene(s) | MOI | Overlapping Clinical
/Laboratory Features | Distinguishing Clinical Features |
|---|
|
Disorders w/overlapping clinical & laboratory features
|
|
SUCLG1-related mtDNA depletion syndrome, encephalomyopathic form w/methylmalonic aciduria
|
SUCLG1
| AR |
| Dystonia, other extrapyramidal features, & basal ganglia signal changes on MRI differentiate this disorder from HBS. |
|
SUCLA2-related mtDNA depletion syndrome, encephalomyopathic form w/methylmalonic aciduria
|
SUCLA2
| AR |
| Dystonia, extrapyramidal features, & basal ganglia signal changes differentiate this disorder from HBS. |
Combined oxidative phosphorylation deficiency 11
(OMIM 614922) |
RMND1
| AR |
| Systemic features incl cardiac abnormalities &
nephropathy differentiate this disorder from HBS. |
|
Hypomyelination and congenital cataract
| HYCC1 (FAM126A) | AR |
| Normal early development & normal hearing distinguish this disorder from HBS. |
Menkes syndrome
(See ATP7A-Related Copper Transport Disorders.) |
ATP7A
| AR |
| Kinky hair, seizures as a predominant manifestation, normal hearing, white matter signal changes on brain MRI, & tortuous blood vessels on brain MR angiogram differentiate this disorder from HBS. |
|
Disorders w/overlapping laboratory features
|
|
Wilson disease
|
ATP7B
| AR |
| Liver disease, movement disorder, & Kayser-Fleischer rings help to differentiate this disorder from HBS. |
| Congenital disorder of glycosylation, type IIo (OMIM 616828) |
CCDC115
| AR | Hypotonia DD ↓ serum ceruloplasmin
| Cataracts & deafness are not characteristic of CDG-IIo. |
| Congenital disorder of glycosylation, type IIp (OMIM 616829) |
TMEM199
| AR | ↓ serum ceruloplasmin | Psychomotor development can be normal, & cataracts & deafness are not characteristic of CDG-IIp. |
| MEDNIK syndrome (OMIM 609313) |
AP1S1
| AR |
| Cutaneous manifestations (icthyosis & keratodermia) & enteropathy differentiate this disorder from HBS. |
AR = autosomal recessive; CDG = congenital disorder of glycosylation; DD = developmental delay; MOI = mode of inheritance
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Huppke-Brendel syndrome (HBS), the following evaluations are recommended if they have not already been completed:
Ophthalmologic evaluation
Assessment of feeding and nutrition
Developmental assessment
Audiologic evaluation including brain stem auditory evoked response and otoacoustic emissions testing
Complete neurologic assessment
Electroencephalography if seizures are suspected
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
The following are indicated:
Cataract extraction in the first few months of life
Early feeding tube placement to manage difficulties with swallowing, ensure adequate nutrition, and reduce the risk of aspiration
Developmental intervention
Physiotherapy to maintain muscle function and prevent contractures
Surveillance
Individuals with HBS require the following periodically:
Developmental and neurologic assessment
Nutritional and growth evaluation
Hearing evaluation
Ophthalmologic evaluation
Orthopedic evaluation because of increased risk for scoliosis and contractures
Evaluation of Relatives at Risk
It is appropriate to evaluate infants at risk for HBS in order to identify as early as possible those who would benefit from prompt removal of cataracts as well as feeding and developmental support. Evaluations can include the following:
Clinical exam, ophthalmologic exam for cataracts, and audiologic evaluation in an at-risk newborn prior to molecular testing or while waiting for molecular results
Note: Serum copper and ceruloplasmin levels may not be informative in a newborn because serum ceruloplasmin and, to a lesser degree, serum copper are very low in normal neonates (even more so in premature infants); further, the depletion of copper stores may not be noticeable in the first few months of life even in individuals with known disorders of copper transport.
Molecular genetic testing if the pathogenic variants in the family are known
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Treatment with copper histidinate in three affected individuals did not result in an increase in serum copper or ceruloplasmin. Clinical improvement was reported in one individual, who died at age four years.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Huppke-Brendel syndrome (HBS) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
The parents of an affected child are obligate heterozygotes (i.e., carriers of one SLC33A1 pathogenic variant).
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. To date, individuals with HBS are not known to reproduce.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an SLC33A1 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the SLC33A1 pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the SLC33A1 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
No specific resources for Huppke-Brendel Syndrome have been identified by GeneReviews staff.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Huppke-Brendel Syndrome: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Gene structure.
SLC33A1 maps to chromosome 3q25.1 and has six coding exons.
Pathogenic variants. To date, only six pathogenic variants have been identified in individuals with Huppke-Brendel syndrome (HBS) (Table 3). Three of the reported variants were homozygous and two others were in compound heterozygous states.
Table 3.
