

Bookshelf ID: NBK65790PMID: 26389232
NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
PDQ Cancer Information Summaries [Internet]. Bethesda (MD): National Cancer Institute (US); 2002-.

PDQ Cancer Information Summaries [Internet].
Show detailsThis PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood liver cancer. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
General Information About Childhood Liver Cancer
Liver cancer is a rare malignancy in children and adolescents and is divided into the following two major histological subgroups:
- Hepatoblastoma.
- Mixed epithelial and fetal histology.
- Small cell undifferentiated histology hepatoblastoma and rhabdoid tumors of the liver.
- Small cell undifferentiated hepatoblastoma (SMARCB1 positive).
- Rhabdoid tumor of liver (SMARCB1 negative).
Other, less common histologies include the following:
- Biliary rhabdomyosarcoma. For more information, see Childhood Rhabdomyosarcoma Treatment.
Cellular Classification of Childhood Liver Cancer
Liver tumors are rare in children. A diagnosis may be challenging, in part because of the lack of consensus regarding a classification system. Systematic central histopathological review of these tumors performed as part of pediatric collaborative therapeutic protocols has allowed the identification of histological subtypes with distinct clinical associations. As a result, histopathology has been incorporated within the Children’s Oncology Group (COG) protocols and, in the United States, as a risk-stratification parameter used for patient management.
The COG Liver Tumor Committee sponsored an International Pathology Symposium in 2011 to discuss the histopathology and classification of pediatric liver tumors (hepatoblastoma, in particular) and develop an International Pediatric Liver Tumors Consensus Classification that would be required for international collaborative projects. The results of this international classification for pediatric liver tumors have been published.[1] This standardized, clinically meaningful classification will allow the integration of new biological parameters and tumor genetics within a common pathologic language to help improve future patient management and outcomes.
For information about the histology of each childhood liver cancer subtype, see the following sections:
References
- López-Terrada D, Alaggio R, de Dávila MT, et al.: Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 27 (3): 472-91, 2014. [PubMed: 24008558]
Tumor Stratification by Imaging and Evans Surgical Staging for Childhood Liver Cancer
Historically, the four major study groups—International Childhood Liver Tumors Strategy Group (previously known as Société Internationale d’Oncologie Pédiatrique–Epithelial Liver Tumor Study Group [SIOPEL]), Children's Oncology Group (COG), Gesellschaft für Pädiatrische Onkologie und Hämatologie (Society for Paediatric Oncology and Haematology), and Japanese Study Group for Pediatric Liver Tumors—have had disparate risk stratification categories, making it difficult to compare outcomes across continents. All groups are now using the PRE-Treatment EXTent of tumor (PRETEXT) grouping system as part of the risk stratification.
Tumor Stratification by Imaging
The main treatment goal for patients with liver cancer is surgical extirpation of the primary tumor. Therefore, the risk grouping depends heavily on factors determined by imaging that are related to safe surgical resection of the tumor, as well as the PRETEXT grouping. These imaging findings include the section or sections of the liver that are involved with the tumor and additional findings, termed annotation factors, that impact surgical decision making and prognosis.
Risk stratification of children with hepatoblastoma depends on the use of high-quality, cross-sectional imaging. Three-phase computed tomography scanning (noncontrast, arterial, and venous) or magnetic resonance imaging (MRI) with contrast agents are used for imaging. MRI with gadoxetate disodium, a gadolinium-based agent that is preferentially taken up and excreted by hepatocytes, is being used with increased frequency and may improve detection of multifocal disease.[1]
PRETEXT and POSTTEXT Group Definitions
The imaging grouping systems used to radiologically define the extent of liver involvement by the tumor are designated as the following:
- PRETEXT (PRE-Treatment EXTent of disease): The extent of liver involvement is defined before therapy.
- POSTTEXT (POST-Treatment EXTent of disease): The extent of liver involvement is defined in response to therapy.
PRETEXT
PRETEXT is used by the major multicenter trial groups as a central component of risk stratification schemes that define treatment of hepatoblastoma. PRETEXT is based on the Couinaud eight-segment anatomic structure of the liver using cross-sectional imaging.
The PRETEXT system divides the liver into four parts, called sections. The left lobe of the liver consists of a lateral section (Couinaud segments I, II, and III) and a medial section (segment IV), whereas the right lobe consists of an anterior section (segments V and VIII) and a posterior section (segments VI and VII) (see Figure 1). PRETEXT groups were devised by the SIOPEL for their first trial, SIOPEL-1,[2] and revised for the SIOPEL-3 trial in 2007.[3]

Figure 1. The liver is divided into four sections: the right posterior section, the right anterior section, the left medial section, and the left lateral section. Each section of the liver is further divided into segments: segments VI and VII make up the right posterior section, segments V and VIII make up the right anterior section, segment IV makes up the left medial section, and segments II and III make up the left lateral section. Segment I is found deep in the left side of the liver, in front of the inferior vena cava and behind the right, middle, and left hepatic veins.
PRETEXT group assignment I, II, III, or IV is determined by the number of uninvolved sections of the liver. PRETEXT is further described by annotation factors. Annotation factors include findings that are important for surgical management and evidence of tumor extension beyond the hepatic parenchyma of the major sections, including metastatic disease. For detailed descriptions of the PRETEXT groups, see Table 1. For descriptions of the annotation factors, see Table 2.
Annotation factors identify the extent of tumor involvement of the major vessels and its effect on venous inflow and outflow, which is critical knowledge for the surgeon and can affect surgical outcomes. There were differences in the definitions of gross vascular involvement used by the COG and major liver surgery centers in the United States compared with SIOPEL definitions used in Europe. These differences have been resolved, and the new definitions are being used in an international trial that began in 2018.[4]
Although PRETEXT can be used to predict tumor resectability, there are limitations. The distinction between real invasion beyond the anatomical border of a given hepatic section and the compression and displacement by the tumor can be difficult, especially at diagnosis. Additionally, it can be difficult to distinguish between vessel encroachment and involvement, particularly if imaging is inadequate. The PRETEXT group assignment has a moderate degree of interobserver variability. In a report published in 2005 using data from the SIOPEL-1 study, the preoperative PRETEXT group aligned with postoperative pathological findings only 51% of the time, with overstaging in 37% of patients and understaging in 12% of patients.[5]
Because distinguishing PRETEXT group assignment is difficult, central review of imaging is critical and is generally performed in all major clinical trials. For patients not enrolled in clinical trials, expert radiological review should be considered in questionable cases in which the PRETEXT group assignment affects choice of treatment.
Table 1. Definitions of PRETEXT and POSTTEXT Groupsa
| PRETEXT and POSTTEXT Groups | Definition | Image | |
|---|---|---|---|
| I | One section involved; three adjoining sections are tumor free. |
 | |
| II | One or two sections involved; two adjoining sections are tumor free. |
 | |
| III | Two or three sections involved; one adjoining section is tumor free. |
 | |
| IV | Four sections involved. |
 | |
aAdapted from Roebuck et al.[3]
Table 2. Annotation Factors for Describing PRETEXT and POSTTEXT Groupsa
| Annotation Factors | Definition | ||
|---|---|---|---|
| Vb | Venous involvement: Vascular involvement of the retrohepatic vena cava or involvement of all three major hepatic veins (right, middle, and left). | ||
| V0 | Tumor within 1 cm. | ||
| V1 | Tumor abutting. | ||
| V2 | Tumor compressing or distorting. | ||
| V3 | Tumor ingrowth, encasement, or thrombus. | ||
| Pb | Portal involvement: Vascular involvement of the main portal vein and/or both right and left portal veins. | ||
| P0 | Tumor within 1 cm. | ||
| P1 | Tumor abutting the main portal vein, the right and left portal veins, or the portal vein bifurcation. | ||
| P2 | Tumor compressing the main portal vein, the right and left portal veins, or the portal vein bifurcation. | ||
| P3 | Tumor ingrowth, encasement (>50% or >180 degrees), or intravascular thrombus within the main portal vein, the right and left portal veins, or the portal vein bifurcation. | ||
| Eb | Extrahepatic spread of disease. Any one of the following criteria is met: | ||
| E1 | Tumor crosses boundaries/tissue planes. | ||
| E2 | Tumor is surrounded by normal tissue more than 180 degrees. | ||
| E3 | Peritoneal nodules (not lymph nodes) are present so that there is at least one nodule measuring ≥10 mm or at least two nodules measuring ≥5 mm. | ||
| Mb | Distant metastases. Any one of the following criteria is met: | ||
| M1 | One noncalcified pulmonary nodule ≥5 mm in diameter. | ||
| M2 | Two or more noncalcified pulmonary nodules, each ≥3 mm in diameter. | ||
| M3 | Pathologically proven metastatic disease. | ||
| C | Tumor involving the caudate. | ||
| F | Multifocality. Two or more discrete hepatic tumors with normal intervening liver tissue. | ||
| Nb | Lymph node metastases. Any one of the following criteria is met: | ||
| N1 | Lymph node with short-axis diameter of >1 cm. | ||
| N2 | Portocaval lymph node with short-axis diameter >1.5 cm. | ||
| N3 | Spherical lymph node shape with loss of fatty hilum. | ||
| Rb | Tumor rupture. Free fluid in the abdomen or pelvis with one or more of the following findings of hemorrhage: | ||
| R1 | Internal complexity/septations within fluid. | ||
| R2 | High-density fluid on CT (>25 HU). | ||
| R3 | Imaging characteristics of blood or blood degradation products on MRI. | ||
| R4 | Heterogeneous fluid on ultrasound with echogenic debris. | ||
| R5 | Visible defect in tumor capsule OR tumor cells are present within the peritoneal fluid OR rupture diagnosed pathologically in patients who have received an upfront resection. | ||
POSTTEXT
The POSTTEXT group is determined after the patients receive chemotherapy. The greatest chemotherapy response, measured as decreases in tumor size and alpha-fetoprotein (AFP) level, occurs after the first two cycles of chemotherapy.[6,7] In addition, a study that evaluated surgical resectability after two versus four cycles of chemotherapy showed that many tumors may be resectable after two cycles.[6]
Evans Surgical Staging for Childhood Liver Cancer (Historical)
The COG/Evans staging system, based on operative findings and surgical resectability, was used for many years in the United States to group and determine treatment for children with liver cancer (see Table 3).[8-10] Currently, other risk stratification systems are used to classify patients and determine treatment strategy. For more information, see Table 5.
Table 3. Definition of Evans Surgical Staging
| Evans Surgical Stage | Definition |
|---|---|
| Stage I | The tumor is completely resected. |
| Stage II | Microscopic residual tumor remains after resection. |
| Stage III | There are no distant metastases and at least one of the following is true: (1) the tumor is either unresectable or the tumor is resected with gross residual tumor; (2) there are positive extrahepatic lymph nodes. |
| Stage IV | There is distant metastasis, regardless of the extent of liver involvement. |
References
- Meyers AB, Towbin AJ, Geller JI, et al.: Hepatoblastoma imaging with gadoxetate disodium-enhanced MRI--typical, atypical, pre- and post-treatment evaluation. Pediatr Radiol 42 (7): 859-66, 2012. [PubMed: 22419052]
- Brown J, Perilongo G, Shafford E, et al.: Pretreatment prognostic factors for children with hepatoblastoma-- results from the International Society of Paediatric Oncology (SIOP) study SIOPEL 1. Eur J Cancer 36 (11): 1418-25, 2000. [PubMed: 10899656]
- Roebuck DJ, Aronson D, Clapuyt P, et al.: 2005 PRETEXT: a revised staging system for primary malignant liver tumours of childhood developed by the SIOPEL group. Pediatr Radiol 37 (2): 123-32; quiz 249-50, 2007. [PMC free article: PMC1805044] [PubMed: 17186233]
- Towbin AJ, Meyers RL, Woodley H, et al.: 2017 PRETEXT: radiologic staging system for primary hepatic malignancies of childhood revised for the Paediatric Hepatic International Tumour Trial (PHITT). Pediatr Radiol 48 (4): 536-554, 2018. [PubMed: 29427028]
- Aronson DC, Schnater JM, Staalman CR, et al.: Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol 23 (6): 1245-52, 2005. [PubMed: 15718322]
- Lovvorn HN, Ayers D, Zhao Z, et al.: Defining hepatoblastoma responsiveness to induction therapy as measured by tumor volume and serum alpha-fetoprotein kinetics. J Pediatr Surg 45 (1): 121-8; discussion 129, 2010. [PMC free article: PMC2852870] [PubMed: 20105591]
- Venkatramani R, Stein JE, Sapra A, et al.: Effect of neoadjuvant chemotherapy on resectability of stage III and IV hepatoblastoma. Br J Surg 102 (1): 108-13, 2015. [PubMed: 25349947]
- Ortega JA, Krailo MD, Haas JE, et al.: Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin chemotherapy: a report from the Childrens Cancer Study Group. J Clin Oncol 9 (12): 2167-76, 1991. [PubMed: 1720452]
- Douglass EC, Reynolds M, Finegold M, et al.: Cisplatin, vincristine, and fluorouracil therapy for hepatoblastoma: a Pediatric Oncology Group study. J Clin Oncol 11 (1): 96-9, 1993. [PubMed: 8380296]
- Ortega JA, Douglass EC, Feusner JH, et al.: Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin Oncol 18 (14): 2665-75, 2000. [PubMed: 10894865]
Treatment Option Overview for Childhood Liver Cancer
Many of the improvements in survival in childhood cancer have been made using new therapies that have attempted to improve on the best available, accepted therapy. Clinical trials in pediatrics are designed to compare potentially better therapy with therapy that is currently accepted as standard. This comparison may be done in a randomized study of two treatment arms or by evaluating a single new treatment and comparing the results with those previously obtained with standard therapy.
