Introduction—Fibroblast Growth Factors (FGFs) and FGF Receptors
The FGFs comprise a large group of structurally similar polypeptide mitogens which currently includes 22 different members. The first members of this family, FGF1 (acidic FGF, aFGF) and FGF2 (basic FGF, bFGF), were described in 1986 by Jaye et al1 and Abraham et al.2 In the meantime, 20 other FGFs have been discovered which were designated FGF3-FGF23. FGF19 has so far only been described in humans and FGF15 only in mice, and it has been suggested that FGF19 is the human ortholog of mouse FGF15. All members of the FGF family range in molecular weight from 17 to 34 kDa in vertebrates, and some of them are glycosylated (for review see ref. 3).
Most of the FGFs have a classical amino-terminal signal sequence and are, therefore, efficiently secreted via the endoplasmic reticulum-Golgi secretory pathway. FGF9, 16 and 20 lack an obvious amino-terminal signal peptide, but are nevertheless secreted. FGFs11–14 are thought to remain intracellular, and secretion of these FGFs has not yet been observed. Although FGF1 and FGF2 lack a signal sequence and are normally located in the cytoplasm or in the nucleus, they can be found on the cell surface and within the extracellular matrix. They are either released from damaged cells or secreted by an alternative exocytotic mechanism that bypasses the endoplasmic reticulum Golgi pathway(for review see refs. 3,4).
FGFs have been shown to interact with three different types of binding partners: heparan sulphate proteoglycans,3–5 a cysteine-rich transmembrane FGFbinding protein which appears to be involved in the regulation of intracellular FGF trafficking,6 and four high-affinity transmembrane FGF receptors of the tyrosine kinase family which are responsible for signal transduction.7 The FGF receptors, FGFR1-FGFR4, are transmembrane protein tyrosine kinases with either two or three immunoglobulin-like domains and a heparin binding sequence in the extracellular part of the receptor. Alternative mRNA splicing generates receptors with different carboxyl-terminal halves of the third immunoglobulin-like domain, designated IIIb or IIIc isoforms. This alternative splicing event is regulated in a cell- and tissue-specific manner and has been shown to dramatically affect ligand-receptor binding specificity.7 The different members of the FGF family bind to the receptor splice variants with different affinities. Most FGFs bind to a specific subset of FGF receptors. FGF1, however, binds to all known receptors, and FGF7 specifically interacts with the IIIb isoform of FGFR2.8
A characteristic feature of FGFs is their interaction with heparin or heparan sulphate proteoglycans. These interactions stabilize FGFs and may limit their diffusion and release into interstitial spaces. Most importantly, the interaction of FGFs with heparin or heparan sulphate proteoglycans is essential for the activation of the signaling receptor.3,5
Most members of the FGF family have a broad mitogenic spectrum. They stimulate proliferation of a variety of cells of mesodermal, ectodermal and also of endodermal origin.3,4 The only known exception is FGF7, which seems to act only on epithelial cells, at least in the adult organism.9 FGFs are not only mitogenic, but they also have the capacity to regulate migration and differentiation in vitro and in vivo. Finally, some of the FGFs have been shown to be cytoprotective (see below).3,9
FGFs and their receptors are expressed at multiple sites in the developing and adult animal, suggesting important roles of FGFs in development and tissue homeostasis. This hypothesis was confirmed by the wide variety of phenotypic abnormalities observed in FGF and FGFR knockout animals.3 Finally, abnormalities in FGF/FGF receptor signaling are associated with human disease, including cancer and various genetic disorders.
Distribution of FGFs in Adult Brain
The brain is a rich source of various FGFs. While the expression pattern of FGF2 has been studied in detail (for review, see refs. 10,11), data on the mRNA expression and the immunoreactivity of the other FGFs is relatively sparse. Both FGF1 and FGF2 have been localized to glial cells and neurons of the CNS. Notably, high levels of FGF1 were found in neurons of brain regions that are at high risk for neurodegenerative diseases such as Alzheimer's disease (magnocellular forebrain cholinergic neurons), Parkinson's disease (substantia nigra neurons) and amyotrophic lateral sclerosis (motor neurons).11 Most, if not all astrocytes of the brain express Fgf2, whereas a prominent neuronal localization appears to be confined mainly to neurons in the CA2 region of the hippocampus.12 In both astrocytes and CA2 neurons, Fgf2 immunoreactivity is localized primarily in the nucleus and to a lesser extent in the cytoplasm and the processes of stained cells.13 Fgf5 mRNA is widely distributed at low levels in the brain, with several loci of Fgf5 expression found in the cerebral cortex, hippocampus and thalamus. At least some of the Fgf5 expressing cells appear to be neurons.14 Fgf7 mRNA could not be detected in any of the postnatal brain regions examined so far.15,16Fgf9 mRNA is moderately to weakly expressed in widespread regions of the brain, with some preference to brain regions involved in motor control (red nucleus, parts of cerebellum, oculomotor nucleus). The cellular localization of Fgf9 mRNA indicated that this FGF family member is mainly produced by neurons.17,18 Expression of the Fgf10 gene is spatially restricted to some regions of the brain, including hippocampus, thalamus, and several nuclei in midbrain and brainstem, with a pre-ferential expression in neurons rather than in glial cells.15 Interestingly, with its predominant expression in the CA1 and CA3, but not in the CA2 region of the hippocampus, the spatial distribution of Fgf10 mRNA is just opposite to that of Fgf2 mRNA, which is largely confined to the CA2 region, suggesting that the two FGFs have distinct functions in the hippocampus. Finally, Fgf14 mRNA was recently detected in the brain, with FGF1–41b as the predominant isoform. The developmental expression pattern suggests that FGF14 plays a significant role in cerebellar development.19
All FGF receptors are expressed in the brain. The Fgfr1 gene is significantly expressed in select neuronal populations, but also in astrocytes and oligodendrocytes.20,21 FGFR2 and FGFR3 are predominantly found in astrocytes and oligodendrocytes,22,23 whereas FGFR4 is almost exclusively expressed in neurons of the medial habenular nucleus.24
FGFs and Neuronal Development
