NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.
Studies performed over the last decade have significantly increased our understanding of the role of Hedgehog (Hh) signalling in brain development. Here, we review the various in vitro and in vivo studies demonstrating the importance of Hh signalling for dorsoventral patterning of the telencephalon. The use of conditional knockouts has been particularly helpful in defining the spatial and temporal requirements of Hh signalling during telencephalic development. We also discuss the primary effectors of Hh signalling, the Gli family of transcription factors, and focus on Gli3, which is particularly important for telencephalic development, as reflected in the severe telencephalic phenotype of Gli3 mutant mice. The presence of some dorsoventral patterning in animals lacking both Shh and Gli3 implies that, although these molecules are major players in patterning the telencephalon, other patterning factors exist.
Introduction
The secreted morphogen, Sonic hedgehog (Shh), is vital for ventral patterning along the entire rostrocaudal extent of the neural tube.1-3 Although most work has concentrated on the role of Shh in patterning of the caudal part of the neural tube, the spinal cord, studies are beginning to elucidate the role that Shh plays in the development of the most rostral part of the neural tube, the telencephalon.
The Hedgehog Signalling Pathway
The Shh gene, along with genes for Indian hedgehog (Ihh) and Desert hedgehog (Dhh), are homologues of the Drosophila gene hedgehog and code for ˜45-kD precursor proteins.4 When Hh binds to the transmembrane receptor, Patched (Ptc), an inhibitory effect on Smoothened (Smo) is relieved and the pathway is activated (for a thorough review of these interactions, see ref. 5). In Drosophila, Hh signalling is transduced by one protein, the zinc-finger transcription factor cubitus interruptus (Ci) (reviewed in refs. 5, 6). In the absence of Hh, Ci is cleaved to form an N-terminal fragment which acts as a transcriptional repressor. When Hh is present, the cleavage of Ci is inhibited and the full-length form of Ci is able to act as a transcriptional activator.
The Hh signalling pathway is more complex in vertebrates. One important difference between Drosophila and vertebrates is that there are three proteins in vertebrates which are homologous to Ci: Gli1, Gli2 and Gli3.7,8 It has been proposed that repressor and activator functions of Ci have been distributed among the three Gli proteins. For instance, expression of Gli1 and Gli2 results in activation of Hh target genes when expressed in Drosophila (similar to the actions of full-length Ci) whereas expression of Gli3 results in the repression of target genes (similar to the actions of cleaved Ci).9,10 Indeed, combined expression of Gli1 and Gli3 is able to substitute for Ci during Drosophila development.9
Based on these results in Drosophila, it is tempting to postulate that Gli1 and Gli2 act as transcriptional activators and Gli3 acts as a transcriptional repressor of Hh target genes in vertebrates. However, the situation is far more complex than this. For instance, although expression of Gli1 results in transcriptional activation of various genes (cyclin D2;11Ptch1;11,12Gli1;13Bcl-2;14Bmp4;15Bmp7;15HNF3 β16,17), it is also able to cause down-regulation of gene transcription (plakoglobin11). Furthermore, although expression of Gli3 can result in transcriptional repression,13,16,18,19 Gli3 has also been shown to mediate Shh-induced activation of the Gli1 promoter13 and expression of Gli3 can result in an increase in transcription of Ptch1,12,20Bmp415 and Bmp7.15 Also, Gli3 has been shown to have activator function in vivo.21-23 Of course, it must be taken into account that many of the studies looking at the transcriptional properties of the Glis have been performed in artificial over-expression systems in vitro and that the transcriptional activities of the Gli proteins may be very different in vivo. Furthermore, the Glis may function differently depending on location and time of action.
Understanding the transcriptional repertoire of the Gli proteins is further complicated by the fact that not all Gli proteins are processed in a similar fashion to Ci. For instance, Gli3, but not Gli1, is cleaved in the absence of Hh.10,13,24-26 Furthermore, the shorter form of Gli3 has been shown to be a more potent repressor of transcription than full-length Gli3.12,24 Because it is unclear in most studies whether Gli3 is cleaved, or the relative amounts of full-length and short forms present in the system, it is difficult to determine whether the transcriptional effects of Gli3 expression are mediated by the full-length or short form of the protein.
Defining the relationship between Shh and the Glis is made even more difficult by the observation that (unlike Hh and Ci in Drosophila) Shh can affect the transcription of Gli1 and Gli3. Shh has been shown to increase Gli116,27 and decrease Gli3 transcription in various systems.19,28-30 Furthermore, it has been suggested that Gli3 represses Shh transcription based on observations of ectopic Shh expression in the limb and spinal cord of Gli3 mutant animals.19,31,32 However, whether Shh and Gli3 are cross-repressive in all tissues is unclear.
Overview of Telencephalic Development
The neural plate is formed from the ectodermal layer of the gastrulating embryo and gives rise to the entire central nervous system (CNS). Neural folds arise in the neural plate (fig. 1A), appose and fuse to form the neural tube (fig. 1B). The brain develops from the most anterior region of the neural tube and is divided into three primary vesicles: the hindbrain vesicle (rhombencephalon), the midbrain vesicle (mesencephalon) and the forebrain vesicle (prosencephalon). The forebrain becomes divided into the diencephalon caudally and telencephalon rostrally. Rapid proliferation of telencephalic cells results in the disproportionate swelling of the telencephalon which forms a pair of fluid-filled vesicles (telencephalic vesicles). The telencephalon eventually differentiates to become the olfactory bulbs anteriorly, the cerebral cortex dorsally and the basal ganglia ventrally.
During the second half of embryogenesis (˜E11 onward in mouse), distinct telencephalic progenitor zones are morphologically apparent (fig. 2). For example, two physically distinguishable eminences are found in the ventral region of the telencephalon: the lateral ganglionic eminence (LGE), the precursor to the adult striatum, and the more ventrally positioned medial ganglionic eminence (MGE), which gives rise to the globus pallidus. The striatum and globus pallidus comprise the basal ganglia, which are important for motor function. Cells from the MGE and LGE, as well as from the recently described caudal ganglionic eminence, produce GABAergic interneurons which migrate to populate a wide range of mature telencephalic structures.33 Around the time the LGE and MGE become physically recognisable, the dorsal midline of the telencephalon invaginates, leading to the separation of the telencephalic vesicles. This dorsal midline structure gives rise to the hippocampus, a structure crucial for learning and memory, as well as choroid plexus, which generates cerebrospinal fluid. The neocortex, which underpins complex cognitive functions, arises from the dorsolateral area of the telencephalon.