Because of the relative rarity of cancer in children, all children with liver cancer should be considered for a clinical trial if available. Treatment planning by a multidisciplinary team of cancer specialists with experience treating tumors of childhood is required to determine and implement optimal treatment.[1]
Surgery
Historically, complete surgical resection of the primary tumor has been essential for cure of malignant liver tumors in children.[2-6]; [7][Level of evidence C1] This approach continues to be the goal of definitive surgical procedures, which are often combined with chemotherapy. The surgeon performs a highly sophisticated liver resection in children and adolescents with primary liver tumors after confirmation of the diagnosis by pathological investigation of intraoperative frozen sections. While complete surgical resection is important for all liver tumors, it is especially important for hepatocellular carcinoma because curative chemotherapy is not available. In patients with advanced hepatoblastoma, postoperative complications are associated with worsened overall survival (OS).[8]
The three surgical options to treat primary pediatric liver cancer include the following:
- Initial surgical resection (alone or with adjuvant chemotherapy).
- Delayed surgical resection (with neoadjuvant chemotherapy).
- Orthotopic liver (cadaveric and living donor) transplant (most often with neoadjuvant chemotherapy).
The decision as to which surgical approach to use (e.g., partial hepatectomy, extended resection, or transplant) depends on many factors, including the following:
- PRE-Treatment EXTent of disease (PRETEXT) group and POST-Treatment EXTent of disease (POSTTEXT) group.
- Size of the primary tumor.
- Presence of multifocal hepatic disease.
- Gross vascular involvement.
- Alpha-fetoprotein (AFP) levels.
- Whether preoperative chemotherapy is potentially likely to convert an unresectable tumor into a resectable tumor.
- Whether hepatic disease meets surgical and histopathological criteria for orthotopic liver transplant.
Timing of the surgical approach is critical. Surgeons who have experience performing pediatric liver resections and transplants are involved early in the decision-making process for determining optimal timing and extent of resection.
Early involvement, preferably at diagnosis, with an experienced pediatric liver surgeon is especially important in patients with PRETEXT group III or IV or involvement of major liver vessels (positive annotation factors V [venous] or P [portal]).[9] Although vascular involvement was initially thought to be a contraindication to resection, experienced liver surgeons are sometimes able to successfully resect the tumor and avoid performing a transplant.[10-12]; [13][Level of evidence C1] Patients with vascular involvement and tumors that have been deemed nonresectable by the pediatric surgical expert should be referred to a transplant center for evaluation to avoid unnecessary delays in evaluation and listing for transplant.
Intraoperative ultrasonography may result in further delineation of tumor extent and location and can affect intraoperative management.[14] Preoperative infusion of indocyanine green, a fluoroactive agent that is concentrated in the liver and retained by abnormal liver tumors, has also been used to provide visual intraoperative guidance to locate the tumor and assess proximity to surgical margins.[15]
If the tumor is determined to be unresectable, measures to reduce the tumor size to make a complete surgical resection possible need to be considered. These measures include preoperative intravenous chemotherapy, transarterial chemotherapy, or transarterial radioactive therapy. These efforts must be carefully coordinated with the surgical team to facilitate planning of resection. Prolonged chemotherapy can lead to unnecessary delays and, in rare cases, tumor progression. If the tumor can be completely excised by an experienced surgical team, less postoperative chemotherapy may be needed. Incomplete resection must be avoided because patients who undergo rescue transplants of incompletely resected tumors have an inferior outcome, compared with patients who undergo transplant as the primary surgical therapy.[16][Level of evidence C1] Accomplishing the appropriate surgery at resection is critical.
The approach taken by the Children's Oncology Group (COG) in North American clinical trials is to perform surgery initially when a complete resection can be done with a simple, negative-margin hemihepatectomy. The COG AHEP0731 (NCT00980460) trial studied the use of PRETEXT and POSTTEXT to determine the optimal approach and timing of surgery. POSTTEXT imaging grouping was performed after two and four cycles of chemotherapy to determine the optimal time for definitive surgery.[6,17] For more information, see the Tumor Stratification by Imaging and Evans Surgical Staging for Childhood Liver Cancer section.
Orthotopic liver transplant
Liver transplants have been associated with significant success in the treatment of children with unresectable hepatic tumors.[18]; [19-21][Level of evidence C1] A review of the world experience has documented a posttransplant survival rate of 70% to 80% for children with hepatoblastomas.[16,22-24] Intravenous vascular invasion, positive lymph nodes, and contiguous extrahepatic spread did not have a significant adverse effect on outcome. Adjuvant chemotherapy after transplant may decrease the risk of tumor recurrence, but its use has not been studied definitively in a randomized clinical trial.[25]
Evidence (orthotopic liver transplant):
- The United Network for Organ Sharing (UNOS) database was queried for all patients younger than 18 years with a primary malignant liver tumor who underwent an orthotopic liver transplant between 1987 and 2012 (N = 544). The patients were diagnosed with hepatoblastoma (n = 376, 70%), hepatocellular carcinoma (n = 84, 15%), and other tumors (n = 84, 15%). Patients with hepatocellular carcinoma were older, more often hospitalized at the time of transplant, and more likely to receive a cadaveric organ than were patients with hepatoblastoma.[26]
- The 5-year patient survival rate was 73%, and the graft survival rate was 74% for the entire cohort, with most deaths resulting from malignancy. On multivariate analysis, independent predictors of 5-year patient and graft survival included the following:
- Diagnosis.
- For the study period of 1987 to 2012, the 5-year survival rate was 76% and the graft survival rate was 77% for patients with hepatoblastoma; the survival and graft survival rates were 63% for patients with hepatocellular carcinoma.
- For the study period of 2009 to 2012, the 3-year survival and graft survival rates were 84% for patients with hepatoblastoma; the survival and graft survival rates were 85% for patients with hepatocellular carcinoma.
- Transplant era.
- The death rate by hazard ratio was 1.0 for the period before 2002, 0.72 for the period of 2002 to 2009, and 0.54 for the period of 2009 to 2012.
- Medical condition at transplant.
- For hepatoblastoma patients, the survival rate by hazard ratio was 1.0 for hospitalized patients versus 1.81 for nonhospitalized patients at the time of transplant.
- For hepatocellular carcinoma patients, the survival rate by hazard ratio was 1.0 for hospitalized patients versus 1.92 for nonhospitalized patients.
- Patients hospitalized in the intensive care unit did not fare worse than patients not in the intensive care unit.
- A report of 149 patients with hepatocellular carcinoma younger than 21 years who underwent transplants between 1987 and 2015 used detailed data collected at all U.S. pediatric transplant centers by the U.S. Scientific Registry of Transplant Recipients.[18]
- The 1-year graft survival rate was about 85%, which did not differ from the survival for patients with hepatoblastoma or biliary atresia. Survival rates continued to decline over time, from 85% at 1 year to 52% at 5 years and 43% at 10 years, a more dramatic decline than what was seen for hepatoblastoma or biliary atresia.
- The survival after transplant did not differ from that of adults who underwent transplant for hepatocellular carcinoma.
- Of the patients with hepatocellular carcinoma, 22 had hepatocellular carcinoma diagnosed after transplant for medical cirrhotic disease such as tyrosinemia. They had a superior outcome, but it was not statistically significant compared with the rest of the 149 patients.
- In a three-institution study of children with hepatocellular carcinoma, the overall 5-year disease-free survival rate was approximately 60%.[28]
Application of the Milan criteria for UNOS selection of recipients of deceased donor livers is controversial.[29,30] The Milan criteria for liver transplant are directed toward adults with cirrhosis and hepatocellular carcinoma. The criteria do not apply to children and adolescents with hepatocellular carcinoma, especially those without cirrhosis.
Cirrhosis is an underlying risk factor for the development of hepatocellular carcinoma in children who suffer from certain diseases or conditions. These diseases include perinatally acquired hepatitis B, hepatorenal tyrosinemia, progressive familial intrahepatic cholestasis, glycogen storage disease, Alagille syndrome, and other conditions. Improvements in screening methodology have allowed for earlier identification and treatment of some of these conditions, as well as monitoring for development of hepatocellular carcinoma. Nevertheless, because of the poor prognosis of patients with hepatocellular carcinoma, liver transplant should be considered for diseases or conditions that have resulted in early findings of cirrhosis, before the development of liver failure or malignancy.[31]
Living-donor liver transplant for hepatic malignancy is more common in children and the outcome is similar to those undergoing cadaveric liver transplant.[32,33] In patients with hepatocellular carcinoma, gross vascular invasion, distant metastases, lymph node involvement, tumor size, and male sex were significant risk factors for recurrence. In one report, 33 patients with hepatoblastoma and 10 patients with hepatocellular carcinoma were treated with living-donor liver transplants. For the hepatoblastoma patients, the 5-year OS rate was 87.4%, and the event-free survival (EFS) rate was 75.8%. The 5-year OS and EFS rates were 75.4% for the patients with hepatocellular carcinoma. The presence of renal vein invasion was associated with an increased incidence of recurrence and death (P = .28).[34][Level of evidence C1]
Surgical resection for metastatic disease
Surgical resection of metastatic disease is often recommended, but the rate of cure in children with hepatoblastoma has not been fully determined. Resection of metastases may be done for areas of locally invasive disease (e.g., diaphragm) and isolated brain metastases. Resection of pulmonary metastases should be considered if the number of metastases is limited.[35-38] In an American study of 38 patients who presented at diagnosis with pulmonary metastases, only nine patients underwent surgical resection. The timing of pulmonary resection in relation to definitive resection of the primary tumor varied (two patients before, five patients simultaneously, and two patients after primary resection). Eight of the nine patients survived. Of 20 children with relapse restricted to the lungs, all patients received salvage chemotherapy, 8 patients had a thoracotomy and pulmonary metastasectomy, and 5 patients had a thoracotomy and biopsy. Among the 13 patients who had surgery, only 4 were long-term survivors, 2 of whom presented with stage I disease and 2 of whom presented with stage IV disease.[37]
Radiofrequency ablation has also been used to treat oligometastatic hepatoblastoma when patients prefer to avoid surgical metastasectomy.[39][Level of evidence C1]
Chemotherapy
Chemotherapy regimens used in the treatment of hepatoblastoma and hepatocellular carcinoma are described in their respective sections. Chemotherapy has been much more successful in the treatment of hepatoblastoma than in hepatocellular carcinoma.[6,27,40] For more information, see the sections on Treatment of Hepatoblastoma and Treatment of Hepatocellular Carcinoma.
The standard of care in the United States is preoperative chemotherapy when the tumor is unresectable and postoperative chemotherapy after complete resection, even if preoperative chemotherapy has already been given.[41] Treatment with preoperative chemotherapy has been shown to benefit children with hepatoblastoma; however, the use of postoperative chemotherapy after definitive surgical resection or liver transplant has not been investigated in a randomized fashion.
Radiation Therapy
Radiation therapy, even in combination with chemotherapy, has not cured children with unresectable hepatic tumors. A study of 154 patients with hepatoblastoma showed that radiation therapy and/or second resection of positive margins may not be necessary in some patients with incompletely resected hepatoblastoma and microscopic residual tumor.[42] Although there is no standard indication, radiation therapy may have a role in the management of patients with incompletely resected hepatoblastomas.[43] Stereotactic body radiation therapy is a safe and effective alternative treatment that has been successfully used in adult patients with hepatocellular carcinoma who are unable to undergo liver ablation/resection.[44] This highly conformal radiotherapeutic technique, when available, may be considered on an individual basis in children with hepatocellular carcinoma.
Other Treatment Approaches
Other treatment approaches include the following:
- Transarterial chemoembolization (TACE): TACE is an image-guided, minimally invasive, nonsurgical procedure that is used to treat malignant lesions in the liver. The procedure uses a catheter to deliver both chemotherapy medication and embolization materials into the blood vessels that lead to the tumor. The arterial catheter route is image guided, most often via the hepatic artery, and perfusion of the tumor by the targeted artery may be confirmed by imaging before therapeutic injection. This procedure allows for the treatment of tumors that are not accessible with conventional surgery or radiation treatments. TACE has been used for patients with inoperable hepatoblastoma.[45-47] This procedure has also been used in a few children to successfully shrink tumors to permit resections.[46]
- Transarterial radioembolization (TARE): TARE is an image-guided, minimally invasive, nonsurgical procedure that delivers radiation therapy to treat tumors in the liver. This procedure delivers radioactive beads and blocks arterial flow within the tumor to keep the radiation inside the tumor. Glass or resin microspheres, coated most commonly with yttrium Y 90 (90Y), are delivered to the tumor via catheters placed in arteries that supply the tumor. Usually, the hepatic artery or its branches are used, but tumors may be partially supplied by parasitized surrounding vessels. Because of the risk of radiation delivery to the nearby lung, technetium Tc 99m microaggregated albumin imaging is performed with delivery via the catheter that is in place before the administration of radioactive beads to carefully measure radiation exposure to the lung. If calculations determine that lung exposure is unsafe, TARE is not pursued. In a report of two patients with unresectable hepatoblastoma, complete remissions were achieved after treatment with TARE using 90Y resin beads, which successfully shrank the tumors and led to complete resections.[48][Level of evidence C3] This approach has also been used for palliation in children with hepatocellular carcinoma.[49] For more information, see Adult Primary Liver Cancer Treatment.