FGFs and their receptors (FGFRs) have been implicated in many aspects of neuronal development, where they promote proliferation, differentiation and axonal branching. For example, FGF8 regulates growth and patterning of the midbrain and the anterior forebrain,25,26 FGF3 plays an essential role in the development of the inner ear,27 and FGF8, FGF14, FGF15, FGF17, and FGF18 are expressed in the developing cerebellum.19,28 Demonstrating the significance of FGFs for appropriate cerebellar development, a phenotype displaying gait defects was observed in FGF17−/− and FGF8Δ2,3n/+ mice, albeit with low penetrance.28 FGF2 appears to be intimately involved in cortical development (for review see refs. 29,30). It supports the survival of neurons from many regions of the fetal rat brain, including hippocampus, cortex, thalamus and striatum.31,32 In addition to its neurotrophic effects, FGF2 acts as a mitogenic factor, stimulating the proliferation of neuronal progenitor cells. Most interestingly in the context of this chapter, FGF2 is capable of activating a latent neurogenic program in neural stem cells from diverse regions of the adult brain and of stimulating neurogenesis in the mature CNS.33–35 FGF2 is present in the telencephalon as early as E9.5 and high levels are found in the cerebral cortex throughout neurogenesis and into adulthood.10 FGF2 appears to exert a dual function on early phases of cortical neuroectoderm cell proliferation and later phases of differentiation. Since astrocytes appear to be the predominant source of FGF2, with only few neuronal populations displaying FGF2 immunoreactivity (see above), glial cells might provide trophic support to neuronal cells.12 Despite accumulating evidence from in vitro studies, which strongly implicate FGF2 as a neurotrophic factor, it was not before the generation of mice lacking FGF2 that the issue of whether endogenous FGF2 is essential for survival of cortical neurons was settled. Neurohistological analysis of FGF2 knockout mice revealed abnormalities in the cytoarchitecture of the neocortex, combined with a significant reduction in neuronal density, most pronounced in layer V.36,37 It was concluded that one important role of FGF2 during cortical neurogenesis may be to amplify the progenitor pool for pyramidal projection neurons.38 Data from several developmental studies indicate that FGF2 also acts as a target-derived factor that promotes axon branching of cortical neurons. Specifically, FGF2 may induce the formation of collateral axon branches by its effects on the morphology and the behavior of the primary growth cone.39 The strong effect of FGF2 on neurite morphogenesis has been linked to the increase by FGF2 of L-type Ca2+ channels in fetal neurons.40
Upregulation of FGFs after Brain Injury
The expression of many neurotrophic factors in CNS neurons is regulated by physiological stimuli, such as afferent synaptic activity, suggesting that these factors participate in functional and/or morphological changes associated with neuronal plasticity. More dramatic alterations in the expression pattern of many neurotrophic factors are induced by several forms of acute brain injury.41 Since the late 1980's, numerous publications have accumulated evidence for a pronounced upregulation of FGFs in the cellular response to acute brain injury. Among the FGF family members, FGF2 clearly emerged as the central player in acute CNS damage, and we will hence focus the following section on this factor. FGF2 upregulation was demonstrated after mechanical brain injury,42–48 after ischemic insults,49,50 and after convulsive seizures.51–54 Three days after of mechanical cortical injury, macrophages are the predominant FGF2 immunoreactive cells at the lesion site. A second phase of increased FGF2 expression, which peaks at about one week post-lesion, is mediated by activated astrocytes and microglia, particularly at the border between the neuronal and scar tissue. Although multiple cells within the lesioned CNS, including astrocytes, microglia, neurons and vascular endothelial cells, might all be able to express FGF2, glial cells appear to be the primary source of the newly synthesized FGF2. In support of this notion, the gradual fall in FGF2 levels one week after injury parallels the concomitant decrease in reactive glia.45 Interestingly, an increase in the intensity of FGF2 immunoreactivity was also detected within the extracellular matrix surrounding the lesion, suggesting that FGF2 was released from astrocytes and becomes available to neurons.46 A similar time course of induction of glial FGF2 synthesis was observed after transient forebrain ischemia.49 In contrast to the slow, but sustained upregulation of FGF2 in traumatic or ischemic lesion models, a faster and transient response was induced by epileptic convulsions not associated with neuronal loss. For example, seizures induced by microinjection of the GABAA receptor antagonist, bicuculline, in the deep prepiriform cortex lead to a significant increase in FGF2 mRNA in the entorhinal cortex, the hippocampus and the olfactory bulb within 5 hours of epileptic activity.51 However, if seizure activity was severe enough to produce neuronal loss and reactive astrocytosis, a long-term induction of FGF2 gene expression for up to 2 weeks was observed in the damaged region.53 Although astrocytes appear to represent the main source of FGF2 in seizure models, elevated FGF2 mRNA signals were also observed in select neuronal populations.54,55
Upregulation of FGF2 also has been implicated in the protective effect of cortical spreading depression (SD) against subsequent ischemic damage.56,57 Cortical SD is a rapid and nearly complete, but reversible depolarization of a large population of neurons, which propagates in a slow wave-like fashion through the gray matter (for review see ref. 58 and Chapter 5 of this book). The protective effect of SD against ischemic insults might last for several days suggesting that some kind of sustained downmodulation of neuronal vulnerability is initiated. Right now, it is not clear whether the upregulation by SD of FGF2 is a pivotal mechanism affording enhanced protection against subsequent stroke. However, with the generation of FGF2 deficient mice (see above), this intriguing issue should be resolved in the near future.
Neuroprotective Effects of FGF2
Among the various growth factors and cytokines studied so far, the neuroprotective and neurotrophic profile of FGF2 is best documented (reviewed in ref. 59). If administered during or within hours of acute injury, systemic or intracerebroventricular (icv.) FGF2 reduces infarct size after stroke,60–66 diminishes histopathologic damage associated with fluid percussion injury,67 affords neuroprotection against N-methyl-D-aspartate (NMDA) and kainate receptor-mediated excitotoxicity, 60,68,69 and prevents the death of axotomized CNS neurons.70–74 It is worth noting that FGF2, which is highly neuroprotective against seizure-induced long-term behavioral deficits, might also act as a convulsant at higher concentrations.75 If administered 1 day after focal cerebral infarction, intracisternal FGF2, although no longer reducing infarct size, is still capable of enhancing behavioral recovery.76 Correspondingly, blockade of FGF2 by neutralizing antibodies retarded recovery of forelimb manipulatory abilities after unilateral suction lesion of the motor cortex.77 Further support for a beneficial role of FGF2 in acute brain injury comes from a study in which mice expressing a bovine FGF2 transgene in the brain showed increased resistance to hypoxemic-ischemic cerebral damage.78 The finding that intravenous FGF2 rapidly crosses the blood brain barrier in adult brain, with the levels in blood plasma and cerebrospinal fluid rising in parallel, bears particular significance for the application of this factor in a clinical setting.34,78A As a result of the outstanding benefits of FGF2 in various animal lesion models and its high cerebral bioavailability after intravenous injection, clinical trials of intravenous FGF2 (Fiblast) in acute stroke were conducted. In a North American phase II/III trial, 302 patients suffering from acute ischemic stroke were enrolled between August 1997 and May 1998, when enrollment was halted by the Data Safety Monitoring Committee following an interim analysis, which revealed an unfavorable risk-to-benefit ratio in stroke patients treated with FGF2 versus those treated with placebo (information according to the Internet Stroke Center at www.strokecenter.org).