In addition to their distinguishable morphology, embryonic telencephalic progenitor domains have unique gene expression profiles (fig. 2). For example, the MGE uniquely expresses the transcription factor, Nkx2.1, whereas transcription factors such as Emx1 and Pax6 are expressed in the cerebral cortex. Characterising these gene expression patterns has facilitated analyses of telencephalic regional specification in various mutant embryos, as described below.
Role of Shh in Telencephalic Dorsoventral Patterning
One of the first studies to implicate Shh in telencephalic regional specification showed that Shh induces the expression of the MGE marker Nkx2.1 in telencephalic neural plate explants.34 Genetic evidence for the involvement of Shh in telencephalic development came from the discovery that humans heterozygous for mutations in the SHH gene suffer from holoprosencephaly (HPE).35,36 Rather than becoming cleaved into distinct left and right hemispheres, the holoprosencephalic telencephalon develops as a single unpaired vesicle and, in extreme cases, ventral structures including the striatum and globus pallidus are completely absent. As a consequence of the lack of ventral diencephalic structures, the optic primordia fail to separate, resulting in a single cyclopic eye.
Around the same time as the human SHH gene was implicated in HPE, researchers generated transgenic mice mutant for Shh.37Shh-/- animals die at birth, have cyclopic eyes, lack olfactory bulbs and exhibit defects in the development of ventral structures along the entire neuraxis. The forebrain is particularly affected and strikingly reminiscent of human HPE. The telencephalon is severely hypoplastic, uninvaginated and the ganglionic eminences are not morphologically identifiable. Consistent with the lack of ventral telencephalic structures, expression of genes characteristic of the most ventral region of the telencephalon, such as Nkx2.1, Lhx6 and Gsh1, is completely absent.38-40 In concert with the reduction of ventral gene expression, genes such as Emx1 and Pax6, normally restricted to the dorsal telencephalon, are expressed throughout the majority of the remaining telencephalic tissue.37,41,42
It has recently been observed, however, that ventral gene expression is not totally absent in the Shh-/- telencephalon. In less severely affected Shh-/- embryos, a small ventral telencephalic domain continues to express genes such as Gsh2, Mash1 and Dlx2.39,40 The gene expression profile of this ventral domain is reminiscent of wild-type LGE. Consequently, whilst providing good evidence for the importance of Shh in setting up a correctly patterned ventral telencephalon, the Shh-/- phenotype demonstrates that Shh is not wholly necessary for the specification of all ventral cell types in the telencephalon.
It remains an open question as to which factors induce the residual ventral gene expression in Shh mutants. It is possible that other Hh homologues can pattern the telencephalon or can partially compensate for the absence of Shh. In support of this possibility, mice mutant for both Shh and Ihh appear to lack all ventral character throughout the CNS.43 This phenotype is essentially indistinguishable from Smo-/- mutants, which are unable to transduce any Hh signal.43 It is also possible that Hh-independent signalling pathways can induce ventral gene expression. Indeed, the ability of both Shh-/- and Smo-/- telencephalic cells to express ventral telencephalic markers when Gli3 is removed39 (see below) strongly supports the idea that Hh signalling is not the only inducer of ventral telencephalic fate. There is evidence that retinoids, acting in a pathway parallel to that of Shh, induce ventral interneurons in the spinal cord.44,45 It is likely that retinoids also play a role in patterning the telencephalon.46-48
Although Shh and Smo mutants clearly demonstrate the importance of these factors during development, they do not define the spatial and temporal requirements of Hh signalling during telencephalic development. For example, because the appearance of MGE-specific gene expression occurs before Shh is expressed in this region,49 it is most likely that sources of Shh outside the telencephalon itself influence its patterning. But exactly where and when is Hh signalling required for telencephalic patterning? A range of cellular and genetic approaches has provided insight into these issues. For instance, Gunhaga et al50 demonstrate that blocking Shh signalling in epiblast explants from gastrula stage embryos results in failure of ventral telencephalic cell specification. Because Shh is expressed in the anterior primitive streak and Henson's node at gastrula stages,4,50,51 it is believed that these sources of Shh are crucial for specification of the MGE.
Shh signalling from the prechordal plate (mesendodermal tissue underlying the prospective rostral diencephalon) (fig. 1),52 may also be required for specification of the ventral telencephalon. Rostral neural plate explants lacking prechordal plate do not express Nkx2.1, whilst transplantation of prechordal plate results in expression of Nkx2.1 and repression of lateral neural plate markers.34,53-55 Furthermore, Shh can induce Nkx2.1 expression in neural explants lacking prechordal plate.34,53,55 Although the prechordal plate does not lie directly under the telencephalon, it may still be an important source of Shh with regard to telencephalic patterning due to the proposed long-range actions of Shh.56-58
Later in telencephalic development, the MGE itself becomes a source of Shh59,60 (fig. 2) and in vitro studies have demonstrated that Shh can induce gene expression characteristic of the LGE and inhibit dorsal marker expression in telencephalic explants.61 Interestingly, even at high concentrations, Shh is unable to induce expression of the MGE marker, Nkx2.1, at this developmental stage.61 These experiments suggest that the role of Shh in telencephalic development is regulated temporally by changes in responsiveness to Shh. Thus, early signalling from extra-telencephalic sources appears to induce MGE fates and later signalling from within the telencephalon itself seems to induce LGE fates. It will be very interesting to determine, at a molecular level, what underlies these changes and whether they involve context-dependent alteration in Gli target genes.
Recent work using conditional gene ablation has also attempted to unravel the temporal and spatial requirements for Hh signalling. Two studies involving the conditional ablation of Smo (in order to abolish all Hh signalling) or Shh reveal strikingly different telencephalic phenotypes depending on the timing of gene excision. Machold et al62 used Cre recombinase under the control of the Nestin promoter to remove ‘floxed’ alleles of either Shh (Shhc/-;NestinCre) or Smo (Smoc/-;NestinCre) in neural progenitors. In these mutants, target gene transcription is reduced by E10.5 and abolished by E12.5. Removal of Shh or Hh signalling by these means results in a surprisingly normal telencephalon, although the olfactory bulbs are reduced in size. In stark contrast to the Shh-/- telencephalon, both the MGE and LGE in Shhc/-;NestinCre and Smoc/-;NestinCre animals are morphologically present and exhibit largely appropriate gene expression. The MGE is variably reduced in size and contains considerably fewer oligodendrocyte precursors, prefiguring the paucity of oligodendrocytes observed later in development. More severe defects were observed postnatally, where there were significantly reduced numbers of progenitors in the neocortical subventricular zone and hippocampal proliferative zones, supporting the idea that Shh is required in adult mammals to maintain telencephalic stem cell niches.63
Fuccillo et al64 used a floxed allele of Smo to ablate Hh signalling earlier in telencephalic development using Cre under the control of the Foxg1 promoter (Smoc/-;Foxg1Cre). Foxg1 is expressed throughout the telencephalic neuroepithelium from its inception at neural plate stages (˜E7.5 in mouse).53 This early ablation of Smo, which is estimated to be complete by E9, results in a much more severe phenotype than animals where Shh or Smo is excised using Nestin-Cre. Smoc/-;Foxg1Cre embryos lack all trace of the ventral telencephalon, as assessed by morphology and gene expression, and all remaining telencephalic tissue expresses dorsal markers. As in the Shhc/-;NestinCre and Smoc/-;NestinCre animals, the olfactory bulbs of Smoc/-;Foxg1Cre embryos are reduced in size. Later in development, the majority of telencephalic GABAergic interneurons and oligodendrocytes, two ventrally-derived cell types, are absent.