- High-intensity focused ultrasonography (HIFU): HIFU is a noninvasive treatment for a wide range of tumors and diseases. HIFU uses an ultrasound transducer, similar to the ones used for diagnostic imaging, but with much higher energy. The transducer focuses sound waves to generate heat at a single point within the body and destroy the target tissue. The tissue can get as hot as 66°C in only 20 seconds. This process is repeated as many times as necessary until the target tissue is destroyed. Magnetic resonance imaging is used to plan the treatment and monitor the amount of heat in real time. A combination of chemotherapy followed by TACE and HIFU showed promising results in China for children with PRETEXT III and PRETEXT IV malignant liver tumors, some of whom had resectable tumors but did not undergo surgery because of parent refusal.[50]
References
- Tiao GM, Bobey N, Allen S, et al.: The current management of hepatoblastoma: a combination of chemotherapy, conventional resection, and liver transplantation. J Pediatr 146 (2): 204-11, 2005. [PubMed: 15689909]
- Czauderna P, Otte JB, Aronson DC, et al.: Guidelines for surgical treatment of hepatoblastoma in the modern era--recommendations from the Childhood Liver Tumour Strategy Group of the International Society of Paediatric Oncology (SIOPEL). Eur J Cancer 41 (7): 1031-6, 2005. [PubMed: 15862752]
- Czauderna P, Mackinlay G, Perilongo G, et al.: Hepatocellular carcinoma in children: results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol 20 (12): 2798-804, 2002. [PubMed: 12065556]
- Meyers RL, Czauderna P, Otte JB: Surgical treatment of hepatoblastoma. Pediatr Blood Cancer 59 (5): 800-8, 2012. [PubMed: 22887704]
- Aronson DC, Meyers RL: Malignant tumors of the liver in children. Semin Pediatr Surg 25 (5): 265-275, 2016. [PubMed: 27955729]
- Murawski M, Weeda VB, Maibach R, et al.: Hepatocellular Carcinoma in Children: Does Modified Platinum- and Doxorubicin-Based Chemotherapy Increase Tumor Resectability and Change Outcome? Lessons Learned From the SIOPEL 2 and 3 Studies. J Clin Oncol 34 (10): 1050-6, 2016. [PubMed: 26811523]
- Allan BJ, Wang B, Davis JS, et al.: A review of 218 pediatric cases of hepatocellular carcinoma. J Pediatr Surg 49 (1): 166-71; discussion 171, 2014. [PubMed: 24439603]
- Becker K, Furch C, Schmid I, et al.: Impact of postoperative complications on overall survival of patients with hepatoblastoma. Pediatr Blood Cancer 62 (1): 24-8, 2015. [PubMed: 25251521]
- D'Antiga L, Vallortigara F, Cillo U, et al.: Features predicting unresectability in hepatoblastoma. Cancer 110 (5): 1050-8, 2007. [PubMed: 17661341]
- Lautz TB, Ben-Ami T, Tantemsapya N, et al.: Successful nontransplant resection of POST-TEXT III and IV hepatoblastoma. Cancer 117 (9): 1976-83, 2011. [PubMed: 21509775]
- Fonseca A, Gupta A, Shaikh F, et al.: Extreme hepatic resections for the treatment of advanced hepatoblastoma: Are planned close margins an acceptable approach? Pediatr Blood Cancer 65 (2): , 2018. [PubMed: 28921939]
- Fuchs J, Cavdar S, Blumenstock G, et al.: POST-TEXT III and IV Hepatoblastoma: Extended Hepatic Resection Avoids Liver Transplantation in Selected Cases. Ann Surg 266 (2): 318-323, 2017. [PubMed: 27501172]
- Baertschiger RM, Ozsahin H, Rougemont AL, et al.: Cure of multifocal panhepatic hepatoblastoma: is liver transplantation always necessary? J Pediatr Surg 45 (5): 1030-6, 2010. [PubMed: 20438949]
- Felsted AE, Shi Y, Masand PM, et al.: Intraoperative ultrasound for liver tumor resection in children. J Surg Res 198 (2): 418-23, 2015. [PubMed: 25940155]
- Rossi G, Tarasconi A, Baiocchi G, et al.: Fluorescence guided surgery in liver tumors: applications and advantages. Acta Biomed 89 (9-S): 135-140, 2018. [PMC free article: PMC6502182] [PubMed: 30561406]
- Otte JB, Pritchard J, Aronson DC, et al.: Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer 42 (1): 74-83, 2004. [PubMed: 14752798]
- Venkatramani R, Stein JE, Sapra A, et al.: Effect of neoadjuvant chemotherapy on resectability of stage III and IV hepatoblastoma. Br J Surg 102 (1): 108-13, 2015. [PubMed: 25349947]
- Vinayak R, Cruz RJ, Ranganathan S, et al.: Pediatric liver transplantation for hepatocellular cancer and rare liver malignancies: US multicenter and single-center experience (1981-2015). Liver Transpl 23 (12): 1577-1588, 2017. [PMC free article: PMC5725660] [PubMed: 28834194]
- Guiteau JJ, Cotton RT, Karpen SJ, et al.: Pediatric liver transplantation for primary malignant liver tumors with a focus on hepatic epithelioid hemangioendothelioma: the UNOS experience. Pediatr Transplant 14 (3): 326-31, 2010. [PubMed: 20051026]
- Malek MM, Shah SR, Atri P, et al.: Review of outcomes of primary liver cancers in children: our institutional experience with resection and transplantation. Surgery 148 (4): 778-82; discussion 782-4, 2010. [PubMed: 20728194]
- Héry G, Franchi-Abella S, Habes D, et al.: Initial liver transplantation for unresectable hepatoblastoma after chemotherapy. Pediatr Blood Cancer 57 (7): 1270-5, 2011. [PubMed: 21910210]
- Suh MY, Wang K, Gutweiler JR, et al.: Safety of minimal immunosuppression in liver transplantation for hepatoblastoma. J Pediatr Surg 43 (6): 1148-52, 2008. [PubMed: 18558198]
- Zsíros J, Maibach R, Shafford E, et al.: Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J Clin Oncol 28 (15): 2584-90, 2010. [PubMed: 20406943]
- Khan AS, Brecklin B, Vachharajani N, et al.: Liver Transplantation for Malignant Primary Pediatric Hepatic Tumors. J Am Coll Surg 225 (1): 103-113, 2017. [PubMed: 28232059]
- Browne M, Sher D, Grant D, et al.: Survival after liver transplantation for hepatoblastoma: a 2-center experience. J Pediatr Surg 43 (11): 1973-81, 2008. [PubMed: 18970927]
- Hamilton EC, Balogh J, Nguyen DT, et al.: Liver transplantation for primary hepatic malignancies of childhood: The UNOS experience. J Pediatr Surg : , 2017. [PubMed: 29108844]
- McAteer JP, Goldin AB, Healey PJ, et al.: Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant 17 (8): 744-50, 2013. [PubMed: 23992390]
- Reyes JD, Carr B, Dvorchik I, et al.: Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr 136 (6): 795-804, 2000. [PubMed: 10839879]
- Otte JB: Should the selection of children with hepatocellular carcinoma be based on Milan criteria? Pediatr Transplant 12 (1): 1-3, 2008. [PubMed: 18086239]
- de Ville de Goyet J, Meyers RL, Tiao GM, et al.: Beyond the Milan criteria for liver transplantation in children with hepatic tumours. Lancet Gastroenterol Hepatol 2 (6): 456-462, 2017. [PubMed: 28497761]
- Khanna R, Verma SK: Pediatric hepatocellular carcinoma. World J Gastroenterol 24 (35): 3980-3999, 2018. [PMC free article: PMC6148423] [PubMed: 30254403]
- Sevmis S, Karakayali H, Ozçay F, et al.: Liver transplantation for hepatocellular carcinoma in children. Pediatr Transplant 12 (1): 52-6, 2008. [PubMed: 18186889]
- Faraj W, Dar F, Marangoni G, et al.: Liver transplantation for hepatoblastoma. Liver Transpl 14 (11): 1614-9, 2008. [PubMed: 18975296]
- Pire A, Tambucci R, De Magnée C, et al.: Living donor liver transplantation for hepatic malignancies in children. Pediatr Transplant 25 (7): e14047, 2021. [PubMed: 34076944]
- Feusner JH, Krailo MD, Haas JE, et al.: Treatment of pulmonary metastases of initial stage I hepatoblastoma in childhood. Report from the Childrens Cancer Group. Cancer 71 (3): 859-64, 1993. [PubMed: 8381705]
- Zsiros J, Brugieres L, Brock P, et al.: Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. Lancet Oncol 14 (9): 834-42, 2013. [PMC free article: PMC3730732] [PubMed: 23831416]
- Meyers RL, Katzenstein HM, Krailo M, et al.: Surgical resection of pulmonary metastatic lesions in children with hepatoblastoma. J Pediatr Surg 42 (12): 2050-6, 2007. [PubMed: 18082706]
- O'Neill AF, Towbin AJ, Krailo MD, et al.: Characterization of Pulmonary Metastases in Children With Hepatoblastoma Treated on Children's Oncology Group Protocol AHEP0731 (The Treatment of Children With All Stages of Hepatoblastoma): A Report From the Children's Oncology Group. J Clin Oncol 35 (30): 3465-3473, 2017. [PMC free article: PMC5648177] [PubMed: 28892430]
- Yevich S, Calandri M, Gravel G, et al.: Reiterative Radiofrequency Ablation in the Management of Pediatric Patients with Hepatoblastoma Metastases to the Lung, Liver, or Bone. Cardiovasc Intervent Radiol 42 (1): 41-47, 2019. [PubMed: 30350173]
- Weeda VB, Murawski M, McCabe AJ, et al.: Fibrolamellar variant of hepatocellular carcinoma does not have a better survival than conventional hepatocellular carcinoma--results and treatment recommendations from the Childhood Liver Tumour Strategy Group (SIOPEL) experience. Eur J Cancer 49 (12): 2698-704, 2013. [PubMed: 23683550]
- Czauderna P, Lopez-Terrada D, Hiyama E, et al.: Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr 26 (1): 19-28, 2014. [PubMed: 24322718]
- Schnater JM, Aronson DC, Plaschkes J, et al.: Surgical view of the treatment of patients with hepatoblastoma: results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer 94 (4): 1111-20, 2002. [PubMed: 11920482]
- Habrand JL, Nehme D, Kalifa C, et al.: Is there a place for radiation therapy in the management of hepatoblastomas and hepatocellular carcinomas in children? Int J Radiat Oncol Biol Phys 23 (3): 525-31, 1992. [PubMed: 1319426]
- Wang PM, Chung NN, Hsu WC, et al.: Stereotactic body radiation therapy in hepatocellular carcinoma: Optimal treatment strategies based on liver segmentation and functional hepatic reserve. Rep Pract Oncol Radiother 20 (6): 417-24, 2015 Nov-Dec. [PMC free article: PMC4661347] [PubMed: 26696781]
- Xianliang H, Jianhong L, Xuewu J, et al.: Cure of hepatoblastoma with transcatheter arterial chemoembolization. J Pediatr Hematol Oncol 26 (1): 60-3, 2004. [PubMed: 14707717]
- Malogolowkin MH, Stanley P, Steele DA, et al.: Feasibility and toxicity of chemoembolization for children with liver tumors. J Clin Oncol 18 (6): 1279-84, 2000. [PubMed: 10715298]
- Hirakawa M, Nishie A, Asayama Y, et al.: Efficacy of preoperative transcatheter arterial chemoembolization combined with systemic chemotherapy for treatment of unresectable hepatoblastoma in children. Jpn J Radiol 32 (9): 529-36, 2014. [PubMed: 24923584]
- Aguado A, Dunn SP, Averill LW, et al.: Successful use of transarterial radioembolization with yttrium-90 (TARE-Y90) in two children with hepatoblastoma. Pediatr Blood Cancer 67 (9): e28421, 2020. [PubMed: 32603027]
- Hawkins CM, Kukreja K, Geller JI, et al.: Radioembolisation for treatment of pediatric hepatocellular carcinoma. Pediatr Radiol 43 (7): 876-81, 2013. [PubMed: 23212597]
- Wang S, Yang C, Zhang J, et al.: First experience of high-intensity focused ultrasound combined with transcatheter arterial embolization as local control for hepatoblastoma. Hepatology 59 (1): 170-7, 2014. [PubMed: 23813416]
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has been slowly increasing since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have multidisciplinary teams of cancer specialists with experience treating pediatric cancers. This multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons and transplant surgeons.
- Radiation oncologists.
- Pediatric medical oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child life professionals.
- Psychologists.
- Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for the role of pediatric cancer centers in the treatment of children and adolescents with cancer.[2] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with current standard therapy. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1] Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PMC free article: PMC4136455] [PubMed: 24853691]
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 8, 2022.
Hepatoblastoma
Incidence
The annual incidence of hepatoblastoma in the United States has increased (more than doubled), from 0.8 (1975–1983) to 2.0 (2018) cases per 1 million children aged 19 years and younger.[1-3] The cause for this increase is unknown, but the improved survival of premature infants with very low birth weight, which is known to be associated with hepatoblastoma, may contribute.[4] In Japan, the risk of hepatoblastoma in children who weighed less than 1,000 g at birth is 15 times the risk in children with normal birth weight.[5] Other data have confirmed the high incidence of hepatoblastoma in premature infants with very low birth weight.[6] Attempts to identify factors resulting from treatment of infants born prematurely have not revealed any suggestive causation of the increased incidence of hepatoblastoma.[4]
The age of onset of liver cancer in children is related to tumor histology. Hepatoblastomas usually occur before the age of 3 years, and approximately 90% of malignant liver tumors in children aged 4 years and younger are hepatoblastomas.[7]
Risk Factors
Conditions associated with an increased risk of hepatoblastoma are described in Table 4.