Data on the possible neuroprotective potential of other FGFs is sparse. Several studies found beneficial effects of FGF1 (acidic FGF) in animal models of acute ischemic or excitotoxic brain damage.79–83 A recent study demonstrated that FGF7 (keratinocyte growth factor) prevents ischemia-induced delayed neuronal death in the hippocampal CA1 region of the gerbil brain.84 Finally, FGF8 was shown to protect cultured hippocampal neurons from oxidative insult.85
FGFs and Glia
Given that FGF2 is mitogenic for oligodendrocytes and astrocytes and stimulates migration and functional differentiation of astrocytes, one might wonder whether, in addition to its neuroprotective effects, FGF2 may also have the undesirable side effect of promoting scar formation (for review see ref. 86 and Chapter 4 of this book). Although the involvement of FGF2 in scar formation appears minor compared to that of other potent fibrotic agents such as TGF-β1, several reports lend credence to the notion that FGF2 is not an innocent player in the glial response to acute brain injury. For example, the reactive gliosis following mechanical or electrolytic lesions in the neonatal and adult brain was significantly augmented and accelerated by FGF2 injected into the lesion site just after the lesion was performed.87–89 Along the same lines, FGF2, if injected into various regions of the noninjured adult rat brain, produced a glial reaction that resembled the reactive gliosis seen after brain injury.89 Supporting its involvement in extracellular matrix remodeling after injury, FGF2 was found to increase the production of tenascin-C mRNA and protein in cultured hippocampal astrocytes.90 It should be pointed out, however, that the effects of FGF2 on glial cells might also indirectly promote survival of select neuronal populations, as FGF2 (and FGF1) were found to stimulate nerve growth factor (NGF) synthesis and secretion by astrocytes.91 Since intercellular communication between astrocytes through gap junctions is essential to many of their functions, the finding that FGF2, FGF5 and FGF9 downregulate astroglial gap junctions and functional coupling in a brain region-specific fashion is of particular interest.92 It is well documented that acute brain injury is associated with downregulation of connexin 43, the predominant component of astroglial gap junctions. One might thus speculate whether the lesion-induced upregulation of FGFs causally linked to the uncoupling of astrocytes.
Neuroprotective Mechanisms of FGF2
Ionotropic Glutamate Receptors
The seminal study by Mattson et al93 in cultured hippocampal neurons provided compelling evidence that FGF2 raises the threshold for glutamate neurotoxicity and reduces the rise in intracellular Ca2+ associated with glutamate receptor activation. Since then, a number of studies by Mattson and coworkers and other groups have elaborated on the mechanisms underlying the protective effect of FGF2 against excitotoxic damage. For example, FGF2 was found to exert differential effects on glutamate receptor subtype expression: Whereas it selectively increases the AMPA receptor subunit GluR1 in hippocampal neurons,94 a functional 71kDA NMDA receptor that mediates Ca2+ influx and neurotoxicity is downregulated by FGF2 in the same preparation.95 In functional terms, this differential effect of FGF2 on glutamate receptor expression translates into an increase in AMPA receptor-mediated, but a decrease in NMDA receptor-mediated Ca2+ elevations, with the latter serving as a significant excitoprotective mechanism.94 Providing further insights into the mechanisms underlying the inhibitory effect of FGF2 on NMDA receptor-mediated Ca2+ influx, a recent study demonstrated that chronic treatment (hours to days) of cultured hippocampal neurons by FGF2 potentiates Ca2+−-dependent inactivation of NMDA receptor currents through a calcineurin-dependent mechanism.96 In marked contrast, short-term exposure of acutely dissociated hippocampal neurons to FGF2 produced a selective enhancement of NMDA receptor-mediated increases in cytosolic Ca2+.97
Ca2+ Homeostasis, Mitochondrial Dysfunction and Reactive Oxygen Species
In addition to reducing Ca2+ influx through NMDA receptors, FGF2 also operates on a second line of defense against the loss of Ca2+ homeostasis and the concurrent mitochondrial dysfunction. For example, FGF2 increases the synthesis of calbindin D28k,98 a Ca2+−-binding protein thought to exert an excitoprotective role in CNS neurons.99 Perhaps even more importantly, FGF2 increases the activity of antioxidant enzymes, such as superoxide dismutase and glutathione reductase.100,101 Extending the beneficial effects of FGF2 into the range of chronic neurodegenerative diseases such as Alzheimer's disease, Mattson and coworkers recently demonstrated that FGF2 also attenuates oxidative stress and mitochondrial dysfunction induced by amyloid peptide Aβ102 and mitigates the enhanced neuronal vulnerability to excitotoxicity in cultured hippocampal neurons from presenilin1 mutant knock-in mice.103
Apoptosis and Neurogenesis
Two recent studies, one performed in vitro and one in vivo, implicated FGF2 in antiapoptotic pathways. In cultured hippocampal neurons, FGF2 prevented apoptosis induced by NO donors. Whereas NO donor-induced apoptosis was typically associated with downregulation of Bcl-2, upregulation of Bax and subsequent caspase-3-like activation, pretreatment with FGF2 abrogated the changes in Bcl-2 and Bax protein levels as well as the caspase-3-like activation.104 Somewhat similar findings were found in an animal stroke model in which permanent occlusion of the right middle cerebral artery (MCAO) causes focal cerebral infarction. In animals receiving intravenous infusion of FGF2 for 3 h, beginning at 30 min after MCAO, FGF2 prevented the reduction of immunoreactivity of the antiapoptotic protein Bcl-2, which is typically observed in untreated stroke animals. In contrast to the in vitro study mentioned above, FGF2 did not alter immunoreactivity to the proapoptotic proteins Bax, caspase-1, and caspase-3. Nevertheless, FGF2 produced a substantial decrease in apoptotic neurons, especially in the border (“penumbra”) of the infarct, which is exactly the zone predominantly spared by FGF2 treatment.105 With respect to the neuroprotective effect of FGF2 in stroke models, it is worth noting that upregulation by FGF2 of endothelial NO synthase (eNOS)106 and subsequent increase in cerebral blood flow (CBF)107 is not the predominant protective mechanism because FGF2 retains its infarct-reducing efficacy in eNOS-deficient mice, in which CBF is not increased by FGF2.108
In addition to preventing lesion-induced apoptosis, FGF2 was also reported to promote neurogenesis in the adult dentate gyrus in response to injury.109 The dentate gyrus belongs to the hippocampal formation and contains into adulthood neuroprogenitor cells that are able to divide and differentiate into granule cells.110,111 Whereas kainate injection or MCAO induced appreciable neurogenesis in the dentate gyrus of adult control mice, the number of newly generated neurons, as indicated by bromodeoxyuridine (BrdU) incorporation into nuclei of dentate granule cells, was significantly decreased in FGF2 deficient mice, but could be restored to control levels after icv. injection of a herpes simplex virus1 amplicon vector carrying the FGF2 gene.109 These data suggest that the lesion-associated upregulation of FGF2 not only promotes survival of neurons exposed to various forms of acute damage, but plays also a critical role in neuronal repair.