From these two studies, we can surmise that early Hh signalling, between the activation of Foxg1-Cre expression (˜E7.5) and Nestin-Cre expression (˜E10.5), is crucial in setting up ventral telencephalic progenitor domains and the cells derived from them. Furthermore, the relatively mild phenotype of the Shhc/-;NestinCre and Smoc/-;NestinCre mice suggests that Hh signalling after approximately E12.5 is not required for the maintenance of ventral telencephalic territories which, as suggested above, are specified earlier in development.
The absence of ventral telencephalic fate specification observed in the Smoc/-;Foxg1Cre mutant would appear to contradict in vitro studies suggesting that early Shh signalling during gastrulation (before significant Foxg1 expression) is necessary and sufficient for induction of ventral telencephalic cell fates.50 The Smoc/-;Foxg1Cre mutant presumably has intact Hh signalling at gastrulation, which should be sufficient for the induction of at least some ventral fate. Some of the contradictions between these studies may simply reflect the inherent differences that exist between in vitro and in vivo studies. It is possible that, whilst very early Hh signalling may indeed specify ventral lineages, maintenance of ventral fates in the absence of persistent Hh signalling can only occur in the rarefied environment of the tissue culture dish. In vivo, continued Hh signalling may be required to maintain ventral fate and removing Hh signalling during this phase may expose ventrally specified cells to dorsalising factors, which are likely to be absent in vitro.
It is also interesting to note that the ventral patterning defects in Smoc/-;Foxg1Cre mutants are more severe than those found in the constitutive Shh knockouts. This might best be explained by activity of other Hh ligands in the embryo. Indeed, a low level of Smo-dependent Hh signalling has been reported to be present in the Shh-/- neural tube.65 The ability of Shh-/- (but not Smo-/-) telencephalic cells to respond to other Hh ligands (if present) might contribute to the different phenotypes of the Shh-/- and Smoc/-;Foxg1Cre mutants. Thus, it will be important to determine whether the ventral telencephalic phenotype of embryos where Shh is excised by Foxg1-Cre is similar to or less severe than that of the Foxg1-Cre excised Smo mutants. It is also possible that heterozygosity at the Foxg1 locus (due to insertion of Cre) synergises with the absence of Smo to contribute to the severe ventral phenotype observed.
The role of Shh in the development of dorsomedial telencephalic structures remains more ambiguous than its role in patterning the ventral telencephalon. In contrast to the increased severity of ventral patterning defects of Smoc/-;Foxg1Cre mutants compared to Shh mutants, the dorsal telencephalic midline of Smoc/-;Foxg1Cre mutants appears to be largely unaffected, whereas it is morphologically absent in Shh mutant mice and holoprosencephalic humans. This discrepancy may suggest that very early Hh signalling (before Foxg1-Cre expression) is required to pattern the dorsal midline. However, Ohkubo et al42 demonstrate that Bmp2 and 7 and Msx1 and 2, genes expressed in the dorsal-most regions of the telencephalon, are still expressed, and may even be overexpressed, in the Shh-/- telencephalon. As such, the requirement for Shh in dorsal midline development may be one of morphological induction rather than cell fate specification. It is also possible that Shh has some Smo-independent activity in this region of the telencephalon.
Role of Shh in Cell Death and Proliferation
In addition to affecting telencephalic dorsoventral patterning, Hh signalling likely influences other processes during telencephalic development. The small size of the telencephalon in various Shh and Smo mutants37,42,62,64,66 suggests that cell death and proliferation may be affected. As Bmps and their effectors, Msx transcription factors, have been shown to mediate cell death in many regions of the developing embryo,67,68 including the brain,42,69 - 72 the increased Bmp and Msx expression in the Shh mutant42 may mediate some of the increase in cell death observed, although it is not known whether Hh signalling is directly required to repress Bmp expression. A more direct mechanism could involve the pro-apoptotic function of Ptc. Thibert et al73 have shown that Ptc induces apoptotic cell death in neuroepithelial cells that can be prevented by binding of Shh to Ptc. In the absence of Shh, this property of Ptc may contribute to the increased cell death observed in the Shh mutant.42 Because this type of Ptc-induced cell death does not involve the Ptc/Smo transducing module,73 the cell death observed in the Smoc/-;Foxg1Cre telencephalon may also be effected by this pathway.64
The role of Shh in proliferation is supported by the observations of decreased proliferation in the Shh-/- telencephalon66 and the increased proliferation of neocortical precursors after Shh treatment in vitro.66 It has also been observed that telencephalic vesicles are enlarged after ectopic expression of Shh in vivo.39,74 One possible mechanism for the mitogenic effect of Shh is its ability to relieve the inhibition of proliferation caused by Ptc's interaction with cyclin B1.75 However, as no obvious proliferation defects have been observed in the telencephalon of Smoc/-;Foxg1Cre,64Shhc/-;NestinCre62 and Smoc/-;NestinCre mice,62 further work is warranted in order to determine when and where Hh signalling is required for telencephalic cell proliferation.