Table 4. Conditions Associated With an Increased Risk of Hepatoblastoma
| Associated Disorder | Clinical Findings |
|---|---|
| Aicardi syndrome [8] | For more information, see the Aicardi syndrome section. |
| Beckwith-Wiedemann syndrome [9,10] | For more information, see the Beckwith-Wiedemann syndrome and hemihyperplasia section. |
| Familial adenomatous polyposis [11-13] | For more information, see the Familial adenomatous polyposis section. |
| Glycogen storage diseases I–IV [14] | Symptoms vary by individual disorder. |
| Low-birth-weight infants [4-6,15,16] | Preterm and small-for-gestation-age neonates. |
| Simpson-Golabi-Behmel syndrome [17] | Macroglossia, macrosomia, renal and skeletal abnormalities, and increased risk of Wilms tumor. |
| Trisomy 18, other trisomies [18] | Trisomy 18: Microcephaly and micrognathia, clenched fists with overlapping fingers, and failure to thrive. Most patients (>90%) die in the first year of life. |
Aicardi syndrome
Aicardi syndrome is presumed to be an X-linked condition reported exclusively in females, leading to the hypothesis that a mutated gene on the X chromosome causes lethality in males. The syndrome is classically defined as agenesis of the corpus callosum, chorioretinal lacunae, and infantile spasms, with a characteristic facies. Additional brain, eye, and costovertebral defects are often found.[8]
Beckwith-Wiedemann syndrome and hemihyperplasia
The incidence of hepatoblastoma increases 1,000-fold to 10,000-fold in infants and children with Beckwith-Wiedemann syndrome.[10,19] The risk of hepatoblastoma also increases in patients with hemihyperplasia, previously termed hemihypertrophy, a condition that results in asymmetry between the right and left side of the body when a body part grows faster than normal.[20,21]
Beckwith-Wiedemann syndrome is most commonly caused by epigenetic changes and is sporadic. The syndrome may also be caused by genetic mutations and be familial. Either mechanism can be associated with an increased incidence of embryonal tumors, including Wilms tumor and hepatoblastoma.[10] The expression of both IGFR2 alleles and ensuing increased expression of insulin-like growth factor 2 (IGF-2) has been implicated in the macrosomia and embryonal tumors seen in patients with Beckwith-Wiedemann syndrome.[10,22] The types of embryonal tumors associated with sporadic Beckwith-Wiedemann syndrome have frequently undergone somatic changes in the Beckwith-Wiedemann syndrome locus and IGF-2.[23,24] The genetics of tumors in children with hemihyperplasia have not been clearly defined.
To detect abdominal malignancies at an early stage, all children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia undergo regular screening for multiple tumor types by abdominal ultrasonography.[21] Screening using alpha-fetoprotein (AFP) levels has also been quite helpful in the early detection of hepatoblastoma in these children.[25] Because the hepatoblastomas that are discovered early are small, treatment may minimize the use of adjuvant therapy after surgery.[19] However, a careful compilation of published data on 1,370 children with (epi)genotyped Beckwith-Wiedemann syndrome demonstrated that the prevalence of hepatoblastoma was 4.7% in those with Beckwith-Wiedemann syndrome caused by chromosome 11p15 paternal uniparental disomy, less than 1% in the two types of alteration in imprinting control regions, and absent in CDKN1C mutation.[26] The authors recommended that only children with Beckwith-Wiedemann syndrome caused by uniparental disomy be screened for hepatoblastoma using abdominal ultrasonography and AFP levels every 3 months from age 3 months to 5 years.
Familial adenomatous polyposis
There is an association between hepatoblastoma and familial adenomatous polyposis (FAP). Children in families that carry the APC gene have an 800-fold increased risk of hepatoblastoma. Screening for hepatoblastoma in members of families with FAP using ultrasonography and AFP levels is controversial because hepatoblastoma has been reported to occur in less than 1% of FAP family members.[11-13,27] However, one study of 50 consecutive children with apparent sporadic hepatoblastoma reported that five children (10%) had APC germline mutations.[27]
Current evidence cannot rule out the possibility that predisposition to hepatoblastoma may be limited to a specific subset of APC mutations. Another study of children with hepatoblastoma found a predominance of the mutation in the 5' region of the gene, but some patients had mutations closer to the 3' region.[28] This preliminary study provides some evidence that screening children with hepatoblastoma for APC mutations and colon cancer may be appropriate.
In the absence of APC germline mutations, childhood hepatoblastomas do not have somatic mutations in the APC gene; however, hepatoblastomas frequently have mutations in the CTNNB1 gene, the function of which is closely related to APC.[29]
Screening children predisposed to hepatoblastoma
An American Association for Cancer Research publication suggested that all children with more-than-a-1% risk of developing hepatoblastoma undergo screening. This includes patients with Beckwith-Wiedemann syndrome, hemihyperplasia, Simpson-Golabi-Behmel syndrome, and trisomy 18 syndrome. Screening is by abdominal ultrasonography and AFP determination every 3 months from birth (or diagnosis) through the fourth birthday, which will identify 90% to 95% of hepatoblastomas that develop in these children.[30]
Genomics of Hepatoblastoma
Molecular features of hepatoblastoma
Genomic findings related to hepatoblastoma include the following:
- The frequency of mutations in hepatoblastoma, as determined by three groups using whole-exome sequencing, was very low (approximately three variants per tumor) in children younger than 5 years.[31-34] A pediatric pan-cancer genomics study found that hepatoblastoma had the lowest gene mutation rate among all childhood cancers studied.[35]
- Hepatoblastoma is primarily a disease of WNT pathway activation. The primary mechanism for WNT pathway activation is CTNNB1 activating mutations/deletions involving exon 3. CTNNB1 mutations have been reported in more than 80% of cases.[31,33,34,36,37] A less common cause of WNT pathway activation in hepatoblastoma is mutations in APC associated with familial adenomatosis polyposis coli.[36]
- Similar NFE2L2 mutations have been found in many types of cancer, including hepatocellular carcinoma. These mutations render NFE2L2 insensitive to KEAP1-mediated degradation, leading to activation of the NFE2L2-KEAP1 pathway, which activates resistance to oxidative stress and is believed to confer resistance to chemotherapy.
- TERT and TP53 mutations, which are common in adults with hepatocellular carcinoma,[38] are uncommon in pediatric hepatoblastoma.[31,33,34,36] Hepatoblastoma cases with TERT mutations occur at a significantly older age, compared with hepatoblastoma cases without TERT mutations (median age at diagnosis, approximately 10 years vs. 1.4 years).[37]
Gene expression and epigenetic profiling have been used to identify biological subtypes of hepatoblastoma and to evaluate the prognostic significance of these subtypes.[33,36,37,40]
- A 16-gene expression signature divided hepatoblastoma cases into two subsets,[37,40] C1 and C2. The C1 subtype included most of the well-differentiated fetal (pure fetal) histology cases. The C2 subtype showed a more immature pattern and was associated with higher rates of metastatic disease at diagnosis. In a study of 174 hepatoblastoma patients, the C2 subtype was a significant predictor of poor outcome in multivariable analysis.[37]
- A second research group also found that gene expression profiling could be used to identify subsets of hepatoblastoma with favorable versus unfavorable prognosis.[33] The unfavorable prognosis group showed elevated expression of genes associated with embryonic stem cell and progenitor cells (e.g., LIN28B, SALL4, and HMGA2). The favorable prognosis group showed elevated expression of genes associated with liver differentiation (e.g., HNF1A).
- A gene expression signature at chromosome 14q32 (e.g., DLK1) was identified, with a stronger expression signal being associated with higher risk of treatment failure.[34] A strong 14q32 expression signature was also observed in fetal liver tissue, further supporting the concept that hepatoblastoma cases with biological characteristics similar to those of hepatic precursor cells have an inferior prognosis.
- Epigenetic profiling of hepatoblastoma has been used to identify molecularly defined hepatoblastoma subtypes. Tumors from 113 patients with hepatoblastoma were evaluated using DNA methylation arrays. Two distinctive subtypes were identified, epigenetic cluster A and B (Epi-CA and Epi-CB).[34] The methylation profile of Epi-CB resembled that of early embryonal/fetal phases of liver development. The methylation profile of Epi-CA was similar to that of late fetal or postnatal liver phases. Event-free survival was significantly lower for patients with the Epi-CB subtype than for those with the Epi-CA subtype.[34]
Delineating the clinical applications of the genomic, transcriptomic, and epigenomic profiling methods described above for the risk classification of patients with hepatoblastoma will require independent validation, which is one of the objectives of the ongoing Paediatric Hepatic International Tumour Trial (PHITT [NCT03017326]).
Diagnosis
Biopsy
A biopsy is always indicated to secure the diagnosis of a pediatric liver tumor, except for the following circumstances:
- Infantile hepatic hemangioma. Biopsy is not indicated for patients with infantile hemangioma of the liver with classic findings on magnetic resonance imaging (MRI). If the diagnosis is in doubt after high-quality imaging, a confirmatory biopsy is done.
- Focal nodular hyperplasia. Biopsy may not be indicated or may be delayed for patients with focal nodular hyperplasia with classic features on MRI using hepatocyte-specific contrast agent. If the diagnosis is in doubt, a confirmatory biopsy is done.
- Children's Oncology Group (COG) surgical guidelines (AHEP0731 [NCT00980460] appendix) recommend tumor resection at diagnosis without preoperative chemotherapy in children with PRE-Treatment EXTent of disease (PRETEXT) group I tumors and PRETEXT group II tumors with greater than 1 cm radiographic margin on the vena cava and middle hepatic and portal veins. Therefore, biopsy is not usually recommended in this circumstance.
- Infantile hepatic choriocarcinoma. In patients with infantile hepatic choriocarcinoma, which can be diagnosed by imaging and markedly elevated beta-human chorionic gonadotropin (beta-hCG), chemotherapy without biopsy is often indicated.[41]
Tumor markers
The AFP and beta-hCG tumor markers are helpful in the diagnosis and management of liver tumors. Although AFP is elevated in most children with hepatic malignancy, it is not pathognomonic for a malignant liver tumor.[42] The AFP level can be elevated with either a benign tumor or a malignant solid tumor. Markedly elevated AFP not caused by the tumor is normal in neonates and steadily falls after birth. The half-life of AFP is 5 to 7 days, and by age 1 year, it should be in the normal range, less than 10 ng/mL.[43,44] Beta-hCG levels may also be elevated in children with hepatoblastoma or hepatocellular carcinoma, which may result in isosexual precocity in boys.[45,46]
Prognosis and Prognostic Factors
Prognosis
The 5-year overall survival (OS) rate for children with hepatoblastoma is 70%.[47,48] Neonates with hepatoblastoma have outcomes comparable to older children up to age 5 years.[49]
A collaborative group consisting of four study groups (International Childhood Liver Tumors Strategy Group [SIOPEL], COG, Gesellschaft für Pädiatrische Onkologie und Hämatologie, and Japanese Study Group for Pediatric Liver Tumor [JPLT]) was named the Children's Hepatic Tumor International Collaboration (CHIC). The CHIC group analyzed survival in a collaborative database of 1,605 patients with hepatoblastoma treated in eight separate multicenter clinical trials, with central review of all tumor imaging and histological details.[50] Patients who underwent orthotopic liver transplant are included in all of the international study results.[51]
Survival rates at 5 years, unrelated to annotation factors, were found to be the following:
- 90% for patients with PRETEXT I group tumors.
- 83% for patients with PRETEXT II group tumors.
- 73% for patients with PRETEXT III group tumors.
- 52% for patients with PRETEXT IV group tumors.
When each annotation factor was examined separately, regardless of the PRETEXT group or other annotation factors present in each patient, the 5-year OS rates were found to be the following:
- 51% for patients with positive V (involvement of all three hepatic veins and/or inferior vena cava).
- 49% for patients with positive P (involvement of both right and left portal veins).
- 53% for patients with positive E (contiguous extrahepatic tumor).
- 52% for patients with positive F (multifocal).
- 51% for patients with positive R (tumor rupture).
- 41% for patients with positive M (distant metastasis).
For more information about PRETEXT grouping and annotation factors, see the PRETEXT and POSTTEXT Group Definitions section.
Hepatoblastoma prognosis by Evans surgical stage. For more information, see the Evans Surgical Staging for Childhood Liver Cancer (Historical) section.
- Stages I and II.Approximately 20% to 30% of children with hepatoblastoma have stage I or II disease. Prognosis varies depending on the subtype of hepatoblastoma:
- Stage III.
- Stage IV.Approximately 10% to 20% of children with hepatoblastoma have stage IV disease. The 3- to 5-year OS rate for children with stage IV hepatoblastoma varies widely, from 20% to approximately 60%, based on published reports.[52,53,56-59] Postsurgical stage IV is equivalent to any PRETEXT group with annotation factor M.[50,60,61]
Prognostic factors
Individual childhood cancer study groups have attempted to define the relative importance of a variety of prognostic factors present at diagnosis and in response to therapy.[62,63] The CHIC study group retrospectively combined data from eight clinical trials (N = 1,605) conducted between 1988 and 2010. They published a univariate analysis of the effect of clinical prognostic factors present at the time of diagnosis on event-free survival (EFS).[50,64] The analysis confirmed many of the statistically significant adverse factors described below:[50]
- Higher PRETEXT group. [50]
- Positive PRETEXT annotation factors: [50]
- V: Involvement of all three hepatic veins and/or intrahepatic inferior vena cava.
