Clinical Description
YIF1B-related neurodevelopmental disorder (YIF1B-NDD) is characterized by severe-to-profound developmental delay / intellectual disability with variable motor abnormalities including axial hypotonia, peripheral hypertonia, dystonia, and dyskinesia; absence of speech in most individuals or very limited speech subject to regression; feeding difficulties; seizures; postnatal microcephaly with nonspecific brain MRI abnormalities; and ophthalmologic involvement (strabismus, nystagmus, optic atrophy, and cortical blindness). Some individuals have hypoventilation. To date, 25 individuals have been identified with biallelic pathogenic variants in YIF1B [AlMuhaizea et al 2020, Diaz et al 2020, Medico Salsench et al 2021, Sanri et al 2023]. The following description of the phenotypic features associated with this condition is based on these reports.
Developmental delay. All individuals with YIF1B-NDD presented with severe-to-profound developmental delay, evident in the first month of life [AlMuhaizea et al 2020, Diaz et al 2020, Medico Salsench et al 2021, Sanri et al 2023]. Normal developmental milestones were most often not achieved. Severe axial hypotonia was seen in most affected individuals, with concurrent peripheral hyperreflexia and spasticity. Head control was only achieved in 5/24 individuals, all of whom had biallelic missense variants; individuals with protein-truncating variants did not achieve head control. Likewise, sitting (4/24), standing (2/24), and walking (2/24) were only observed in individuals with YIF1B missense variants. No speech development occurred in individuals with biallelic YIF1B protein-truncating variants (0/18), with limited speech development only in 4/6 individuals with biallelic YIF1B missense variants; subsequent progressive loss of speech was observed in 3/4 individuals. Feeding problems and difficulties in swallowing occurred in 20/24 individuals early in life, often requiring tube feeding.
Intellectual disability. Although for the majority of affected individuals currently no formal IQ testing results are available, based on clinical estimates the level of intellectually disability of all affected individuals is in the severe-to-profound range [AlMuhaizea et al 2020, Diaz et al 2020, Medico Salsench et al 2021, Sanri et al 2023].
Movement disorders observed in YIF1B-NDD include dystonia and dyskinesia or tremor [AlMuhaizea et al 2020, Diaz et al 2020, Medico Salsench et al 2021, Sanri et al 2023]. For those few individuals for which detailed information is available, movement disorders had an onset around age two to three years with the development of choreiform limb movements, which evolved into axial and limb dystonia with dyskinesia by age four to eight years [AlMuhaizea et al 2020]. In those individuals, dystonia was unresponsive to levodopa or carbidopa but partially improved on trihexyphenidyl [AlMuhaizea et al 2020].
Epilepsy. Seizures occurred in 15/23 affected individuals, and included myoclonic, generalized tonic-clonic, and infantile epileptic spasms of variable degrees of severity [AlMuhaizea et al 2020, Diaz et al 2020, Medico Salsench et al 2021, Sanri et al 2023]. Most seizures had onset before age two years, with EEGs showing frequent multifocal and epileptiform discharges and abnormal background indicating diffuse dysfunction. For some individuals, seizure control could be achieved using phenobarbital, levetiracetam, and valproate, whereas others presented with medically refractory generalized epilepsy.
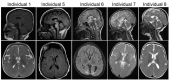
Growth. Growth parameters at birth were normal for those individuals for which information is available. Postnatal microcephaly occurred in 16/24 individuals. Currently, information on postnatal growth and weight is sparse, although a few individuals have been reported to present with poor weight gain and growth deficiency [Diaz et al 2020, Sanri et al 2023].
Ophthalmologic involvement included strabismus, nystagmus, and optic atrophy. Cataract or retinal involvement have not been reported. Cortical blindness was reported in one individual.
Central hypoventilation has been observed in 6/19 reported individuals and seems to be correlated with the level of brain stem atrophy [AlMuhaizea et al 2020, Diaz et al 2020, Medico Salsench et al 2021, Sanri et al 2023]. At least three individuals became ventilation dependent.
Neurobehavioral/psychiatric manifestations. Autism spectrum disorder and autistic behaviors have been reported in 4/14 individuals [AlMuhaizea et al 2020, Diaz et al 2020, Medico Salsench et al 2021, Sanri et al 2023]. Anxiety and aggression have been reported in a single individual [Diaz et al 2020].
Facial features. No consistent recognizable facial features have been reported in individuals affected with YIF1B-NDD [AlMuhaizea et al 2020, Diaz et al 2020, Medico Salsench et al 2021, Sanri et al 2023] (see ).
Images of Individuals 4–7 and 8 at ages 1 year, 27 years, 7.5 years, 11 months, and 4.5 years, respectively. No consistent dysmorphic features were observed. Note the neurologic posture in Individuals 4, 5, and 7. Reproduced with permission from (more...)
Prognosis. Premature death (before age 2 years) has been reported in five individuals, all with protein-truncating variants. The precise cause of death has not been reported.
Three individuals were older than age 25 years (the oldest reported individual was age 37 years) at the time of publication, demonstrating that survival into adulthood is possible [Diaz et al 2020, Medico Salsench et al 2021].