Role of Shh in Cell Type Specification
Hh signalling is important for the specification of two cell types derived from the ventral telencephalon: oligodendrocytes and GABAergic interneurons. As mentioned above, oligodendrocytes are depleted in Nestin-Cre and Foxg1-Cre excised Hh signalling mutants62,64 and the Shh-/- telencephalon lacks oligodendrocyte precursors altogether.76 Although various in vitro and in vivo studies suggest that Shh is necessary and sufficient for telencephalic oligodendrocyte generation,59,76-78 the ability of Shh-/- telencephalic tissue to generate oligodendrocytes in vitro59 suggests that Shh is not required in vitro for oligodendrocyte generation and/or that there exists a pathway parallel to that of Shh for oligodendrogenesis. Shh also plays a role in the generation of GABAergic interneurons. As mentioned earlier, most GABAergic interneurons are absent in the Smoc/-;Foxg1Cre mutant.64 Moreover, Shh induces dorsomedial telencephalic cells to produce more GABAergic interneurons than normal in vitro.79
Telencephalic Phenotypes of Gli Mutants
Given the strong telencephalic phenotype of mice mutant for Shh or Smo and that the Gli proteins are transducers of Hh signalling in vertebrates, it is reasonable to assume that mice mutant for Glis would also have strong telencephalic phenotypes. Interestingly, mice mutant for Gli1 do not show any obvious abnormalities,80,81 demonstrating that Gli1 is dispensable for normal development and/or may be compensated for by the presence of other Glis. Mice mutant for Gli2 were initially reported to have a grossly normal telencephalon,81,82 although, on an outbred background, these mice display a variably penetrant incidence of exencephaly.83 In nonexencephalic Gli2-/- mice, the telencephalic vesicles are expanded but have a thinner proliferative zone.83
The Gli3 mouse mutant has the most dramatic telencephalic phenotype of all three Gli mutants. Gli3 is widely expressed very early in mouse development in both the mesoderm and ectoderm.8 It is then expressed throughout the telencephalon, with high expression in the cortex and LGE and lower expression in the MGE (fig. 2).8,84 The most widely studied strain of mice with mutation in the Gli3 gene is referred to as extra toes (Xt) due to heterozygotes demonstrating polydactyly.85,86 The Xt deletion results in a Gli3 transcript lacking the sequence coding for the DNA binding element,87,88 presumably resulting in a functionally null Gli3 allele. Mice homozygous for the Xt allele die perinatally, display extreme polydactyly and are often exencephalic.86 In nonexencephalic Gli3Xt/Xt mice, the telencephalon is highly abnormal. Gli3Xt/Xt embryos have no olfactory bulbs and do not develop dorsomedial telencephalic structures such as the hippocampus and choroid plexus.39,86,89-92 The tissue of the putative neocortex is severely disorganised and heterotopic clusters of cells are observed in this area.90
Gene expression patterns in the Gli3Xt/Xt dorsal telencephalon are distinctly abnormal. Genes such as Emx1and 2 have been reported to be reduced or absent in the Gli3Xt/Xt dorsal telencephalon, although the telencephalon retains dorsal character as reflected by the perdurance of some dorsal marker expression.41,90,92-94 Furthermore, the genes Dlx2 and Mash1, which are normally ventrally-restricted, are expressed in the dorsal region of the telencephalon, particularly rostrally.92,94 The boundary between the dorsal telencephalon and the ventral telencephalon is also compromised.39,92 Gene expression within the ventral telencephalon appears relatively normal with Nkx2.1 being expressed in the area of the MGE and Gli1 expressed at the boundary between the MGE and LGE.90,92,94
Based on studies observing ectopic expression of Shh in Gli3Xt/Xt limbs (and with lower penetrance in the Gli3Xt/Xt spinal cord19),31,32 it was thought that ectopic expression of Shh might be observed in the dorsal telencephalon of Gli3Xt/Xt mice, contributing to some of the telencephalic defects present in these embryos. Somewhat surprisingly, Shh expression appears to be normal in the Gli3Xt/Xt ventral telencephalon.41,90,92 Hh target genes, such as Gli1 and Ptc1, also appear to be normally expressed,90,92 providing further evidence that Hh signalling is not aberrantly activated in the dorsal region of the Gli3Xt/Xt telencephalon.
Abnormal expression of genes for signalling molecules other than Shh is, however, observed in the Gli3Xt/Xt telencephalon. For instance, expression of various Bmps are decreased or absent in the dorsal telencephalon90,92 and the cortical hem, a Bmp- and Wnt-rich signalling center in the dorsal midline important for formation of the hippocampus and choroid plexus, does not form in the Gli3Xt/Xt mutant.91 Furthermore, Fgf8 expression is expanded in the anterior neural ridge41 and dorsomedial telencephalon.94 Because these signalling molecules are crucial for the proper development of the telencephalon,95-97 it is likely that the abnormal expression of these molecules contributes to the severe phenotype of the Gli3Xt/Xt telencephalon.
Role of Gli3 in Cell Death
Whereas increased cell death is observed in Hh signalling mutants, decreased cell death is observed in the forebrain of Gli3 mutants.41 As Bmps mediate cell death in many regions of the developing embryo, one possible mechanism through which Gli3 might regulate cell death is via modulation of Bmp signalling. This is supported by findings that expression of several Bmps is lost or reduced in the Gli3Xt/Xt telencephalon.90,92 Reduced expression of Bmp genes is consistent with the ability of Gli3 to enhance promoter activity of Bmp4 and Bmp7.15 Gli3 may also decrease cell death by directly affecting genes such as the anti-apoptotic factor Bcl2.98 Because the repressor form of Gli3 is able to inhibit transactivation of the Bcl2 gene in vitro,14 it is possible that loss of Gli3 function results in an overall increase in Bcl2 activity, resulting in decreased levels of cell death.
Loss of Gli3 Partially Rescues Shh-/- Telencephalic Phenotypes
Based on work in the limb25,99 and spinal cord,65,100 it has been proposed that Shh acts to antagonise the actions of Gli3. For example, Litingtung and Chiang100 were the first to demonstrate that many of the ventral spinal cord defects found in Shh-/- animals were partially rescued in Shh-/-; Gli3Xt/+ animals and further rescued in Shh-/-; Gli3Xt/+ animals. Based on these findings, it was suggested that Gli3 normally represses ventral fates and that Shh is required to counteract Gli3 function in order to allow ventral fate specification in the spinal cord. Could Hh signalling play a similar role with respect to Gli3 in the telencephalon?