- P: Involvement of both left and right portal veins.
- E: Contiguous extrahepatic tumor extensions (e.g., diaphragm, adjacent organs).
- F: Multifocal tumors.
- R: Tumor rupture.
- M: Distant metastases, usually lung.
- Low AFP level (<100 ng/mL or 100–1,000 ng/mL to account for infants with elevated AFP levels). [64]
- Older age. Patients aged 3 to 7 years have a worse outcome in the PRETEXT IV group.[50] Patients aged 8 years and older have a worse outcome than do younger patients in all PRETEXT groups. In a subsequent report from the CHIC group, risk of an event increased with advancing age throughout all age cohorts.[65][Level of evidence C1] Increasing age attenuated the effect of other risk factors, including metastasis, AFP level less than 100 ng/mL, tumor rupture, and the presence of one annotation factor.In contrast, in the SIOPEL-2 and -3 studies, infants younger than 6 months had PRETEXT, annotation factors, and outcomes similar to that of older children undergoing the same treatment.[66][Level of evidence C1]
In the CHIC study, sex, prematurity, birth weight, and Beckwith-Wiedemann syndrome had no effect on EFS.[50]
A multivariate analysis of these prognostic factors was published to help develop a new risk group classification for hepatoblastoma.[64] This classification was used to generate a risk stratification schema to be used in international clinical trials. For more information, see the International risk classification model section.
Other studies observed the following factors that affected prognosis:
- PRETEXT group: In SIOPEL studies, having a low PRETEXT group at diagnosis (PRETEXT I, II, and III tumors) is a good prognostic factor, whereas PRETEXT IV is a poor prognostic factor.[50] For more information, see the Tumor Stratification by Imaging and Evans Surgical Staging for Childhood Liver Cancer section.
- Tumor stage: In COG studies, patients with stage I tumors that were resected at diagnosis with usual hepatoblastoma histology have a favorable outcome when treated with limited chemotherapy. Patients with tumors that have well-differentiated fetal histology have an excellent prognosis. These tumors are not generally treated with chemotherapy. Patients with tumors of other stages and histologies are treated more aggressively.[50]
- Treatment-related factors:Chemotherapy: Chemotherapy often decreases the size and extent of hepatoblastoma, allowing complete resection.[53,56,67-69] Favorable response of the primary tumor to chemotherapy, defined as either a 30% decrease in tumor size by Response Evaluation Criteria In Solid Tumors (RECIST) or 90% or greater decrease in AFP levels, predicted the resectability of the tumor. In turn, this favorable response predicted OS among all CHIC risk groups treated with neoadjuvant chemotherapy in the JPLT-2 Japanese national clinical trial.[70][Level of evidence B4]Surgery: Cure of hepatoblastoma requires gross tumor resection. Hepatoblastoma is most often unifocal, so resection may be possible. If a hepatoblastoma is completely removed, most patients survive. However, because of vascular or other involvement, less than one-third of patients have lesions that are amenable to complete resection at diagnosis.[50] It is critically important that a child with probable hepatoblastoma be evaluated by a pediatric surgeon who is experienced in the techniques of extreme liver resection with vascular reconstruction. The child should also have access to a liver transplant program. In advanced tumors, surgical treatment of hepatoblastoma is a demanding procedure. Postoperative complications in high-risk patients decrease the OS rate.[71]Orthotopic liver transplant: Orthotopic liver transplant is an additional treatment option for patients whose tumor remains unresectable after preoperative chemotherapy;[51,72] however, the presence of microscopic residual tumor at the surgical margin does not preclude a favorable outcome.[73,74] This may result from the additional courses of chemotherapy that are administered before or after resection.[67,68,73]For more information about the outcomes associated with specific chemotherapy regimens, see Table 6.
- Tumor marker–related factors:Ninety percent of children with hepatoblastoma and two-thirds of children with hepatocellular carcinoma exhibit elevated levels of the serum tumor marker AFP, which parallels disease activity. The level of AFP at diagnosis and rate of decrease in AFP levels during treatment are compared with the age-adjusted normal range. Lack of a significant decrease in AFP levels with treatment may predict a poor response to therapy.[75] In an exploratory study of 34 children with hepatoblastoma, the rate of decrease in AFP and tumor volume, but not in RECIST I measurements, following two courses of treatment after diagnosis was predictive of EFS and OS.[76]Absence of elevated AFP levels at diagnosis (AFP <100 ng/mL) occurs in a small percentage of children with hepatoblastoma and appears to be associated with very poor prognosis, as well as with the small cell undifferentiated variant of hepatoblastoma.[50] Some of these variants do not express SMARCB1 and may be considered rhabdoid tumors of the liver, which require alternative therapy. All small cell undifferentiated hepatoblastomas are tested for loss of SMARCB1 expression by immunohistochemistry to determine those that should be treated as a hepatoblastoma versus those that should be treated as rhabdoid tumors of the liver.[52,55,58,59,77,78]
- Tumor histology:For more information, see the Histology section in the Hepatoblastoma section.
Other variables have been proposed to be poor prognostic factors, but the relative importance of their prognostic significance has been difficult to define. In the SIOPEL-1 study, a multivariate analysis of prognosis after positive response to chemotherapy showed that only one variable, PRETEXT, predicted OS, while metastasis and PRETEXT predicted EFS.[77] In an analysis of the U.S. intergroup study from the time of diagnosis, well-differentiated fetal histology, small cell undifferentiated histology, and AFP less than 100 ng/mL were prognostic in a log rank analysis. PRETEXT was prognostic among patients designated group III, but not group IV.[52,79] The CHIC study incorporated detailed hepatoblastoma patient data from multiple groups, establishing a solid foundation of risk factors.[79]
Histology
Hepatoblastoma arises from precursors of hepatocytes and can have several morphologies, including the following:[80]
- Small cells that reflect neither epithelial nor stromal differentiation. It is critical to discriminate between small cell undifferentiated hepatoblastoma expressing SMARCB1 and rhabdoid tumor of the liver, which lacks the SMARCB1 gene and SMARCB1 expression. Both diseases may share similar histology. Optimal treatment of rhabdoid tumor of the liver and small cell undifferentiated hepatoblastoma may require different approaches and different chemotherapy. For a more extensive discussion on the differences of these two diseases, see the Small cell undifferentiated histology hepatoblastoma and rhabdoid tumors of the liver section.
- Embryonal epithelial cells resembling the liver epithelium at 6 to 8 weeks of gestation.
- Well-differentiated fetal hepatocytes morphologically indistinguishable from normal fetal liver cells.
Most often the tumor consists of a mixture of epithelial hepatocyte precursors. About 20% of tumors have stromal derivatives such as osteoid, chondroid, and rhabdoid elements. Occasionally, neuronal, melanocytic, squamous, and enteroendocrine elements are found. The following histological subtypes have clinical relevance:
- Mixed fetal and embryonal epithelial cells.
- Small cell undifferentiated histology hepatoblastoma and rhabdoid tumors of the liver.
- Small cell undifferentiated hepatoblastoma (SMARCB1 positive).
- Rhabdoid tumor of liver (SMARCB1 negative).
Well-differentiated fetal (pure fetal) histology hepatoblastoma
An analysis of patients with initially resected hepatoblastoma tumors (before receiving chemotherapy) has suggested that patients with well-differentiated fetal (previously termed pure fetal) histology tumors have a better prognosis than do patients with an admixture of more primitive and rapidly dividing embryonal components or other undifferentiated tissues. Studies have reported the following:
- A study of patients with hepatoblastoma and well-differentiated fetal histology tumors observed the following:[53]
- In a COG study (COG-P9645), 16 patients with well-differentiated fetal histology hepatoblastoma with two or fewer mitoses per 10 high-power fields were not treated with chemotherapy. Retrospectively, their PRETEXT groups were group I (n = 4), group II (n = 6), and group III (n = 2).[54]
- The survival rate was 100% for these patients who did not receive chemotherapy.
- All 16 patients were alive with no evidence of disease at a median follow-up of 4.9 years (range, 9 months to 9.2 years).
Thus, complete resection of a well-differentiated fetal hepatoblastoma may preclude the need for chemotherapy.
Small cell undifferentiated histology hepatoblastoma and rhabdoid tumors of the liver
Small cell undifferentiated hepatoblastoma (SMARCB1 positive) is an uncommon hepatoblastoma variant that represents several percent of all hepatoblastomas. It tends to occur at a younger age (6–10 months) than do other cases of hepatoblastoma [52,83] and is associated with AFP levels that are normal for age at presentation.[55,83]
Histologically, small cell undifferentiated hepatoblastoma is typified by a diffuse population of small cells with scant cytoplasm resembling neuroblasts.[84]
Small cell undifferentiated hepatoblastoma may be difficult to distinguish from malignant rhabdoid tumor of the liver, which has been conflated with small cell undifferentiated hepatoblastoma in past studies. A characteristic shared by small cell undifferentiated hepatoblastoma and malignant rhabdoid tumors is the poor prognosis associated with each.[52,83,85] They can be distinguished by the following characteristic abnormalities:
- Lack of SMARCB1 expression. Lack of detection of SMARCB1 by immunohistochemistry is characteristic of malignant rhabdoid tumors.[83]
The ongoing Paediatric Hepatic International Tumour Trial (PHITT) designates any childhood liver tumor as rhabdoid tumor of the liver if it contains cells that lack SMARCB1 expression. Patients with SMARCB1-negative tumors, which are presumed to be related to rhabdoid tumors, may not be enrolled in the international trial, which addresses treatment of hepatoblastoma that includes small cell undifferentiated histology, hepatocellular carcinoma, and hepatic malignancy of childhood, not otherwise specified (NOS), but not rhabdoid tumor of the liver. In this trial, all patients with histology consistent with pure small cell undifferentiated hepatoblastoma, as assessed by the institutional pathologist, are required to have testing for SMARCB1 by immunohistochemistry according to the practices at the institution. In addition, presence of a blastemal component indicates conventional hepatoblastoma.[80] For more information about the AHEP1531 trial, see the Treatment Options Under Clinical Evaluation for Hepatoblastoma section.
If SMARCB1 is maintained but small cell undifferentiated histology is present, the current literature suggests a worse outcome for these patients. However, because small cell undifferentiated hepatoblastoma and rhabdoid tumor of the liver have not been discriminated in past studies, some of the prognostic features attributed to the former may have been contributed in part by the latter. Published studies of prognostic features related to small cell undifferentiated histology include the following:
- In 2009, the results of a study of 11 young children with low AFP levels and small cell morphology were reported. Ten children died of disease progression, and one child died of complications. Six of six children tested were SMARCB1 negative, but only one child had any rhabdoid morphology. This finding suggests that many or all liver tumors with small cell morphology and very low AFP levels in young children may be rhabdoid tumors of the liver. These tumors have a poor prognosis that is associated with the driver mutation.[83]
- A single-institution study of seven children with small cell morphology liver tumors found that all retained expression of SMARCB1. Six children survived, and one child died of complications from liver transplant.[87]
- A study of 23 liver tumors from the Kiel tumor bank found 12 tumors with small cell morphology. Nine tumors had malignant rhabdoid tumor classic histology, and two tumors had mixed small cell and rhabdoid histologies. Outcomes were not provided, but it was noted that rhabdoid brain tumors had small cell, not classic, rhabdoid histology.[88]
- In a single-institution study of six children with SMARCB1-negative liver tumors, two children with small cell morphology died. The remaining four children with classic rhabdoid histology were not treated with cisplatin-based therapy; three children survived, and one child died of complications from transplant.[89]
The outcomes of the CHIC trial of childhood liver tumors may clarify some of the questions regarding these different histological and genetic findings.
Patients with small cell undifferentiated hepatoblastoma whose tumors are unresectable have an especially poor prognosis.[83] Patients with stage I tumors appear to have increased risk of treatment failure when small cell elements are present.[90] For this reason, completely resected tumors composed of well-differentiated fetal histology or of mixed fetal and embryonal cells must have a thorough histological examination because small foci of undifferentiated small cell histology indicates a need for aggressive chemotherapy.[90] Aggressive treatment for this histology was investigated in the completed COG AHEP0731 (NCT00980460) study, and all tumors were tested for SMARCB1 expression by immunohistochemistry. In this study, hepatoblastoma that would otherwise be considered very low or low risk was upgraded to intermediate risk if any small cell undifferentiated elements were found. For more information, see Table 5.
Risk Stratification
There are significant differences among childhood cancer study groups in risk stratification used to determine treatment, making it difficult to compare results of the different treatments administered. Table 5 demonstrates the variability in the definitions of risk groups.