Pathogenic Variants in SLC33A1
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.328G>C | p.Ala110Pro |
NM_004733.3
NP_004724.1
|
| c.542_543delTG | p.Val181GlyfsTer6 |
| c.614dupT | p.Leu205PhefsTer32 |
| c.1098C>G | p.Tyr366Ter |
| c.1267-1G>A | |
| c.1474_1482+9del | |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
SLC33A1 encodes acetyl-CoA transporter 1 (AT-1) protein. AT-1 is a multiple transmembrane domain protein that consists of 549 amino acids and has a mass of 60.9 kd [Kanamori et al 1997]. The encoded protein is predicted to contain 6-10 transmembrane domains and a leucine zipper motif in transmembrane domain III.
AT-1 is an endoplasmic reticulum (ER) membrane transporter that regulates the influx of acetyl CoA into the ER lumen [Jonas et al 2010], a key step for the intraluminal acetylation of ER resident and transiting proteins. This is essential for the induction of tightly controlled autophagy-dependent ER-associated degradation (ERAD-II), which allows the cell to recover from the resulting transient accumulation of protein aggregates [Pehar et al 2012].
Abnormal gene product:
SLC33A1 pathogenic variants cause severe loss of protein function. The functional consequences have been studied in a number of pathogenic variants. The p.Ala110Pro variant results in abnormal function of the first and second transmembrane domain [Huppke et al 2012].
Downregulation of AT-1 in zebrafish has been shown to cause embryonic lethality and severe morphologic defects as well as defective neuronal outgrowth in the surviving animals [Lin et al 2008]. In cultured cells, downregulation of AT-1 results in engulfment and enlargement of the ER followed by autophagic cell death [Jonas et al 2010].
The relationship between AT-1 and low serum copper and ceruloplasmin levels is unclear at present. One theory is that ceruloplasmin, which is a glycoprotein, requires AT-1-dependent transient acetylation for proper function. Studies in HepG2 cells with reduced AT-1 expression demonstrated 30% less ceruloplasmin excretion consistent with AT-1 deficiency causing reduced ceruloplasmin levels [Huppke et al 2012]. Individuals with HBS display no signs of copper toxicity as occurs in individuals with Wilson disease with hepatic cirrhosis. Copper analysis in tissues in a few affected individuals did not reveal any abnormality.
References
Literature Cited
Chiplunkar
S, Bindu
PS, Nagappa
M, Bineesh
C, Govindaraj
P, Gayathri
N, Bharath
MM, Arvinda
HR, Mathuranath
PS, Sinha
S, Taly
AB. Huppke-Brendel syndrome in a seven months old boy with a novel 2-bp deletion in SLC33A1.
Metab Brain Dis.
2016;31:1195-8.
[
PubMed: 27306358]
Horváth
R, Freisinger
P, Rubio
R, Merl
T, Bax
R, Mayr
JA, Shawan, Müller-Höcker J, Pongratz D, Moller LB, Horn N, Jaksch M. Congenital cataract, muscular hypotonia, developmental delay and sensorineural hearing loss associated with a defect in copper metabolism.
J Inherit Metab Dis.
2005;28:479-92.
[
PubMed: 15902551]
Huppke
P, Brendel
C, Kalscheuer
V, Korenke
GC, Marquardt
I, Freisinger
P, Christodoulou
J, Hillebrand
M, Pitelet
G, Wilson
C, Gruber-Sedlmayr
U, Ullmann
R, Haas
S, Elpeleg
O, Nürnberg
G, Nürnberg
P, Dad
S, Møller
LB, Kaler
SG, Gärtner
J. Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin.
Am J Hum Genet.
2012; 90: 61-8. Erratum in: Am J Hum Genet. 2012;90:378.
[
PMC free article: PMC3257879] [
PubMed: 22243965]
Kanamori
A, Nakayama
J, Fukuda
MN, Stallcup
WB, Sasaki
K, Fukuda
M, Hirabayashi
Y. Expression cloning and characterization of a cDNA encoding a novel membrane protein required for the formation of O-acetylated ganglioside: a putative acetyl-CoA transporter.
Proc Natl Acad Sci U S A.
1997;94:2897-902.
[
PMC free article: PMC20294] [
PubMed: 9096318]
Lin
P, Li
J, Liu
Q, Mao
F, Li
J, Qiu
R, Hu
H, Song
Y, Yang
Y, Gao
G, Yan
C, Yang
W, Shao
C, Gong
Y.
A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42).
Am J Hum Genet.
2008;83:752-9.
[
PMC free article: PMC2668077] [
PubMed: 19061983]
Richards
S, Aziz
N, Bale
S, Bick
D, Das
S, Gastier-Foster
J, Grody
WW, Hegde
M, Lyon
E, Spector
E, Voelkerding
K, Rehm
HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med.
2015;17:405-24.
[
PMC free article: PMC4544753] [
PubMed: 25741868]