It has been shown that loss of Gli3 can partially rescue the telencephalic phenotype of Shh-/- mutants.39 For example, formation of two telencephalic vesicles is restored when one copy of Gli3 is removed from Shh-/- embryos. Furthermore, correct regional expression of ventral markers Mash1, Dlx2 and Gsh2 appears to be restored in the Shh-/-; Gli3Xt/+ mutant compared to the aberrant expression of these genes in the Shh-/- mutant. There is even a small amount of Nkx2.1 expression present in the Shh-/-; Gli3Xt/+ mutant, which is never seen in the Shh-/- telencephalon, suggesting that some MGE character is restored in the Shh-/-; Gli3Xt/+ mutant. Unfortunately, the high incidence of exencephaly in double homozygous mutants precluded a thorough analysis of the dorsoventral patterning of these animals. However, it appears that the MGE is more fully specified in the Shh-/-; Gli3Xt/+ mutant than in the Shh-/-; Gli3Xt/+ and Shh-/- mutants. Thus, ventral patterning is able to occur in the absence of both Shh and Gli3, demonstrating that other pathways are capable of dorsoventral patterning in the telencephalon.
Conclusion
It has been a decade since Hh signalling was first implicated in development of the telencephalon and the Hh pathway now has a unique and undisputed position as a key regulator of ventral fate specification. Nevertheless, many issues remain to be addressed regarding the mechanism of the Hh-Gli signalling pathway. For example, it will be important to define what the respective contributions of the full-length and cleaved forms of the Gli3 protein are during telencephalic development. One group has already begun to address this by generating a mouse (Gli3Δ699/Δ699 mutant) that only expresses a truncated Gli3 protein similar to the cleaved form of Gli3.101 This mutant form of Gli3 would thus have DNA binding capability, unlike the potential protein product resulting from the Xt allele. Interestingly, these Gli3Δ699/Δ699 mutants exhibit a very different phenotype to that of Gli3Xt/Xt mice. They exhibit a variety of defects, such as imperforate anus and absence of adrenal glands,101 which are not present in Gli3Xt/Xt mice. Furthermore, Gli3Δ699/Δ699 mice do not display the spinal cord defects found in Gli3Xt/Xt mice.102 Although analysis of the rostral portion of the nervous system in these mice has not been published, it appears unlikely that these mice have a similar telencephalic phenotype to Gli3Xt/Xt mice. If the telencephalon of the Gli3Δ699/Δ699 mutant is correctly patterned, it would suggest that full-length Gli3 is either not necessary for telencephalic development, or other proteins, presumably Gli1 or Gli2, are able to compensate for its absence. The generation of a mouse expressing a cleavage-resistant form of Gli3 would be of great help in defining the relative importance of full-length and cleaved forms of Gli3 during development.
Questions regarding the relationship between Hh ligands and the Gli proteins also remain. For example, do Hhs have Gli-independent action in telencephalic development? Conversely, to what extent do Gli proteins have roles independent of their Hh transducing functions? With regard to the first issue, the identification of a Shh-response element in the COUP-TFII promoter that is distinct from the Gli-response element suggests that factors other than Gli can transduce the Shh signal.103 This is particularly relevant to telencephalic development since COUP-TFII is thought to be involved in the migration of neurons from the ventral telencephalon.104 Furthermore, the Hh receptor Ptc has been shown to modulate both cell death73 and proliferation75 independent of the Ptc/Smo/Gli transducing module and these actions are regulated by binding of Shh, adding further support to the notion that Shh can act without Gli proteins. Regarding whether Gli proteins have roles independent of their Hh transducing functions, there is evidence that C-terminally truncated Gli3 is able to interact with Smads,105 transducers of Bmp signalling. This, in addition to the ability of Glis to activate the Bmp4 and Bmp7 promoters,15 suggests that Glis are able to influence Bmp signalling at both a transcriptional and post-translational level. Thus, it is important to keep in mind that not all functions of Shh and Gli proteins are confined to the well-described linear Shh-Smo-Gli pathway and that future models will need to accommodate these actions.
Acknowledgments
The authors are supported by the National Institutes of Health (NEI individual NRSA postdoctoral fellowship, PAZ), the Wellcome Trust (BM, DJP), the Medical Research Council (DJP) and the Biotechnology and Biological Sciences Research Council (DJP).
References
- 1.
- Ericson J, Muhr J, Jessell TM. et al. Sonic hedgehog: A common signal for ventral patterning along the rostrocaudal axis of the neural tube. Int J Dev Biol. 1995;39(5):809–816. [PubMed: 8645565]
- 2.
- Lumsden A, Krumlauf R. Patterning the vertebrate neuraxis. Science. 1996;274(5290):1109–1115. [PubMed: 8895453]
- 3.
- Patten I, Placzek M. The role of Sonic hedgehog in neural tube patterning. Cell Mol Life Sci. 2000;57(12):1695–1708. [PubMed: 11130176]
- 4.
- Echelard Y, Epstein DJ, St-Jacques B. et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75(7):1417–1430. [PubMed: 7916661]
- 5.
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001;15(23):3059–3087. [PubMed: 11731473]
- 6.
- Lum L, Beachy PA. The Hedgehog response network: Sensors, switches, and routers. Science. 2004;304(5678):1755–1759. [PubMed: 15205520]
- 7.
- Walterhouse D, Ahmed M, Slusarski D. et al. Gli, a zinc finger transcription factor and oncogene, is expressed during normal mouse development. Dev Dyn. 1993;196(2):91–102. [PubMed: 8364225]
- 8.
- Hui CC, Slusarski D, Platt KA. et al. Expression of three mouse homologs of the Drosophila segment polarity gene cubitus interruptus, Gli, Gli-2, and Gli-3, in ectoderm- and mesoderm-derived tissues suggests multiple roles during postimplantation development. Dev Biol. 1994;162(2):402–413. [PubMed: 8150204]
- 9.
- von MeringC, Basler K. Distinct and regulated activities of human Gli proteins in Drosophila. Curr Biol. 1999;9(22):1319–1322. [PubMed: 10574767]
- 10.
- Aza-Blanc P, Lin HY, Ruiz i Altaba A. et al. Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development. 2000;127(19):4293–4301. [PubMed: 10976059]
- 11.
- Yoon JW, Kita Y, Frank DJ. et al. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem. 2002;277(7):5548–5555. [PubMed: 11719506]
- 12.
- Agren M, Kogerman P, Kleman MI. et al. Expression of the PTCH1 tumor suppressor gene is regulated by alternative promoters and a single functional Gli-binding site. Gene. 2004;330:101–114. [PubMed: 15087129]
- 13.
- Dai P, Akimaru H, Tanaka Y. et al. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem. 1999;274(12):8143–8152. [PubMed: 10075717]
- 14.
- Bigelow RL, Chari NS, Unden AB. et al. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J Biol Chem. 2004;279(2):1197–1205. [PubMed: 14555646]
- 15.
- Kawai S, Sugiura T. Characterization of human bone morphogenetic protein (BMP)-4 and -7 gene promoters: Activation of BMP promoters by Gli, a sonic hedgehog mediator. Bone. 2001;29(1):54–61. [PubMed: 11472891]
- 16.