Table 5. A Comparison of the Use of PRETEXT in Risk Stratification Schemes for Hepatoblastomaa,b
| COG (AHEP-0731) | SIOPEL (SIOPEL-3, -3HR, -4, -6) | GPOH | JPLT (JPLT-2 and -3) | |
|---|---|---|---|---|
| Very low risk | PRETEXT I or II; well-differentiated fetal histology; primary resection at diagnosis | |||
| Low risk/standard risk | PRETEXT I or II of any histology with primary resection at diagnosis | PRETEXT I, II, or III | PRETEXT I, II, or III | PRETEXT I, II, or III |
| Intermediate riskb | PRETEXT II, III, or IV unresectable at diagnosis; or V+c, P+, E+; SCU histology | PRETEXT IV or any PRETEXT with rupture; or N1, P2, P2a, V3, V3a; or multifocal | ||
| High riskb | Any PRETEXT with M+; AFP level <100 ng/mL | Any PRETEXT; V+, P+, E+, M+; SCU histology; AFP level <100 ng/mL; tumor rupture | Any PRETEXT with V+, E+, P+, M+ or multifocal | Any PRETEXT with M1 or N2; or AFP level <100 ng/mL |
AFP = alpha-fetoprotein; COG = Children's Oncology Group; GPOH = Gesellschaft für Pädiatrische Onkologie und Hämatologie (Society for Paediatric Oncology and Haematology); JPLT = Japanese Study Group for Pediatric Liver Tumor; PRETEXT = PRE-Treatment EXTent of disease; SCU = small cell undifferentiated; SIOPEL = International Childhood Liver Tumors Strategy Group.
aAdapted from Czauderna et al.[79]
bFor more information about the annotations used in PRETEXT, see Table 2.
cThe COG and PRETEXT definitions of vascular involvement differ.
International risk classification model
The CHIC group developed a novel risk stratification system for use in international clinical trials on the basis of prognostic features present at diagnosis. CHIC unified the disparate definitions and staging systems used by pediatric cooperative multicenter trial groups, enabling the comparison of studies conducted by heterogeneous groups in different countries.[64] Original detailed clinical patient data were extracted from eight published clinical trials using central review of imaging and histology, and prognostic factors were identified by univariate analysis.[50]
Based on the initial univariate analysis of the data combined with historical clinical treatment patterns and data from previous large clinical trials, five backbone groups were selected, which allowed for further risk stratification. Subsequent multivariate analysis on the basis of these backbone groups defined the following clinical prognostic factors: PRETEXT group (I, II, III, or IV), presence of metastasis (yes or no), and AFP (≤100 ng/mL). The backbone groups are as follows:[64]
- Backbone 1: PRETEXT I/II, not metastatic, AFP greater than 100 ng/mL.
- Backbone 2: PRETEXT III, not metastatic, AFP greater than 100 ng/mL.
- Backbone 3: PRETEXT IV, not metastatic, AFP greater than 100 ng/mL.
- Backbone 4: Any PRETEXT group, metastatic disease at diagnosis, AFP greater than 100 ng/mL.
- Backbone 5: Any PRETEXT group, metastatic or not, AFP less than or equal to 100 ng/mL at diagnosis.
Other diagnostic factors (e.g., age) were queried for each of the backbone categories, including the presence of at least one of the following PRETEXT annotations (defined as VPEFR+, see Table 2) or AFP less than or equal to 100 ng/mL:[64]
- V: Involvement of vena cava or all three hepatic veins, or both.
- P: Involvement of portal bifurcation or both right and left portal veins, or both.
- E: Extrahepatic contiguous tumor extension.
- F: Multifocal liver tumor.
- R: Tumor rupture at diagnosis.
An assessment of surgical resectability at diagnosis was added for PRETEXT I and II patients. Patients in each of the five backbone categories were stratified on the basis of backwards stepwise elimination multivariable analysis of additional patient characteristics, including age and presence or absence of PRETEXT annotation factors (V, P, E, F, and R). Each of these subcategories received one of four risk designations (very low, low, intermediate, or high). The result of the multivariate analysis was used to assign patients to very low-, low-, intermediate-, and high-risk categories, as shown in Figure 2. For example, the finding of an AFP level of 100 to 1,000 ng/mL was significant only among patients younger than 8 years in the backbone PRETEXT III group. The analysis enables prognostically similar risk groups to be assigned to the appropriate treatment groups on upcoming international protocols.[64]
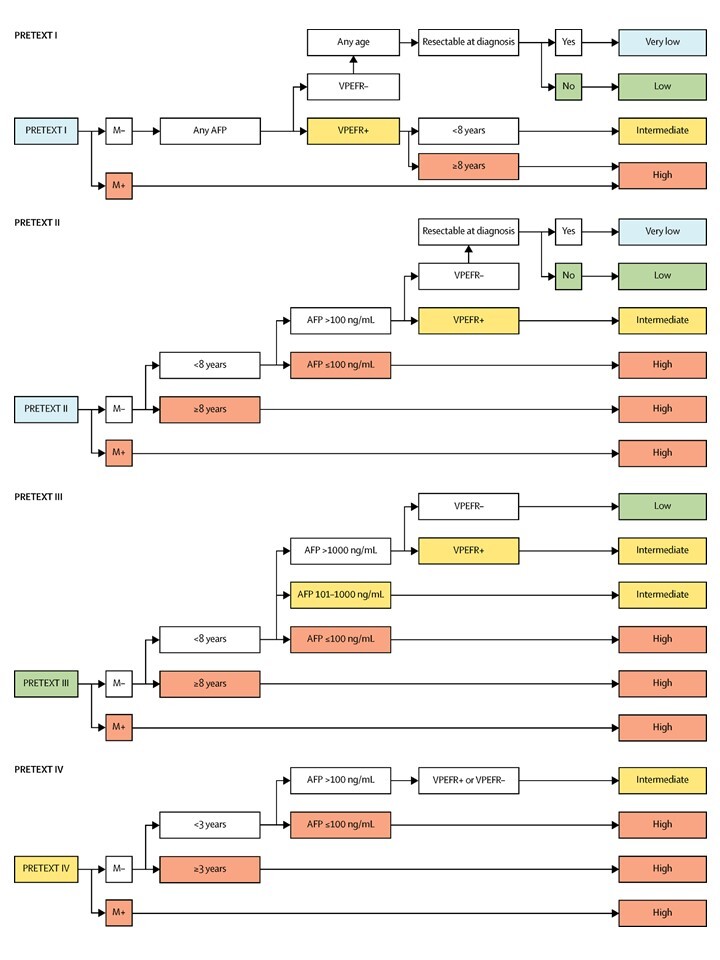
Figure 2. Risk stratification trees for the Children’s Hepatic tumors International Collaboration—Hepatoblastoma Stratification (CHIC-HS). Very low-risk group and low-risk group are separated only by their resectability at diagnosis, which has been defined by international consensus as part of the surgical guidelines for the upcoming collaborative trial, Paediatric Hepatic International Tumour Trial (PHITT). Separate risk stratification trees are used for each of the four PRETEXT groups. AFP = alpha-fetoprotein. M = metastatic disease. PRETEXT = PRETreatment EXTent of disease.[64] Reprinted from The Lancet Oncology, Volume 18, Meyers RL, Maibach R, Hiyama E, Häberle B, Krailo M, Rangaswami A, Aronson DC, Malogolowkin MH, Perilongo G, von Schweinitz D, Ansari M, Lopez-Terrada D, Tanaka Y, Alaggio R, Leuschner I, Hishiki T, Schmid I, Watanabe K, Yoshimura K, Feng Y, Rinaldi E, Saraceno D, Derosa M, Czauderna P, Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration, Pages 122–131, Copyright (2017), with permission from Elsevier.
Treatment of Hepatoblastoma
Treatment options for newly diagnosed hepatoblastoma depend on the following:
- Whether the cancer is resectable at diagnosis.
- The tumor histology.
- How the cancer responds to chemotherapy.
- Whether the cancer has metastasized.
Cisplatin-based chemotherapy has resulted in a survival rate of more than 90% for children with PRETEXT and POST-Treatment EXTent (POSTTEXT) I and II resectable disease before or after chemotherapy.[56,58,68]
Chemotherapy regimens used in the treatment of hepatoblastoma and their respective outcomes are described in Table 6. For information describing each stage, see the Tumor Stratification by Imaging and Evans Surgical Staging for Childhood Liver Cancer section.
Table 6. Outcomes for Hepatoblastoma Multicenter Trialsa
| Study | Chemotherapy Regimen | Number of Patients | Outcomes |
|---|---|---|---|
| INT0098 (CCG/POG) 1989–1992 | C5V vs. CDDP/DOXO | Stage I/II: 50 | 4-Year EFS/OS: |
| I/II = 88%/100% vs. 96%/96% | |||
| Stage III: 83 | III = 60%/68% vs. 68%/71% | ||
| Stage IV: 40 | IV = 14%/33% vs. 37%/42% | ||
| P9645 (COG)b 1999–2002 | C5V vs. CDDP/CARBO | Stage I/II: Pending publication | 1-Year EFS: |
| I/II: Pending publication | |||
| Stage III: 38 | III/IV: C5V = 51%; CDDP/CARBO = 37% | ||
| Stage IV: 50 | |||
| AHEP0731 (COG) 2010–2014 [92][Level of evidence C1] | LR: C5V (2 cycles) | LR (stage I/II): 49 | 5-year EFS: 88%; 5-year OS: 91% |
| HB 94 (GPOH) 1994–1997 | I/II: IFOS/CDDP/DOXO | Stage I: 27 | 4-Year EFS/OS: |
| I = 89%/96% | |||
| Stage II: 3 | II = 100%/100% | ||
| III/IV: IFOS/CDDP/DOXO + VP/CARBO | Stage III: 25 | III = 68%/76% | |
| Stage IV: 14 | IV = 21%/36% | ||
| HB 99 (GPOH) 1999–2004 | SR: IPA | SR: 58 | 3-Year EFS/OS: |
| SR = 90%/88% | |||
| HR: CARBO/VP16 | HR: 42 | HR = 52%/55% | |
| SIOPEL-2 1994–1998 | SR: PLADO | PRETEXT I: 6 | 3-Year EFS/OS: |
| SR: 73%/91% | |||
| PRETEXT II: 36 | |||
| PRETEXT III: 25 | |||
| HR: CDDP/CARBO/DOXO | PRETEXT IV: 21 | HR: IV = 48%/61% | |
| Metastases: 25 | HR: metastases = 36%/44% | ||
| SIOPEL-3 1998–2006 | SR: CDDP vs. PLADO | SR: PRETEXT I: 18 | 3-Year EFS/OS: |
| SR: CDDP = 83%/95%; PLADO = 85%/93% | |||
| PRETEXT II: 133 | |||
| PRETEXT III: 104 | |||
| HR: SUPERPLADO | HR: PRETEXT IV: 74 | HR: Overall = 65%/69% | |
| VPE+: 70 | |||
| Metastases: 70 | Metastases = 57%/63% | ||
| AFP <100 ng/mL: 12 | |||
| SIOPEL-4 2005–2009 | HR: Block A: Weekly; CDDP/3 weekly DOXO; Block B: CARBO/DOXO | PRETEXT I: 2 | 3-Year EFS/OS: |
| All HR = 76%/83% | |||
| PRETEXT II: 17 | |||
| PRETEXT III: 27 | |||
| PRETEXT IV: 16 | HR: IV = 75%/88% | ||
| Metastases: 39 | HR: Metastases = 77%/79% | ||
| JPLT-1 1991–1999 | I/II: CDDP(30)/THP-DOXO | Stage I: 9 | 5-Year EFS/OS: |
| I = NR/100% | |||
| Stage II: 32 | II = NR/76% | ||
| III/IV: CDDP(60)/THP-DOXO | Stage IIIa: 48 | IIIa = NR/50% | |
| Stage IIIb: 25 | IIIb = NR/64% | ||
| Stage IV: 20 | IV = NR/77% | ||
| JPLT-2 1999–2010 [93][Level of evidence C1] | Initial surgery and 2 cycles of CITA | Stratum 1: PRETEXT I/II, 0 annotation factors except H+ (n = 40) | 5-Year EFS/OS: |
| 74.2%/89.9% | |||
| 2 cycles of CITA followed by surgery and 2–4 cycles of CITA | Stratum 2: PRETEXT II with multifocality (n = 80) | 84.8%/90.8% | |
| 2 cycles of CITA followed by 2 cycles of CITA (responders); attempted surgery including transplant | Stratum 3: PRETEXT I/II (annotation factors present) and III/IV (n = 176) responders | 71.6%/85.9% | |
| 2 cycles of CITA followed by 2 cycles of ITEC (nonresponders); attempted surgery including transplant | Stratum 4: PRETEXT I/II (annotation factors present) and III/IV (n = 59) nonresponders | 59.1%/67.3% |
AFP = alpha-fetoprotein; C5V = cisplatin, fluorouracil (5-FU), and vincristine; CARBO = carboplatin; CCG = Children’s Cancer Group; CDDP = cisplatin; CITA = pirarubicin-cisplatin; COG = Children's Oncology Group; DOXO = doxorubicin; EFS = event-free survival; GPOH = Gesellschaft für Pädiatrische Onkologie und Hämatologie (Society for Paediatric Oncology and Haematology); H+ = rupture or intraperitoneal hemorrhage; HR = high risk; IFOS = ifosfamide; IPA = ifosfamide, cisplatin, and doxorubicin; ITEC = ifosfamide, pirarubicin, etoposide, and carboplatin; JPLT = Japanese Study Group for Pediatric Liver Tumor; LR = low risk; NR = not reported; OS = overall survival; PLADO = cisplatin and doxorubicin; POG = Pediatric Oncology Group; PRETEXT = PRE-Treatment EXTent of disease; SIOPEL = International Childhood Liver Tumors Strategy Group; SR = standard risk; SUPERPLADO = cisplatin, doxorubicin, and carboplatin; THP = tetrahydropyranyl-adriamycin (pirarubicin); VP = vinorelbine and cisplatin; VPE+ = venous, portal, and extrahepatic involvement; VP16 = etoposide.
bStudy closed early because of inferior results in the CDDP/CARBO arm.