Interaction Between FGF2 and Activin A
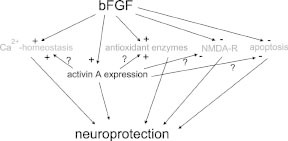
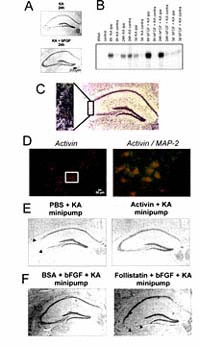
In a collaborative study between our laboratories, we made the intriguing observation that induction of activin A, a member of the transforming growth factor-β (TGF-β) superfamily (see next chapter), is essential for the neuroprotective action of FGF2 in vivo.112 Like the other members of the TGF-β superfamily, activins are dimeric proteins, consisting of two βA subunits (activin A), two bB subunits (activin B) or a βA and a βB subunit (activin AB). In addition, βC, βD, and βE subunits have been identified, although the corresponding proteins have not been characterized in detail. The βA and βB subunits can also dimerize with a homologous a subunit, leading to the formation of inhibin A (αβA) or inhibin B (αβB). Previous findings from our and other laboratories already pointed to a possible involvement of activin A in the early neuronal response to injury (Fig. 1C).113–117 Since activin A promotes survival of midbrain and hippocampal neurons in vitro,118,119 reduces ischemic brain injury in infant rats,120 and protects striatal and midbrain neurons against neurotoxic damage,118,121 we speculated that the lesion-induced upregulation of activin A might serve an excitoprotective function. A first indication for a possible interaction between FGF2 and activin A came from a comparison between the activin βA mRNA expression pattern in lesioned and in FGF2-protected hippocampi. In our lesion model, exogenous FGF2 abolished the neuronal damage in the CA3 region of the ipsilateral hippocampus which is typically seen after intracerebroventricular (icv.) application of this excitotoxin (Fig. 1A). Unexpectedly, FGF2 also produced a strikingly stronger upregulation of βA mRNA than KA injection alone (Fig. 1B). FGF2 substantially augmented βA mRNA expression on the ipsilateral side at 6h and 24h postlesion, without appreciably influencing the time course of signal elevation. Two independent sets of experiments corroborated the hypothesis that activin A is crucially involved in the neuroprotective effects ascribed to FGF2. First, recombinant activin A was as effective as exogenous FGF2 in preventing excitotoxic neuronal loss (Fig. 1E). Second, the activin-binding protein follistatin, which neutralizes activin in vitro and in vivo,122,123 consistently abrogated the beneficial action of FGF2 in the KA lesion model (Fig. 1F). Control experiments dispelled concerns that follistatin caused neuronal damage by a mechanism other than its activin-neutralizing action. A different type of interaction between FGF2 and the TGF-β superfamily was reported from cultured midbrain dopaminergic neurons, in which the neurotrophic effect of bFGF was mediated by TGF-β1–3.124 In contrast to activin A, which is of neuronal origin in lesioned hippocampus, the effect of the TGF-βs was mediated by cocultured glial cells.
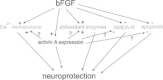
The main neuroprotective mechanisms of FGF2 are summarized in Fig. 2. As it is becoming evident that induction of activin A is an essential step in the signaling cascade affording neuroprotection after application of FGF2, it remains to be determined which of the effects, originally attributed to FGF2, are directly mediated by this growth factor, and which require upregulation of activin A.
Summary and Conclusions
Several members of the FGF family, in particular FGF2, are intimately involved in neuronal protection and repair after ischemic, metabolic or traumatic brain injury. Expression of Fgf2 mRNA and protein is strongly upregulated after neuronal damage, with glial cells as the predominant source. Given its survival-promoting effects on cultured neurons, exogenous FGF2 was tested in several animal models of stroke and excitotoxic damage, in which it consistently proved protective against neuronal loss. FGF2 affords neuroprotection by interfering with a number of signaling pathways, including expression and gating of NMDA receptors, maintenance of Ca2+ homeostasis and regulation of ROS detoxifying enzymes. FGF2 prevents apoptosis by strengthening antiapoptotic pathways and promotes neurogenesis in adult hippocampus after injury. The protective action of FGF2 has been linked to its augmenting effect on the lesioninduced upregulation of activin A, a member of the TGF-β superfamily. Despite the well-documented benefits of FGF2 in animal models of stroke, there is currently no clinical development in stroke, after a phase II/III trial with FGF2 in acute stroke patients was discontinued because of an unfavorable risk-to-benefit ratio. As the molecular targets of FGF2 are going to be unraveled over the next years, new therapeutic strategies will hopefully emerge that enable us to influence the various protective mechanisms of FGF2 in a more specific fashion.
Acknowledgments
The authors thank David Ornitz for comments on the manuscript. The work of the authors was supported by the Deutsche Forschungsgemeinschaft (DFG), the Bundesministerium für Bildung und Forschung (Bmb+f), and the Human Frontier Science Program.
References
- 1.
- Jaye M, Howk R, Burgess W. et al. Human endothelial cell growth factor: cloning, nucleotide sequence, and chromosome localization. Science. 1986;233:541–545. [PubMed: 3523756]
- 2.
- Abraham JA, Mergia A, Whang JL. et al. Nucleotide sequence of a bovine clone encoding the angiogenic protein, basic fibroblast growth factor. Science. 1986;233:545–548. [PubMed: 2425435]
- 3.
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:REVIEWS3005. [PMC free article: PMC138918] [PubMed: 11276432]
- 4.
- Basilico C, Moscatelli D. The FGF family of growth factors and oncogenes. Adv Cancer Res. 1992;59:115–165. [PubMed: 1381547]
- 5.
- Ornitz DM. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. BioEssays. 2000;22:108–112. [PubMed: 10655030]
- 6.
- Zuber ME, Zhou Z, Burrus LW. et al. Cysteine-rich FGF receptor regulates intracellular FGF1 and FGF2 levels. J Cell Physiol. 1997;170:217–227. [PubMed: 9066777]
- 7.
- Johnson DE, Williams LT. Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res. 1993;60:1–41. [PubMed: 8417497]
- 8.
- Ornitz DM, Xu J, Colvin JS. et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. [PubMed: 8663044]
- 9.
- Werner S. Keratinocyte growth factor: a unique player in epithelial repair processes. Cytokine Growth Factor Rev. 1998;9:153–165. [PubMed: 9754709]
- 10.
- Baird A. Fibroblast growth factors: activities and significance of non neurotrophin neurotrophic growth factors. Curr Opin Neurobiol. 1994;4:78–86. [PubMed: 8173328]
- 11.
- Eckenstein FP, Andersson C, Kuzis K. et al. Distribution of acidic and basic fibroblast growth factors in the mature, injured and developing rat nervous system. Prog Brain Res. 1994;103:55–64. [PubMed: 7533916]
- 12.