- Sasaki H, Hui C, Nakafuku M. et al. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124(7):1313–1322. [PubMed: 9118802]
- 17.
- Hynes M, Stone DM, Dowd M. et al. Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron. 1997;19(1):15–26. [PubMed: 9247260]
- 18.
- Sasaki H, Nishizaki Y, Hui C. et al. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126(17):3915–3924. [PubMed: 10433919]
- 19.
- Ruiz i Altaba A. Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development. 1998;125(12):2203–2212. [PubMed: 9584120]
- 20.
- Shin SH, Kogerman P, Lindstrom E. et al. GLI3 mutations in human disorders mimic Drosophila cubitus interruptus protein functions and localization. Proc Natl Acad Sci USA. 1999;96(6):2880–2884. [PMC free article: PMC15863] [PubMed: 10077605]
- 21.
- Motoyama J, Milenkovic L, Iwama M. et al. Differential requirement for Gli2 and Gli3 in ventral neural cell fate specification. Dev Biol. 2003;259(1):150–161. [PubMed: 12812795]
- 22.
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6(1):103–115. [PubMed: 14723851]
- 23.
- Tyurina OV, Guner B, Popova E. et al. Zebrafish Gli3 functions as both an activator and a repressor in Hedgehog signaling. Dev Biol. 2005;277(2):537–556. [PubMed: 15617692]
- 24.
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–434. [PubMed: 10693759]
- 25.
- Litingtung Y, Dahn RD, Li Y. et al. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418(6901):979–983. [PubMed: 12198547]
- 26.
- Bastida MF, Delgado MD, Wang B. et al. Levels of Gli3 repressor correlate with Bmp4 expression and apoptosis during limb development. Dev Dyn. 2004;231(1):148–160. [PubMed: 15305295]
- 27.
- Lee J, Platt KA, Censullo P. et al. Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development. 1997;124(13):2537–2552. [PubMed: 9216996]
- 28.
- Marigo V, Johnson RL, Vortkamp A. et al. Sonic hedgehog differentially regulates expression of GLI and GLI3 during limb development. Dev Biol. 1996;180(1):273–283. [PubMed: 8948590]
- 29.
- Takahashi M, Tamura K, Buscher D. et al. The role of Alx-4 in the establishment of anteroposterior polarity during vertebrate limb development. Development. 1998;125(22):4417–4425. [PubMed: 9778501]
- 30.
- Schweitzer R, Vogan KJ, Tabin CJ. Similar expression and regulation of Gli2 and Gli3 in the chick limb bud. Mech Dev. 2000;98(1-2):171–174. [PubMed: 11044624]
- 31.
- Buscher D, Bosse B, Heymer J. et al. Evidence for genetic control of Sonic hedgehog by Gli3 in mouse limb development. Mech Dev. 1997;62(2):175–182. [PubMed: 9152009]
- 32.
- Masuya H, Sagai T, Moriwaki K. et al. Multigenic control of the localization of the zone of polarizing activity in limb morphogenesis in the mouse. Dev Biol. 1997;182(1):42–51. [PubMed: 9073443]
- 33.
- Corbin JG, Nery S, Fishell G. Telencephalic cells take a tangent: Nonradial migration in the mammalian forebrain. Nat Neurosci. 2001;(4 Suppl):1177–1182. [PubMed: 11687827]
- 34.
- Ericson J, Muhr J, Placzek M. et al. Sonic hedgehog induces the differentiation of ventral forebrain neurons: A common signal for ventral patterning within the neural tube. Cell. 1995;81(5):747–756. [PubMed: 7774016]
- 35.
- Roessler E, Belloni E, Gaudenz K. et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14(3):357–360. [PubMed: 8896572]
- 36.
- Belloni E, Muenke M, Roessler E. et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14(3):353–356. [PubMed: 8896571]
- 37.
- Chiang C, Litingtung Y, Lee E. et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383(6599):407–413. [PubMed: 8837770]
- 38.
- Pabst O, Herbrand H, Takuma N. et al. NKX2 gene expression in neuroectoderm but not in mesendodermally derived structures depends on sonic hedgehog in mouse embryos. Dev Genes Evol. 2000;210(1):47–50. [PubMed: 10603087]
- 39.
- Rallu M, Machold R, Gaiano N. et al. Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development. 2002;129(21):4963–4974. [PubMed: 12397105]
- 40.
- Corbin JG, Rutlin M, Gaiano N. et al. Combinatorial function of the homeodomain proteins Nkx2.1 and Gsh2 in ventral telencephalic patterning. Development. 2003;130(20):4895–4906. [PubMed: 12930780]
- 41.
- Aoto K, Nishimura T, Eto K. et al. Mouse GLI3 regulates Fgf8 expression and apoptosis in the developing neural tube, face, and limb bud. Dev Biol. 2002;251(2):320–332. [PubMed: 12435361]
- 42.
- Ohkubo Y, Chiang C, Rubenstein JL. Coordinate regulation and synergistic actions of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience. 2002;111(1):1–17. [PubMed: 11955708]
- 43.
- Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell. 2001;105(6):781–792. [PubMed: 11440720]
- 44.
- Pierani A, Brenner-Morton S, Chiang C. et al. A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell. 1999;97(7):903–915. [PubMed: 10399918]
- 45.
- Novitch BG, Wichterle H, Jessell TM. et al. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron. 2003;40(1):81–95. [PubMed: 14527435]
- 46.
- LaMantia AS, Colbert MC, Linney E. Retinoic acid induction and regional differentiation prefigure olfactory pathway formation in the mammalian forebrain. Neuron. 1993;10(6):1035–1048. [PubMed: 8318228]
- 47.
- Toresson H, Mata de Urquiza A, Fagerstrom C. et al. Retinoids are produced by glia in the lateral ganglionic eminence and regulate striatal neuron differentiation. Development. 1999;126(6):1317–1326. [PubMed: 10021349]
- 48.
- Schneider RA, Hu D, Rubenstein JL. et al. Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development. 2001;128(14):2755–2767. [PubMed: 11526081]
- 49.
- Rubenstein JL, Shimamura K, Martinez S. et al. Regionalization of the prosencephalic neural plate. Annu Rev Neurosci. 1998;21:445–477. [PubMed: 9530503]
- 50.
- Gunhaga L, Jessell TM, Edlund T. Sonic hedgehog signaling at gastrula stages specifies ventral telencephalic cells in the chick embryo. Development. 2000;127(15):3283–3293. [PubMed: 10887084]
- 51.