Treatment options for hepatoblastoma that is resectable at diagnosis
Approximately 20% to 30% of children with hepatoblastoma have resectable disease at diagnosis. COG surgical guidelines (AHEP0731 [NCT00980460] appendix) recommend tumor resection at diagnosis without preoperative chemotherapy in children with PRETEXT I tumors and PRETEXT II tumors with greater than 1 cm radiographic margin on the vena cava and middle hepatic and portal veins. Outcomes for patients after undergoing a complete resection at diagnosis, compared with patients who had positive microscopic margins found at resection, are similar after receiving chemotherapy.[58,59,73]; [94][Level of evidence C1]
Prognosis varies depending on the histological subtype, as follows:
Treatment options for hepatoblastoma resectable at diagnosis showing non–well-differentiated fetal histology include the following:
- Resection followed by two to four cycles of chemotherapy.[50]
Re-resection of positive microscopic margins may not be necessary. Conclusive evidence is lacking for tumors with resection at diagnosis compared with those with positive microscopic margins resected after preoperative chemotherapy.
Evidence (gross surgical resection, with or without microscopic margins, and postoperative chemotherapy):
- In the COG AHEP0731 (NCT00980460) trial, 49 of 51 patients with stage I or stage II hepatoblastoma (without pure fetal histology) received two cycles of adjuvant chemotherapy consisting of cisplatin, fluorouracil, and vincristine.[92][Level of evidence C1]
- The 5-year EFS rate was 88%, and the 5-year OS rate was 91%.
- This outcome is comparable to the outcomes for children treated with four cycles after initial resection, and to the outcomes for children treated with two cycles of neoadjuvant chemotherapy before resection followed by two cycles of chemotherapy after resection.
- There is no reliable data for local recurrence risk in patients with a positive microscopic margin status who underwent resection at diagnosis.[69] SIOPEL studies suggest that in patients who received preoperative chemotherapy, positive microscopic margin did not increase risk of local recurrence.[58,59,73]; [94][Level of evidence C1]
- In a European study conducted between 1990 and 1994, 11 patients had tumor found at the surgical margins after hepatic resection and two patients died, neither of whom had a local recurrence. None of the 11 patients underwent a second resection, and only one patient received radiation therapy postoperatively. All of the patients were treated with four courses of cisplatin and doxorubicin before surgery and received two courses of postoperative chemotherapy.[73]
- In another European study of high-risk hepatoblastoma, 11 patients had microscopic residual tumor remaining after initial surgery and received two to four postoperative cycles of chemotherapy with no additional surgery. Of these 11 patients, 9 survived.[59]
- In the SIOPEL-2 study, 13 of 13 patients with microscopic positive resection margins survived.[58]
- An unplanned retrospective study of the SIOPEL-2 and SIOPEL-3 trials found that after four courses of cisplatin for standard-risk patients and seven courses of cisplatin alternating with doxorubicin/carboplatin for high-risk patients, resection was performed where imaging suggested it would be safe. Of the 431 children treated in these trials, 58 patients had positive microscopic tumor margins, and 371 patients were in complete remission. There were no statistically significant differences in the rates of local recurrence, EFS, or OS between the two groups.[94][Level of evidence C1]
- A randomized clinical trial demonstrated comparable efficacy with postoperative cisplatin/vincristine/fluorouracil and cisplatin/doxorubicin in the treatment of patients with hepatoblastoma.[53]
- Although survival outcomes were nominally higher for the children who received cisplatin/doxorubicin, this difference was not statistically significant.
- The combination of cisplatin/vincristine/fluorouracil was significantly less toxic than were the doses of cisplatin/doxorubicin.
Results of chemotherapy clinical trials are described in Table 6.
Treatment options for hepatoblastoma of well-differentiated fetal (pure fetal) histology resectable at diagnosis include the following:
- Complete surgical resection followed by watchful waiting or chemotherapy.[54]
Evidence (complete surgical resection followed by watchful waiting or chemotherapy):
- In a COG prospective clinical trial (INT0098), nine children with stage I (completely resected) well-differentiated fetal histology and fewer than two mitoses per high-power field were treated with four cycles of adjuvant doxorubicin.[53]
- At a median follow-up of 5.1 years, the EFS and OS rates were 100% for all nine children.
- In the COG P9645 (NCT00003994) study, 16 patients with stage I (completely resected) tumors had well-differentiated fetal histology and received no adjuvant chemotherapy. In a retrospective PRETEXT classification of 21 of these 25 patients with adequate data, PRETEXT I, II, and III tumors were found in 7, 10, and 4 patients, respectively.[54]
- The EFS and OS rates were 100% for patients with stage I well-differentiated fetal histology, including one patient who had a second surgery to address a positive tumor margin.
- Treatment of a small focus of undifferentiated small cell histology within an otherwise well-differentiated fetal histology tumor with aggressive chemotherapy has been reported in the following small series, suggesting the importance of a thorough histological examination of apparent well-differentiated fetal histology.[90]A retrospective study of 16 patients with well-differentiated fetal histology treated at multiple institutions had complete surgical resections, but also had elements of (or, in some case, predominance of) small cell histology found in the resected tumor.[90]
- Despite receiving postoperative chemotherapy, hepatoblastoma recurred in 10 of 16 patients, and 5 of these patients died.
Treatment options for hepatoblastoma that is not resectable or not resected at diagnosis
Approximately 70% to 80% of children with hepatoblastoma have tumors that are not resected at diagnosis. COG surgical guidelines (AHEP0731 [NCT00980460] appendix) recommend a diagnostic biopsy without an attempt to resect the tumor in children with PRETEXT II tumors with less than 1 cm radiographic margin on the vena cava and middle hepatic vein and in all children with PRETEXT III and IV tumors.
Treatment options for hepatoblastoma that is not resectable or is not resected at diagnosis include the following:
- Chemotherapy followed by reassessment of surgical resectability and complete surgical resection.
Tumor rupture at presentation, resulting in major hemorrhage that can be controlled by transcatheter arterial embolization or partial resection to stabilize the patient, does not preclude a favorable outcome when followed by chemotherapy and definitive surgery.[105]
In recent years, most children with hepatoblastoma have been treated with chemotherapy. In European cancer centers, children with resectable hepatoblastoma at diagnosis are treated with preoperative chemotherapy, which may reduce the incidence of surgical complications at the time of resection.[56,58,73] Treatment with preoperative chemotherapy has been shown to benefit children with hepatoblastoma. In contrast, an American intergroup study of treatment of children with hepatoblastoma encouraged resection at the time of diagnosis for all tumors amenable to resection without undue risk. The study (COG-P9645) did not treat children with stage I tumors of well-differentiated fetal histology with preoperative or postoperative chemotherapy unless they developed progressive disease.[54] In this study, most patients with PRETEXT III and all PRETEXT IV tumors were treated with chemotherapy before resection or transplant.
Patients whose tumors remain unresectable after chemotherapy should be considered for liver transplant.[51,56,96-100] In the presence of features predicting unresectability, early coordination with a pediatric liver transplant service is critical.[78] In the COG AHEP0731 (NCT00980460) study, early referral (i.e., based on imaging done after the second cycle of chemotherapy) to a liver specialty center with transplant capability was recommended for patients with POSTTEXT III tumors with positive V or P and POSTTEXT IV tumors with positive F.
Evidence (chemotherapy followed by reassessment of surgical resectability and complete surgical resection or liver transplant):
- In the SIOPEL-1 study, preoperative chemotherapy (doxorubicin and cisplatin) was given to all children with hepatoblastoma with or without metastases. After chemotherapy, and excluding those who underwent a liver transplant (<5% of patients), complete resection was performed.[56]
- The chemotherapy was well tolerated.
- Complete resection was obtained in 87% of children.
- This strategy resulted in an OS rate of 75% at 5 years after diagnosis.
- Identical results were seen in a follow-up international study (SIOPEL-2).[58]
- The SIOPEL-3 study compared cisplatin alone with cisplatin and doxorubicin in patients with preoperative standard-risk hepatoblastoma. Standard risk was defined as tumor confined to the liver and not involving more than three sectors.[95][Level of evidence A1]
- The resection rates and OS rates were similar for the cisplatin (95%) and cisplatin/doxorubicin (93%) groups.
- In a pilot study, SIOPEL-3HR, cisplatin alternating with carboplatin/doxorubicin was administered in a dose-intensive fashion to high-risk patients with hepatoblastoma.[59]
- In 74 patients with PRETEXT IV tumors, 22 of whom also had metastases, 31 patients had tumors that became resectable, and 26 patients underwent transplant. The 3-year OS rate of this group was 69% (± 11%).
- Of the 70 patients with metastases enrolled in the trial, the 3-year EFS rate was 56%, and the OS rate was 62%. Of patients with lung metastases, 50% were able to achieve complete remission of metastases with chemotherapy alone (without lung surgery).
- SIOPEL-4 (NCT00077389) was a multinational feasibility trial of dose-dense cisplatin/doxorubicin chemotherapy and radical surgery for a group of children with high-risk hepatoblastoma. Surgical removal of all remaining tumor lesions after chemotherapy was performed if feasible (including liver transplant and metastasectomy, if needed). Patients whose tumors were resected or whose livers were transplanted after three cycles of chemotherapy subsequently received two postoperative cycles of carboplatin and doxorubicin. Patients whose tumors remained unresectable after three cycles of chemotherapy received two cycles of very intensive carboplatin and doxorubicin before surgery. The primary tumor masses were identified as PRETEXT II (27%), III (44%), and IV (26%).[74][Level of evidence B4]
- Ninety-seven percent of patients (60 of 61) had a partial response with chemotherapy.
- Eighty-five percent of patients (53) underwent complete macroscopic resection; tumor was microscopically present in five patients, all of whom are disease-free survivors.
- Two patients died postoperatively.
- There were 37 partial hepatectomies and 16 liver transplants.
- The study had a total of 62 high-risk patients; 74% of patients (62%–84%) underwent resection.
- The 3-year disease-free survival (DFS) rate was 76% (95% CI, 65%–87%).
- The 3-year OS rate was 83% (95% CI, 73%–93%).
- Of the 16 PRETEXT IV patients, 11 were downstaged after chemotherapy—6 patients to PRETEXT III, 4 patients to PRETEXT II, and 1 patient to PRETEXT I. Twelve tumors became resectable; of these, four patients underwent a partial hepatectomy and eight patients underwent a liver transplant. For patients who presented with PRETEXT IV disease:
- The 3-year DFS rate was 73% (95% CI, 51%–96%).
- The 3-year OS rate was 80% (95% CI, 60%–100%).
- In approximately 75% of children and adolescents with initially unresectable hepatoblastoma, tumors can be rendered resectable with cisplatin-based preoperative chemotherapy, and 60% to 65% of patients will survive disease-free.[106]
In the United States, unresectable tumors have been treated with chemotherapy before resection or transplant.[53,54,67,68] On the basis of radiographic imaging, most stage III and IV hepatoblastomas are rendered resectable after two cycles of chemotherapy.[107] A combination of ifosfamide, cisplatin, and doxorubicin followed by postinduction resection has also been used in the treatment of advanced-stage disease.[108] Some centers have also used extended resection of selected POSTTEXT III and IV tumors rather than liver transplant.[78,109-112] Other options, such as TARE and TACE, have been used to shrink residual tumor mass. TARE may also facilitate surgical resection by tumor shrinkage when added to chemotherapy.[104]
The COG conducted a single-arm phase III trial (AHEP0731 [NCT00980460]) for patients with intermediate-risk hepatoblastoma. The study included 93 patients with unresectable nonmetastatic disease and 9 patients with a complete resection at diagnosis. All of the tumors had small cell undifferentiated histology. The addition of doxorubicin to standard treatment (cisplatin, fluorouracil, and vincristine) was assessed for feasibility and efficacy. In the 93 patients with initially unresectable disease, the 5-year EFS rate was 85% (95% confidence interval [CI], 79%–93%), and the OS rate was 95% (95% CI, 87%–98%).[113]
Chemotherapy followed by TACE, then high-intensity focused ultrasound, showed promising results in China for patients with PRETEXT III and IV tumors, some of which were resectable, but patients did not undergo surgical resection because of parent refusal.[114]
Treatment options for hepatoblastoma with metastases at diagnosis
The outcomes of patients with metastatic hepatoblastoma at diagnosis are poor, but long-term survival and cure are possible.[53,67,68] Survival rates at 3 to 5 years range from 20% to 79%.[57,59,74,115] To date, the best outcomes for children with metastatic hepatoblastoma resulted from treatment with dose-dense cisplatin and doxorubicin, although significant toxicity was also noted (SIOPEL-4 [NCT00077389] trial).[74][Level of evidence B4]
Treatment options for hepatoblastoma with metastases at diagnosis include the following:
- Chemotherapy followed by reassessment of surgical resectability.
- If the primary tumor and extrahepatic disease (usually pulmonary nodules) are resectable after chemotherapy, surgical resection is followed by additional chemotherapy.
- If extrahepatic metastatic disease is in complete remission after chemotherapy and/or surgical resection of lung nodule but the primary tumor remains unresectable, orthotopic liver transplant is warranted.