- Woodward WR, Nishi R, Meshul CK. et al. Nuclear and cytoplasmic localization of basic fibroblast growth factor in astrocytes and CA2 hippocampal neurons. J Neurosci. 1992;12:142–152. [PMC free article: PMC6575685] [PubMed: 1729432]
- 13.
- Williams TE, Meshul CK, Cherry NJ. et al. Characterization and distribution of basic fibroblast growth factor containing cells in the rat hippocampus. J Comp Neurol. 1996;370:147–158. [PubMed: 8808727]
- 14.
- Haub O, Drucker B, Goldfarb M. Expression of the murine fibroblast growth factor 5 gene in the adult central nervous system. Proc Natl Acad Sci USA. 1990;87:8022–8026. [PMC free article: PMC54884] [PubMed: 1700424]
- 15.
- Hattori Y, Yamasaki M, Konishi M. et al. Spatially restricted expression of fibroblast growth factor 10 mRNA in the rat brain. Brain Res Mol Brain Res. 1997;47:139–146. [PubMed: 9221911]
- 16.
- Mason IJ, Fuller-Pace F, Smith R. et al. FGF7 (keratinocyte growth factor) expression during mouse development suggests roles in myogenesis, forebrain regionalisation and epithelial mesenchymal interactions. Mech Dev. 1994;45:15–30. [PubMed: 8186145]
- 17.
- Tagashira S, Ozaki K, Ohta M. et al. Localization of fibroblast growth factor 9 mRNA in the rat brain. Brain Res Mol Brain Res. 1995;30:233–241. [PubMed: 7637574]
- 18.
- Colvin JS, Feldman B, Nadeau JH. et al. Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev Dyn. 1999;216:72–88. [PubMed: 10474167]
- 19.
- Wang Q, McEwen DG, Ornitz DM. Subcellular and developmental expression of alternatively spliced forms of fibroblast growth factor 14. Mech Dev. 2000;90:283–287. [PubMed: 10640713]
- 20.
- Gonzalez AM, Berry M, Maher PA. et al. A comprehensive analysis of the distribution of FGF2 and FGFR1 in the rat brain. Brain Res. 1995;701:201–226. [PubMed: 8925285]
- 21.
- Beer HD, Vindevoghel L, Gait MJ. et al. Fibroblast growth factor (FGF) receptor 1IIIb is a naturally occurring functional receptor for FGFs that is preferentially expressed in the skin and the brain. J Biol Chem. 2000;275:16091–16097. [PubMed: 10821861]
- 22.
- Peters K, Ornitz D, Werner S. et al. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol. 1993;155:423–430. [PubMed: 8432397]
- 23.
- Miyake A, Hattori Y, Ohta M. et al. Rat oligodendrocytes and astrocytes preferentially express fibroblast growth factor receptor2 and 3 mRNAs. J Neurosci Res. 1996;45:534–541. [PubMed: 8875318]
- 24.
- Itoh N, Yazaki N, Tagashira S. et al. Rat FGF receptor 4 mRNA in the brain is expressed preferentially in the medial habenular nucleus. Brain Res Mol Brain Res. 1994;21:344–348. [PubMed: 8170355]
- 25.
- Crossley PH, Martinez S, Martin GR. Midbrain development induced by FGF8 in the chick embryo. Nature. 1996;380:66–68. [PubMed: 8598907]
- 26.
- Martinez S, Crossley PH, Cobos I. et al. FGF8 induces formation of an ectopic isthmic organizer and isthmocerebellar development via a repressive effect on Otx2 expression. Development. 1999;126:1189–1200. [PubMed: 10021338]
- 27.
- Represa J, Leon Y, Miner C. et al. The int2 protooncogene is responsible for induction of the inner ear. Nature. 1991;353:561–563. [PubMed: 1922362]
- 28.
- Xu J, Liu Z, Ornitz DM. Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development. 2000;127:1833–1843. [PubMed: 10751172]
- 29.
- Temple S, Qian X. bFGF, neurotrophins, and the control or cortical neurogenesis. Neuron. 1995;15:249–252. [PubMed: 7646883]
- 30.
- Vaccarino FM, Schwartz ML, Raballo R. et al. Fibroblast growth factor signaling regulates growth and morphogenesis at multiple steps during brain development. Curr Top Dev Biol. 1999;46:179–200. [PubMed: 10417880]
- 31.
- Walicke P, Cowan WM, Ueno N. et al. Fibroblast growth factor promotes survival of dissociated hippocampal neurons and enhances neurite extension. Proc Natl Acad Sci USA. 1986;83:3012–3016. [PMC free article: PMC323437] [PubMed: 3458259]
- 32.
- Walicke PA, Baird A. Trophic effects of fibroblast growth factor on neural tissue. Prog Brain Res. 1988;78:333–338. [PubMed: 3073416]
- 33.
- Palmer TD, Ray J, Gage FH. FGF2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol Cell Neurosci. 1995;6:474–486. [PubMed: 8581317]
- 34.
- Wagner JP, Black IB, DiCicco-Bloom E. Stimulation of neonatal and adult brain neurogenesis by subcutaneous injection of basic fibroblast growth factor. J Neurosci. 1999;19:6006–6016. [PMC free article: PMC6783097] [PubMed: 10407038]
- 35.
- Palmer TD, Markakis EA, Willhoite AR. et al. Fibroblast growth factor 2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19:8487–8497. [PMC free article: PMC6783019] [PubMed: 10493749]
- 36.
- Ortega S, Ittmann M, Tsang SH. et al. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci USA. 1998;95:5672–5677. [PMC free article: PMC20437] [PubMed: 9576942]
- 37.
- Dono R, Texido G, Dussel R. et al. Impaired cerebral cortex development and blood pressure regulation in FGF2-deficient mice. EMBO J. 1998;17:4213–4225. [PMC free article: PMC1170755] [PubMed: 9687490]
- 38.
- Raballo R, Rhee J, LynCook R. et al. Basic fibroblast growth factor (FGF2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci. 2000;20:5012–5023. [PMC free article: PMC6772267] [PubMed: 10864959]
- 39.
- Szebenyi G, Dent EW, Callaway JL. et al. Fibroblast growth factor 2 promotes axon branching of cortical neurons by influencing morphology and behavior of the primary growth cone. J Neurosci. 2001;21:3932–3941. [PMC free article: PMC6762708] [PubMed: 11356881]
- 40.
- Shitaka Y, Matsuki N, Saito H. et al. Basic fibroblast growth factor increases functional L-type Ca2+ channels in fetal rat hippocampal neurons: implications for neurite morphogenesis in vitro. J Neurosci. 1996;16:6476–6489. [PMC free article: PMC6578905] [PubMed: 8815926]
- 41.
- Isackson PJ. Trophic factor response to neuronal stimuli or injury. Curr Opin Neurobiol. 1995;5:350–357. [PubMed: 7580158]
- 42.