- Epstein DJ, McMahon AP, Joyner AL. Regionalization of Sonic hedgehog transcription along the anteroposterior axis of the mouse central nervous system is regulated by Hnf3-dependent and -independent mechanisms. Development. 1999;126(2):281–292. [PubMed: 9847242]
- 52.
- Kiecker C, Niehrs C. The role of prechordal mesendoderm in neural patterning. Curr Opin Neurobiol. 2001;11(1):27–33. [PubMed: 11179869]
- 53.
- Shimamura K, Rubenstein JL. Inductive interactions direct early regionalization of the mouse forebrain. Development. 1997;124(14):2709–2718. [PubMed: 9226442]
- 54.
- Pera EM, Kessel M. Patterning of the chick forebrain anlage by the prechordal plate. Development. 1997;124(20):4153–4162. [PubMed: 9374411]
- 55.
- Dale JK, Vesque C, Lints TJ. et al. Cooperation of BMP7 and SHH in the induction of forebrain ventral midline cells by prechordal mesoderm. Cell. 1997;90(2):257–269. [PubMed: 9244300]
- 56.
- Briscoe J, Chen Y, Jessell TM. et al. A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol Cell. 2001;7(6):1279–1291. [PubMed: 11430830]
- 57.
- Lewis PM, Dunn MP, McMahon JA. et al. Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell. 2001;105(5):599–612. [PubMed: 11389830]
- 58.
- Zeng X, Goetz JA, Suber LM. et al. A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature. 2001;411(6838):716–720. [PubMed: 11395778]
- 59.
- Nery S, Wichterle H, Fishell G. Sonic hedgehog contributes to oligodendrocyte specification in the mammalian forebrain. Development. 2001;128(4):527–540. [PubMed: 11171336]
- 60.
- Platt KA, Michaud J, Joyner AL. Expression of the mouse Gli and Ptc genes is adjacent to embryonic sources of hedgehog signals suggesting a conservation of pathways between flies and mice. Mech Dev. 1997;62(2):121–135. [PubMed: 9152005]
- 61.
- Kohtz JD, Baker DP, Corte G. et al. Regionalization within the mammalian telencephalon is mediated by changes in responsiveness to Sonic Hedgehog. Development. 1998;125(24):5079–5089. [PubMed: 9811591]
- 62.
- Machold R, Hayashi S, Rutlin M. et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39(6):937–950. [PubMed: 12971894]
- 63.
- Palma V, Lim DA, Dahmane N. et al. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132(2):335–344. [PMC free article: PMC1431583] [PubMed: 15604099]
- 64.
- Fuccillo M, Rallu M, McMahon AP. et al. Temporal requirement for hedgehog signaling in ventral telencephalic patterning. Development. 2004;131(20):5031–5040. [PubMed: 15371303]
- 65.
- Wijgerde M, McMahon JA, Rule M. et al. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16(22):2849–2864. [PMC free article: PMC187482] [PubMed: 12435628]
- 66.
- Dahmane N, Sanchez P, Gitton Y. et al. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development. 2001;128(24):5201–5212. [PubMed: 11748155]
- 67.
- Graham A, Koentges G, Lumsden A. Neural crest apoptosis and the establishment of craniofacial pattern: An honorable death. Mol Cell Neurosci. 1996;8(2-3):76–83.
- 68.
- Merino R, Ganan Y, Macias D. et al. Bone morphogenetic proteins regulate interdigital cell death in the avian embryo. Ann NY Acad Sci. 1999;887:120–132. [PubMed: 10668469]
- 69.
- Golden JA, Bracilovic A, McFadden KA. et al. Ectopic bone morphogenetic proteins 5 and 4 in the chicken forebrain lead to cyclopia and holoprosencephaly. Proc Natl Acad Sci USA. 1999;96(5):2439–2444. [PMC free article: PMC26803] [PubMed: 10051661]
- 70.
- Furuta Y, Piston DW, Hogan BL. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 1997;124(11):2203–2212. [PubMed: 9187146]
- 71.
- Mabie PC, Mehler MF, Kessler JA. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci. 1999;19(16):7077–7088. [PMC free article: PMC6782885] [PubMed: 10436062]
- 72.
- Israsena N, Kessler JA. Msx2 and p21(CIP1/WAF1) mediate the proapoptotic effects of bone morphogenetic protein-4 on ventricular zone progenitor cells. J Neurosci Res. 2002;69(6):803–809. [PubMed: 12205674]
- 73.
- Thibert C, Teillet MA, Lapointe F. et al. Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science. 2003;301(5634):843–846. [PubMed: 12907805]
- 74.
- Gaiano N, Kohtz JD, Turnbull DH. et al. A method for rapid gain-of-function studies in the mouse embryonic nervous system. Nat Neurosci. 1999;2(9):812–819. [PubMed: 10461220]
- 75.
- Barnes EA, Kong M, Ollendorff V. et al. Patched1 interacts with cyclin B1 to regulate cell cycle progression. Embo J. 2001;20(9):2214–2223. [PMC free article: PMC125436] [PubMed: 11331587]
- 76.
- Lu QR, Yuk D, Alberta JA. et al. Sonic hedgehog—regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron. 2000;25(2):317–329. [PubMed: 10719888]
- 77.
- Tekki-Kessaris N, Woodruff R, Hall AC. et al. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development. 2001;128(13):2545–2554. [PubMed: 11493571]
- 78.
- Spassky N, Heydon K, Mangatal A. et al. Sonic hedgehog-dependent emergence of oligodendrocytes in the telencephalon: Evidence for a source of oligodendrocytes in the olfactory bulb that is independent of PDGFRalpha signaling. Development. 2001;128(24):4993–5004. [PubMed: 11748136]
- 79.
- Gulacsi A, Lillien L. Sonic hedgehog and bone morphogenetic protein regulate interneuron development from dorsal telencephalic progenitors in vitro. J Neurosci. 2003;23(30):9862–9872. [PMC free article: PMC6740884] [PubMed: 14586015]
- 80.
- Matise MP, Epstein DJ, Park HL. et al. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development. 1998;125(15):2759–2770. [PubMed: 9655799]
- 81.
- Park HL, Bai C, Platt KA. et al. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127(8):1593–1605. [PubMed: 10725236]
- 82.
- Ding Q, Motoyama J, Gasca S. et al. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development. 1998;125(14):2533–2543. [PubMed: 9636069]
- 83.
- Palma V, Ruiz i Altaba A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development. 2004;131(2):337–345. [PubMed: 14681189]
- 84.