- Finklestein SP, Apostolides PJ, Caday CG. et al. Increased basic fibroblast growth factor (bFGF) immunoreactivity at the site of focal brain wounds. Brain Res. 1988;460:253–259. [PubMed: 3224261]
- 43.
- Frautschy SA, Walicke PA, Baird A. Localization of basic fibroblast growth factor and its mRNA after CNS injury. Brain Res. 1991;553:291–299. [PMC free article: PMC4352091] [PubMed: 1933286]
- 44.
- Logan A, Frautschy SA, Baird A. Basic fibroblast growth factor and central nervous system injury. Ann N Y Acad Sci. 1991;638:474–476. [PMC free article: PMC4086467] [PubMed: 1785823]
- 45.
- Logan A, Frautschy SA, Gonzalez AM. et al. A time course for the focal elevation of synthesis of basic fibroblast growth factor and one of its high-affinity receptors (flg) following a localized cortical brain injury. J Neurosci. 1992;12:3828–3837. [PMC free article: PMC4086626] [PubMed: 1403086]
- 46.
- GomezPinilla F, Lee JW, Cotman CW. Basic FGF in adult rat brain: cellular distribution and response to entorhinal lesion and fimbria-fornix transection. J Neurosci. 1992;12:345–355. [PMC free article: PMC6575703] [PubMed: 1309575]
- 47.
- GomezPinilla F, Cotman CW. Transient lesion-induced increase of basic fibroblast growth factor and its receptor in layer VIb (subplate cells) of the adult rat cerebral cortex. Neuroscience. 1992;49:771–780. [PubMed: 1436480]
- 48.
- Reilly JF, Kumari VG. Alterations in fibroblast growth factor receptor expression following brain injury. Exp Neurol. 1996;140:139–150. [PubMed: 8690057]
- 49.
- Takami K, Iwane M, Kiyota Y. et al. Increase of basic fibroblast growth factor immunoreactivity and its mRNA level in rat brain following transient forebrain ischemia. Exp Brain Res. 1992;90:1–10. [PubMed: 1521598]
- 50.
- Lippoldt A, Andbjer B, Rosen L. et al. Photochemically induced focal cerebral ischemia in rat: time-dependent and global increase in expression of basic fibroblast growth factor mRNA. Brain Res. 1993;625:45–56. [PubMed: 8242399]
- 51.
- Riva MA, Gale K, Mocchetti I. Basic fibroblast growth factor mRNA increases in specific brain regions following convulsive seizures. Brain Res Mol Brain Res. 1992;15:311–318. [PubMed: 1331686]
- 52.
- van der Wal EA, Gomez-Pinilla F, Cotman CW. Seizure-associated induction of basic fibroblast growth factor and its receptor in the rat brain. Neuroscience. 1994;60:311–323. [PubMed: 8072686]
- 53.
- Riva MA, Donati E, Tascedda F. et al. Short- and long-term induction of basic fibroblast growth factor gene expression in rat central nervous system following kainate injection. Neuroscience. 1994;59:55–65. [PubMed: 8190272]
- 54.
- Gall CM, Berschauer R, Isackson PJ. Seizures increase basic fibroblast growth factor mRNA in adult rat forebrain neurons and glia. Brain Res Mol Brain Res. 1994;21:190–205. [PubMed: 8170344]
- 55.
- Bugra K, Pollard H, Charton G. et al. aFGF, bFGF and flg mRNAs show distinct patterns of induction in the hippocampus following kainate-induced seizures. Eur J Neurosci. 1994;6:58–66. [PubMed: 7510570]
- 56.
- Matsushima K, Schmidt-Kastner R, Hogan MJ. et al. Cortical spreading depression activates trophic factor expression in neurons and astrocytes and protects against subsequent focal brain ischemia. Brain Res. 1998;807:47–60. [PubMed: 9756993]
- 57.
- Kawahara N, Ruetzler CA, Mies G. et al. Cortical spreading depression increases protein synthesis and upregulates basic fibroblast growth factor. Exp Neurol. 1999;158:27–36. [PubMed: 10448415]
- 58.
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression like depolarization. Physiol Rev. 2001;81:1065–1096. [PubMed: 11427692]
- 59.
- Ay H, Ay I, Koroshetz WJ. et al. Potential usefulness of basic fibroblast growth factor as a treatment for stroke. Cerebrovasc Dis. 1999;9:131–135. [PubMed: 10207203]
- 60.
- Nozaki K, Finklestein SP, Beal MF. Basic fibroblast growth factor protects against hypoxia-ischemia and NMDA neurotoxicity in neonatal rats. J Cereb Blood Flow Metab. 1993;13:221–228. [PubMed: 8436614]
- 61.
- Koketsu N, Berlove DJ, Moskowitz MA. et al. Pretreatment with intraventricular basic fibroblast growth factor decreases infarct size following focal cerebral ischemia in rats. Ann Neurol. 1994;35:451–457. [PubMed: 8154872]
- 62.
- Fisher M, Meadows ME, Do T. et al. Delayed treatment with intravenous basic fibroblast growth factor reduces infarct size following permanent focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1995;15:953–959. [PubMed: 7593356]
- 63.
- Tanaka R, Miyasaka Y, Yada K. et al. Basic fibroblast growth factor increases regional cerebral blood flow and reduces infarct size after experimental ischemia in a rat model. Stroke. 1995;26:2154–2158. [PubMed: 7482665]
- 64.
- Jiang N, Finklestein SP, Do T. et al. Delayed intravenous administration of basic fibroblast growth factor (bFGF) reduces infarct volume in a model of focal cerebral ischemia/reperfusion in the rat. J Neurol Sci. 1996;139:173–179. [PubMed: 8856649]
- 65.
- Bethel A, Kirsch JR, Koehler RC. et al. Intravenous basic fibroblast growth factor decreases brain injury resulting from focal ischemia in cats. Stroke. 1997;28:609–615. [PubMed: 9056620]
- 66.
- Nakata N, Kato H, Kogure K. Protective effects of basic fibroblast growth factor against hippocampal neuronal damage following cerebral ischemia in the gerbil. Brain Res. 1993;605:354–356. [PubMed: 8481788]
- 67.
- Dietrich WD, Alonso O, Busto R. et al. Posttreatment with intravenous basic fibroblast growth factor reduces histopathological damage following fluid-percussion brain injury in rats. J Neurotrauma. 1996;13:309–316. [PubMed: 8835798]
- 68.
- Kirschner PB, Henshaw R, Weise J. et al. Basic fibroblast growth factor protects against excitotoxicity and chemical hypoxia in both neonatal and adult rats. J Cereb Blood Flow Metab. 1995;15:619–623. [PubMed: 7790410]
- 69.
- Liu Z, Holmes GL. Basic fibroblast growth factor is highly neuroprotective against seizure-induced long-term behavioural deficits. Neuroscience. 1997;76:1129–1138. [PubMed: 9027873]
- 70.