- Shinozaki K, Yoshida M, Nakamura M. et al. Emx1 and Emx2 cooperate in initial phase of archipallium development. Mech Dev. 2004;121(5):475–489. [PubMed: 15147765]
- 85.
- Schimmang T, Lemaistre M, Vortkamp A. et al. Expression of the zinc finger gene Gli3 is affected in the morphogenetic mouse mutant extra-toes (Xt). Development. 1992;116(3):799–804. [PubMed: 1289066]
- 86.
- Hui CC, Joyner AL. A mouse model of greig cephalopolysyndactyly syndrome: The extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet. 1993;3(3):241–246. [PubMed: 8387379]
- 87.
- Buscher D, Grotewold L, Ruther U. The XtJ allele generates a Gli3 fusion transcript. Mamm Genome. 1998;9(8):676–678. [PubMed: 9680393]
- 88.
- Maynard TM, Jain MD, Balmer CW. et al. High-resolution mapping of the Gli3 mutation extra-toes reveals a 51.5-kb deletion. Mamm Genome. 2002;13(1):58–61. [PubMed: 11773971]
- 89.
- Franz T. Extra-toes (Xt) homozygous mutant mice demonstrate a role for the Gli-3 gene in the development of the forebrain. Acta Anat (Basel). 1994;150(1):38–44. [PubMed: 7976186]
- 90.
- Theil T, Alvarez-Bolado G, Walter A. et al. Gli3 is required for Emx gene expression during dorsal telencephalon development. Development. 1999;126(16):3561–3571. [PubMed: 10409502]
- 91.
- Grove EA, Tole S, Limon J. et al. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development. 1998;125(12):2315–2325. [PubMed: 9584130]
- 92.
- Tole S, Ragsdale CW, Grove EA. Dorsoventral patterning of the telencephalon is disrupted in the mouse mutant extra-toes(J). Dev Biol. 2000;217(2):254–265. [PubMed: 10625551]
- 93.
- Theil T, Aydin S, Koch S. et al. Wnt and Bmp signalling cooperatively regulate graded Emx2 expression in the dorsal telencephalon. Development. 2002;129(13):3045–3054. [PubMed: 12070081]
- 94.
- Kuschel S, Ruther U, Theil T. A disrupted balance between Bmp/Wnt and Fgf signaling underlies the ventralization of the Gli3 mutant telencephalon. Dev Biol. 2003;260(2):484–495. [PubMed: 12921747]
- 95.
- Rubenstein JL, Beachy PA. Patterning of the embryonic forebrain. Curr Opin Neurobiol. 1998;8(1):18–26. [PubMed: 9568388]
- 96.
- Monuki ES, Walsh CA. Mechanisms of cerebral cortical patterning in mice and humans. Nat Neurosci. 2001;(4 Suppl):1199–1206. [PubMed: 11687830]
- 97.
- Zaki PA, Quinn JC, Price DJ. Mouse models of telencephalic development. Curr Opin Genet Dev. 2003;13(4):423–437. [PubMed: 12888017]
- 98.
- Roth KA, D'Sa C. Apoptosis and brain development. Ment Retard Dev Disabil Res Rev. 2001;7(4):261–266. [PubMed: 11754520]
- 99.
- te WelscherP, Zuniga A, Kuijper S. et al. Progression of vertebrate limb development through SHH-mediated counteraction of GLI3. Science. 2002;298(5594):827–830. [PubMed: 12215652]
- 100.
- Litingtung Y, Chiang C. Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat Neurosci. 2000;3(10):979–985. [PubMed: 11017169]
- 101.
- Bose J, Grotewold L, Ruther U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum Mol Genet. 2002;11(9):1129–1135. [PubMed: 11978771]
- 102.
- Persson M, Stamataki D, te Welscher P. et al. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002;16(22):2865–2878. [PMC free article: PMC187477] [PubMed: 12435629]
- 103.
- Krishnan V, Pereira FA, Qiu Y. et al. Mediation of Sonic hedgehog-induced expression of COUP-TFII by a protein phosphatase. Science. 1997;278(5345):1947–1950. [PubMed: 9395397]
- 104.
- Tripodi M, Filosa A, Armentano M. et al. The COUP-TF nuclear receptors regulate cell migration in the mammalian basal forebrain. Development. 2004;131(24):6119–6129. [PubMed: 15548577]
- 105.
- Liu F, Massague J, Ruiz i Altaba A. Carboxy-terminally truncated Gli3 proteins associate with Smads. Nat Genet. 1998;20(4):325–326. [PubMed: 9843199]
Figures
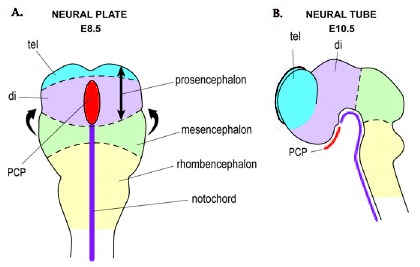
Figure 1
Neural plate and neural tube stages in mouse. A) The anterior neural plate at around E8.5. The neural plate folds in the direction of the arrows to form the neural tube. B) The brain viewed from the side after neural tube closure (at around E10.5). The brain vesicles are the prosencephalon (comprised of the telencephalon (tel) and diencephalon (di)), mesencephalon and rhombencephalon. The prechordal plate (pcp) underlies the rostral part of the neural tube (at the level of the diencephalon) whereas the notochord underlies the caudal neural tube.
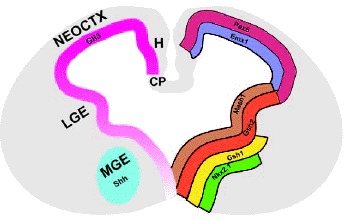
Figure 2
Coronal section of a midgestional (E12.5) mouse telencephalon illustrating major telencephalic subdivisions, a selection of gene expression patterns which are regionally restricted and the expression pattern of Shh and Gli3. Genes such as Pax6 and Emx1 are expressed in the dorsal telencephalon, which will give rise to the neocortex (neoctx), hippocampus (H) and choroid plexus (CP). The ventral telencephalon contains the precursors for the adult striatum (lateral ganglionic eminence, LGE) and globus pallidus (medial ganglionic eminence, MGE). Genes such as Mash2 and Gsh2 are expressed in both the LGE and MGE whereas genes such as Gsh1 and Nkx2.1 are primarily restricted to the MGE. Shh expression is confined to the MGE, while Gli3 is expressed throughout the entire telencephalon, with high levels in the dorsal telencephalon and LGE and lower levels in the MGE.