- Anderson KJ, Dam D, Lee S. et al. Basic fibroblast growth factor prevents death of lesioned cholinergic neurons in vivo. Nature. 1988;332:360–361. [PubMed: 3352734]
- 71.
- Peterson DA, Lucidi-Phillipi CA, Murphy DP. et al. Fibroblast growth factor 2 protects entorhinal layer II glutamatergic neurons from axotomy-induced death. J Neurosci. 1996;16:886–898. [PMC free article: PMC6578815] [PubMed: 8558257]
- 72.
- Otto D, Frotscher M, Unsicker K. Basic fibroblast growth factor and nerve growth factor administered in gel foam rescue medial septal neurons after fimbria fornix transection. J Neurosci Res. 1989;22:83–91. [PubMed: 2926842]
- 73.
- Cummings BJ, Yee GJ, Cotman CW. bFGF promotes the survival of entorhinal layer II neurons after perforant path axotomy. Brain Res. 1992;591:271–276. [PubMed: 1446240]
- 74.
- Himmelseher S, Pfenninger E, Georgieff M. Effects of basic fibroblast growth factor on hippocampal neurons after axonal injury. J Trauma. 1997;42:659–664. [PubMed: 9137254]
- 75.
- Liu Z, Holmes GL. Basic fibroblast growth factor-induced seizures in rats. Neurosci Lett. 1997;233:85–88. [PubMed: 9350838]
- 76.
- Kawamata T, Alexis NE, Dietrich WD. et al. Intracisternal basic fibroblast growth factor (bFGF) enhances behavioral recovery following focal cerebral infarction in the rat. J Cereb Blood Flow Metab. 1996;16:542–547. [PubMed: 8964792]
- 77.
- Rowntree S, Kolb B. Blockade of basic fibroblast growth factor retards recovery from motor cortex injury in rats. Eur J Neurosci. 1997;9:2432–2441. [PubMed: 9464937]
- 78.
- MacMillan V, Judge D, Wiseman A. et al. Mice expressing a bovine basic fibroblast growth factor transgene in the brain show increased resistance to hypoxemic-ischemic cerebral damage. Stroke. 1993;24:1735–1739. [PubMed: 8236350]
- 78A.
- Cuevas P, Carceller F, Munoz-Willery I. et al. Intravenous fibroblast growth factor penetrates the blood-brain barrier and protects hippocampal neurons against ischemia-reperfusion injury. Surg Neurol. 1998;49:77–83. [PubMed: 9428898]
- 79.
- Sasaki K, Oomura Y, Suzuki K. et al. Acidic fibroblast growth factor prevents death of hippocampal CA1 pyramidal cells following ischemia. Neurochem Int. 1992;21:397–402. [PubMed: 1284624]
- 80.
- MacMillan V, WaltonRoche K, Davis J. Acidic fibroblast growth factor infusion reduces ischemic CA1 hippocampal damage in the gerbil. Can J Neurol Sci. 1993;20:37–40. [PubMed: 7682146]
- 81.
- Cuevas P, Carceller F, Reimers D. et al. Acidic fibroblast growth factor rescues gerbil hippocampal neurons from ischemic apoptotic death. Neurol Res. 1998;20:271–274. [PubMed: 9583591]
- 82.
- Hossain MA, Fielding KE, Trescher WH. et al. Human FGF1 gene delivery protects against quinolinateinduced striatal and hippocampal injury in neonatal rats. Eur J Neurosci. 1998;10:2490–2499. [PubMed: 9767380]
- 83.
- Li AJ, Oomura Y, Sasaki K. et al. Protective effect of acidic fibroblast growth factor against ischemia-induced learning and memory deficits in two tasks in gerbils. Physiol Behav. 1999;66:577–583. [PubMed: 10386900]
- 84.
- Sadohara T, Sugahara K, Urashima Y. et al. Keratinocyte growth factor prevents ischemiainduced delayed neuronal death in the hippocampal CA1 field of the gerbil brain. NeuroReport. 2001;12:71–76. [PubMed: 11201095]
- 85.
- Mark RJ, Fuson KS, KeaneLazar K. et al. Fibroblast growth factor8 protects cultured rat hippocampal neurons from oxidative insult. Brain Res. 1999;830:88–93. [PubMed: 10350562]
- 86.
- Logan A, Berry M. Transforming growth factor-beta 1 and basic fibroblast growth factor in the injured CNS. Trends Pharmacol Sci. 1993;14:337–342. [PubMed: 8249155]
- 87.
- Eclancher F, Perraud F, Faltin J. et al. Reactive astrogliosis after basic fibroblast growth factor (bFGF) injection in injured neonatal rat brain. Glia. 1990;3:502–509. [PubMed: 2148552]
- 88.
- Menon VK, Landerholm TE. Intralesion injection of basic fibroblast growth factor alters glial reactivity to neural trauma. Exp Neurol. 1994;129:142–154. [PubMed: 7925836]
- 89.
- Eclancher F, Kehrli P, Labourdette G. et al. Basic fibroblast growth factor (bFGF) injection activates the glial reaction in the injured adult rat brain. Brain Res. 1996;737:201–214. [PubMed: 8930367]
- 90.
- Mahler M, Ferhat L, Gillian A. et al. Tenascin C mRNA and tenascin C protein immunoreactivity increase in astrocytes after activation by bFGF. Cell Adhes Commun. 1996;4:175–186. [PubMed: 8969863]
- 91.
- Yoshida K, Gage FH. Fibroblast growth factors stimulate nerve growth factor synthesis and secretion by astrocytes. Brain Res. 1991;538:118–126. [PubMed: 1708303]
- 92.
- Reuss B, Hertel M, Werner S. et al. Fibroblast growth factors 5 and 9 distinctly regulate expression and function of the gap junction protein connexin43 in cultured astroglial cells from different brain regions. Glia. 2000;30:231–241. [PubMed: 10756073]
- 93.
- Mattson MP, Murrain M, Guthrie PB. et al. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9:3728–3740. [PMC free article: PMC6569923] [PubMed: 2585052]
- 94.
- Cheng B, Furukawa K, O'Keefe JA. et al. Basic fibroblast growth factor selectively increases AMPA receptor subunit GluR1 protein level and differentially modulates Ca2+ responses to AMPA and NMDA in hippocampal neurons. J Neurochem. 1995;65:2525–2536. [PubMed: 7595547]
- 95.
- Mattson MP, Kumar KN, Wang H. et al. Basic FGF regulates the expression of a functional 71 kDa NMDA receptor protein that mediates calcium influx and neurotoxicity in hippocampal neurons. J Neurosci. 1993;13:4575–4588. [PMC free article: PMC6576350] [PubMed: 7901348]
- 96.
- Boxer AL, Moreno H, Rudy B. et al. FGF2 potentiates Ca2+-dependent inactivation of NMDA receptor currents in hippocampal neurons. J Neurophysiol. 1999;82:3367–3377. [PubMed: 10601468]
- 97.
- Abe K, Saito H. Selective enhancement by basic fibroblast growth factor of NMDA receptor-mediated increase of intracellular Ca2+ concentration in hippocampal neurons. Brain Res. 1992;595:128–132. [PubMed: 1467948]
- 98.
- Collazo D, Takahashi H, McKay RD. Cellular targets and trophic functions of neurotrophin 3 in the developing rat hippocampus. Neuron. 1992;9:643–656. [PubMed: 1389181]
- 99.
- Mattson MP, Rychlik B, Chu C. et al. Evidence for calcium-reducing and excitoprotective roles for the calcium-binding protein calbindin D28k in cultured hippocampal neurons. Neuron. 1991; 6:41–51. [PubMed: 1670921]
- 100.
- Mattson MP, Lovell MA, Furukawa K. et al. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem. 1995;65:1740–1751. [PubMed: 7561872]
- 101.
- Hou JG, Cohen G, Mytilineou C. Basic fibroblast growth factor stimulation of glial cells protects dopamine neurons from 6-hydroxydopamine toxicity: involvement of the glutathione system. J Neurochem. 1997;69:76–83. [PubMed: 9202296]
- 102.
- Mark RJ, Keller JN, Kruman I. et al. Basic FGF attenuates amyloid-beta peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na/KATPase activity in hippocampal neurons. Brain Res. 1997;756:205–214. [PubMed: 9187334]
- 103.
- Guo Q, Sebastian L, Sopher BL. et al. Neurotrophic factors [activity-dependent neurotrophic factor (ADNF) and basic fibroblast growth factor (bFGF)] interrupt excitotoxic neurodegenerative cascades promoted by a PS1 mutation. Proc Natl Acad Sci USA. 1999;96:4125–4130. [PMC free article: PMC22431] [PubMed: 10097174]
- 104.
- Tamatani M, Ogawa S, Nunez G. et al. Growth factors prevent changes in Bcl-2 and Bax expression and neuronal apoptosis induced by nitric oxide. Cell Death Differ. 1998;5:911–919. [PubMed: 10203697]
- 105.
- Ay I, Sugimori H, Finklestein SP. Intravenous basic fibroblast growth factor (bFGF) decreases DNA fragmentation and prevents downregulation of Bcl-2 expression in the ischemic brain following middle cerebral artery occlusion in rats. Brain Res Mol Brain Res. 2001;87:71–80. [PubMed: 11223161]
- 106.
- Rosenblatt S, Irikura K, Caday CG. et al. Basic fibroblast growth factor dilates rat pial arterioles. J Cereb Blood Flow Metab. 1994;14:70–74. [PubMed: 8263060]
- 107.
- Tanaka R, Miyasaka Y, Yada K. et al. Basic fibroblast growth factor increases regional cerebral blood flow and reduces infarct size after experimental ischemia in a rat model. Stroke. 1995;26:2154–2158. [PubMed: 7482665]
- 108.
- Huang Z, Chen K, Huang PL. et al. bFGF ameliorates focal ischemic injury by blood flow-independent mechanisms in eNOS mutant mice. Am J Physiol. 1997;272:H1401–H1405. [PubMed: 9087617]
- 109.
- Yoshimura S, Takagi Y, Harada J. et al. FGF2 regulation of neurogenesis in adult hippocampus after brain injury. Proc Natl Acad Sci USA. 2001;98:5874–5879. [PMC free article: PMC33306] [PubMed: 11320217]
- 110.
- Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. [PubMed: 5861717]
- 111.
- Kaplan MS, Hinds JW. Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science. 1977;197:1092–1094. [PubMed: 887941]
- 112.
- Tretter YP, Hertel M, Munz B. et al. Induction of activin A is essential for the neuroprotective action of basic fibroblast growth factor in vivo. Nat Med. 2000;6:812–815. [PubMed: 10888932]
- 113.
- Tretter YP, Munz B, Hübner G. et al. Strong induction of activin expression after hippocampal lesion. NeuroReport. 1996;7:1819–1823. [PubMed: 8905672]
- 114.
- Lai M, Sirimanne E, Williams CE. et al. Sequential patterns of inhibin subunit gene expression following hypoxicischemic injury in the rat brain. Neuroscience. 1996;70:1013–1024. [PubMed: 8848164]
- 115.
- Lai M, Gluckman P, Dragunow M. et al. Focal brain injury increases activin bA mRNA expression in hippocampal neurons. NeuroReport. 1997;8:2691–2694. [PubMed: 9295102]
- 116.
- Hubner G, Alzheimer C, Werner S. Activin: a novel player in tissue repair processes. Histol Histopathol. 1999;14:295–304. [PubMed: 9987674]
- 117.
- Munz B, Hubner G, Tretter Y. et al. A novel role of activin in inflammation and repair. J Endocrinol. 1999;161:187–193. [PubMed: 10320815]
- 118.
- Krieglstein K, Suter-Crazzolara C, Fischer WH. et al. TGF-β superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J. 1995;14:736–742. [PMC free article: PMC398139] [PubMed: 7882977]
- 119.
- Iwahori Y, Saito H, Torii K. et al. Activin exerts a neurotrophic effect on cultured hippocampal neurons. Brain Res. 1997;760:52–58. [PubMed: 9237517]
- 120.
- Wu DD, Lai M, Hughes PE. et al. Expression of the activin axis and neuronal rescue effects of recombinant activin A following hypoxic-ischemic brain injury in the infant rat. Brain Res. 1999;835:369–378. [PubMed: 10415398]
- 121.
- Hughes PE, Alexi T, Williams CE. et al. Administration of recombinant human activin A has powerful neurotrophic effects on select striatal phenotypes in the quinolinic acid lesion model of Huntington's disease. Neuroscience. 1999;92:197–209. [PubMed: 10392842]
- 122.
- Nakamura T, Takio K, Eto Y. et al. Activin-binding protein from rat ovary is follistatin. Science. 1990;247:836–838. [PubMed: 2106159]
- 123.
- De Winter JP, Ten Dijke P, De Vries C J M. et al. Follistatins neutralize activin bioactivity by inhibition of activin binding to its type II receptors. Mol Cell Endocrinol. 1996;116:105–114. [PubMed: 8822271]
- 124.
- Krieglstein K, Reuss B, Maysinger D. et al. Transforming growth factor-beta mediates the neurotrophic effect of fibroblast growth factor 2 on midbrain dopaminergic neurons. Eur J Neurosci. 1998;10:2746–2750. [PubMed: 9767406]
Publication Details
Author Information and Affiliations
Authors
Christian Alzheimer and Sabine Werner.Copyright
Publisher
Landes Bioscience, Austin (TX)
NLM Citation
Alzheimer C, Werner S. Fibroblast Growth Factors. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.