- If extrahepatic metastatic disease is not resectable or the patient is not a transplant candidate, additional chemotherapy, TACE, TARE, or radiation therapy may be indicated.[104]
The standard combination chemotherapy regimen in North America is four courses of cisplatin/vincristine/fluorouracil [53] or doxorubicin/cisplatin,[54,56,57] followed by attempted complete tumor resection. If the tumor is completely removed, two postoperative courses of the same chemotherapy are usually given. Study results for different chemotherapy regimens have been reported. For more information, see Table 6.
High-dose chemotherapy with stem cell rescue does not appear to be more effective than standard multiagent chemotherapy.[116]
Evidence (chemotherapy to treat metastatic disease at diagnosis followed by surgery):
- A subset of 39 patients presenting with metastases were entered on the SIOPEL-4 (NCT00077389) trial, a multinational feasibility trial of dose-dense cisplatin/doxorubicin chemotherapy and radical surgery for a group of children with high-risk hepatoblastoma. Patients whose tumors were resected or livers transplanted after three cycles of chemotherapy subsequently received two postoperative cycles of carboplatin and doxorubicin. Patients whose tumors were unresectable after three cycles of chemotherapy received two additional cycles of very intensive carboplatin and doxorubicin before surgery.[74][Level of evidence B4]
- After three cycles of chemotherapy, there was a complete response (only in the metastases) in 20 of 39 patients and a partial response in 18 of 39 patients. Nineteen of the patients who achieved a complete response were alive without disease 3 years after diagnosis.
- Of the patients who achieved a partial response, seven patients underwent metastasectomy near the time of resection or liver transplant, with an OS rate of 100%. An additional seven patients with residual small pulmonary nodules underwent resection without metastasectomy; of those, six patients did well and one patient had a recurrence.
- Two patients with initial metastases eventually experienced a recurrence.
- Liver transplant, rather than resection alone, was needed to treat 7 of the 39 patients who presented with metastases.
- For the subset of 39 patients presenting with metastases, the 3-year DFS rate was 77% (95% CI, 63%–90%), and the OS rate was 79% (95% CI, 66%–92%).
In patients with resected primary tumors, any remaining pulmonary metastases should be surgically removed, if possible.[57] Resection of pulmonary metastases may be facilitated by computed tomography needle localization or preoperative indocyanine green administration with intraoperative fluorescence localization.[117] A review of patients treated on a U.S. intergroup trial suggested that resection of metastasis may be done at the time of resection of the primary tumor.[115][Level of evidence C1]
If extrahepatic disease is in complete remission after chemotherapy, and the primary tumor remains unresectable, an orthotopic liver transplant may be performed.[54,59,74,108]
The outcome results are discrepant for patients with lung metastases at diagnosis who undergo orthotopic liver transplant after complete resolution of lung disease in response to pretransplant chemotherapy. Some studies have reported favorable outcomes for these patients,[59,74,100,108] while others have noted high rates of hepatoblastoma recurrence.[51,96,99,102] All of these studies are limited by small patient numbers. Additional studies are needed to better define outcomes for this subset of patients. Recent clinical trials have resulted in few pulmonary recurrences in children who presented with metastatic disease and underwent liver transplants.[59,60,74]
If extrahepatic disease is not resectable after chemotherapy or the patient is not a transplant candidate, alternative treatment approaches include the following:
- Other chemotherapy agents. Chemotherapy agents such as irinotecan, high-dose cisplatin/etoposide, or continuous-infusion doxorubicin have been used.[118-120]; [121][Level of evidence C1]
- Radiation therapy.[123]
Treatment of Progressive or Recurrent Hepatoblastoma
The prognosis for a patient with progressive or recurrent hepatoblastoma depends on several factors, including the following:[124]
- Site of recurrence.
- Previous treatment.
- Individual patient considerations.
Treatment options for progressive or recurrent hepatoblastoma include the following:
- Surgical resection. In patients with hepatoblastoma that is completely resected at initial diagnosis, aggressive surgical treatment of isolated pulmonary metastases that develop in the course of the disease, in conjunction with an overall strategy that includes chemotherapy, may make extended DFS possible.[115,124,125]If possible, isolated metastases are resected completely in patients whose primary tumor is controlled.[126] A retrospective study of patients in SIOPEL studies 1, 2, and 3 showed a 12% incidence of recurrence after complete remission by imaging and AFP levels. Outcome after recurrence was best if the tumor was amenable to surgery. Of patients who underwent chemotherapy and surgery, the 3-year EFS rate was 34%, and the OS rate was 43%.[124][Level of evidence C1]Enrollment in a clinical trial should be considered if all of the recurrent disease cannot be surgically removed. Phase I and phase II clinical trials may be appropriate.
- Chemotherapy. Analysis of survival after recurrence demonstrated that some patients treated with cisplatin/vincristine/fluorouracil could be salvaged with doxorubicin-containing regimens, but patients treated with doxorubicin/cisplatin could not be salvaged with vincristine/fluorouracil.[127] The addition of doxorubicin to vincristine/fluorouracil/cisplatin was clinically evaluated in the COG study AHEP0731 (NCT00980460).Combined vincristine/irinotecan and single-agent irinotecan have been used with some success.[121]; [120][Level of evidence C1]A review of COG phase I and II studies found no promising agents for relapsed hepatoblastoma.[128]
- Percutaneous ablation. Percutaneous radiofrequency ablation has been used as an alternative to surgical resection of oligometastatic hepatoblastoma.[129][Level of evidence C1] Percutaneous ablation techniques may also be considered for palliation.[130]
Treatment Options Under Clinical Evaluation for Hepatoblastoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- AHEP1531 (NCT03533582) (Cisplatin and Combination Chemotherapy in Treating Children and Young Adults with Hepatoblastoma or Liver Cancer After Surgery):This is the COG's participation in a large international trial (PHITT) of treatment of all stages of hepatoblastoma and hepatocellular carcinoma in children.
- Very low-risk hepatoblastoma is defined as either: 1) a well-differentiated fetal mass completely resected at diagnosis, and patients receive no chemotherapy; or 2) a non–well-differentiated fetal mass or incompletely resected well-differentiated fetal mass, and patients are treated with two cycles of cisplatin chemotherapy.
- Low-risk hepatoblastoma is defined as a PRETEXT I to III tumor without any positive VPEFR annotation factors (venous involvement, portal involvement, extrahepatic spread, multifocality, and tumor rupture). These patients are treated with two cycles of cisplatin chemotherapy and then undergo resection (if possible), followed by randomization between two and four more cycles of cisplatin. If the tumor is unresectable, the patient receives two more cycles of cisplatin chemotherapy, and the tumor's resectability is reassessed. If it is still unresectable, patients undergo liver transplant.
- Intermediate-risk hepatoblastoma is defined as a PRETEXT I to III primary tumor with a positive VPEFR annotation factor but without metastasis. Patients are randomly assigned to either four 14-day cycles of cisplatin or four 21-day cycles of C5VD (cisplatin, fluorouracil, vincristine, and doxorubicin). Transplant teams are consulted early as needed. Patients in both arms then undergo resection and receive two more cycles of their assigned chemotherapy.
- High-risk hepatoblastoma is defined as presence of distant metastasis, AFP less than 100, or age 8 years and older. All patients are treated with three cycles of induction chemotherapy per the SIOPEL-4 trial (dose-intensive cisplatin, doxorubicin, and carboplatin). The patients aged 8 years and older or with AFP less than 100 and those whose metastases have cleared receive three cycles of carboplatin and doxorubicin. Patients with metastases that have not cleared by end of induction are randomly assigned to receive either carboplatin/doxorubicin cycles alternating with carboplatin/etoposide for six cycles or carboplatin/doxorubicin alternating with vincristine/irinotecan for six cycles.
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified in a patient's tumor (refractory or recurrent). Children and adolescents aged 1 to 21 years are eligible for the trial.Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the NCI website and ClinicalTrials.gov website.
References
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012, Section 29. Also available online. Last accessed August 11, 2022.
- Bulterys M, Goodman MT, Smith MA, et al.: Hepatic tumors. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 91-98. Also available online. Last accessed August 11, 2022.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 6, 2022.
- Turcotte LM, Georgieff MK, Ross JA, et al.: Neonatal medical exposures and characteristics of low birth weight hepatoblastoma cases: a report from the Children's Oncology Group. Pediatr Blood Cancer 61 (11): 2018-23, 2014. [PMC free article: PMC4287257] [PubMed: 25044669]
- Tanimura M, Matsui I, Abe J, et al.: Increased risk of hepatoblastoma among immature children with a lower birth weight. Cancer Res 58 (14): 3032-5, 1998. [PubMed: 9679968]
- McLaughlin CC, Baptiste MS, Schymura MJ, et al.: Maternal and infant birth characteristics and hepatoblastoma. Am J Epidemiol 163 (9): 818-28, 2006. [PubMed: 16510543]
- Darbari A, Sabin KM, Shapiro CN, et al.: Epidemiology of primary hepatic malignancies in U.S. children. Hepatology 38 (3): 560-6, 2003. [PubMed: 12939582]
- Kamien BA, Gabbett MT: Aicardi syndrome associated with hepatoblastoma and pulmonary sequestration. Am J Med Genet A 149A (8): 1850-2, 2009. [PubMed: 19610089]
- DeBaun MR, Tucker MA: Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132 (3 Pt 1): 398-400, 1998. [PubMed: 9544889]
- Weksberg R, Shuman C, Smith AC: Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 137C (1): 12-23, 2005. [PubMed: 16010676]
- Iwama T, Mishima Y: Mortality in young first-degree relatives of patients with familial adenomatous polyposis. Cancer 73 (8): 2065-8, 1994. [PubMed: 8156512]
- Garber JE, Li FP, Kingston JE, et al.: Hepatoblastoma and familial adenomatous polyposis. J Natl Cancer Inst 80 (20): 1626-8, 1988. [PubMed: 2848134]
- Giardiello FM, Petersen GM, Brensinger JD, et al.: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 39 (96): 867-9, 1996. [PMC free article: PMC1383462] [PubMed: 9038672]
- Ito E, Sato Y, Kawauchi K, et al.: Type 1a glycogen storage disease with hepatoblastoma in siblings. Cancer 59 (10): 1776-80, 1987. [PubMed: 3030527]
- Ikeda H, Hachitanda Y, Tanimura M, et al.: Development of unfavorable hepatoblastoma in children of very low birth weight: results of a surgical and pathologic review. Cancer 82 (9): 1789-96, 1998. [PubMed: 9576303]
- Spector LG, Birch J: The epidemiology of hepatoblastoma. Pediatr Blood Cancer 59 (5): 776-9, 2012. [PubMed: 22692949]
- Buonuomo PS, Ruggiero A, Vasta I, et al.: Second case of hepatoblastoma in a young patient with Simpson-Golabi-Behmel syndrome. Pediatr Hematol Oncol 22 (7): 623-8, 2005 Oct-Nov. [PubMed: 16166055]
- Tan ZH, Lai A, Chen CK, et al.: Association of trisomy 18 with hepatoblastoma and its implications. Eur J Pediatr 173 (12): 1595-8, 2014. [PubMed: 23975412]
- Trobaugh-Lotrario AD, Venkatramani R, Feusner JH: Hepatoblastoma in children with Beckwith-Wiedemann syndrome: does it warrant different treatment? J Pediatr Hematol Oncol 36 (5): 369-73, 2014. [PubMed: 24608075]
- Hoyme HE, Seaver LH, Jones KL, et al.: Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet 79 (4): 274-8, 1998. [PubMed: 9781907]
- Clericuzio CL, Martin RA: Diagnostic criteria and tumor screening for individuals with isolated hemihyperplasia. Genet Med 11 (3): 220-2, 2009. [PMC free article: PMC3111026] [PubMed: 19367194]
- Algar EM, St Heaps L, Darmanian A, et al.: Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 67 (5): 2360-5, 2007. [PubMed: 17325026]
- Steenman M, Westerveld A, Mannens M: Genetics of Beckwith-Wiedemann syndrome-associated tumors: common genetic pathways. Genes Chromosomes Cancer 28 (1): 1-13, 2000. [PubMed: 10738297]
- Albrecht S, Hartmann W, Houshdaran F, et al.: Allelic loss but absence of mutations in the polyspecific transporter gene BWR1A on 11p15.5 in hepatoblastoma. Int J Cancer 111 (4): 627-32, 2004. [PubMed: 15239143]
- Clericuzio CL, Chen E, McNeil DE, et al.: Serum alpha-fetoprotein screening for hepatoblastoma in children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia. J Pediatr 143 (2): 270-2, 2003. [PubMed: 12970646]
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016. [PubMed: 27372391]
- Aretz S, Koch A, Uhlhaas S, et al.: Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer 47 (6): 811-8, 2006. [PubMed: 16317745]
- Hirschman BA, Pollock BH, Tomlinson GE: The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 147 (2): 263-6, 2005. [PubMed: 16126064]
- Koch A, Denkhaus D, Albrecht S, et al.: Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 59 (2): 269-73, 1999. [PubMed: 9927029]
- Kalish JM, Doros L, Helman LJ, et al.: Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin Cancer Res 23 (13): e115-e122, 2017. [PMC free article: PMC5538793] [PubMed: 28674120]
- Eichenmüller M, Trippel F, Kreuder M, et al.: The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 61 (6): 1312-20, 2014. [PubMed: 25135868]
- Jia D, Dong R, Jing Y, et al.: Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 60 (5): 1686-96, 2014. [PubMed: 24912477]
- Sumazin P, Chen Y, Treviño LR, et al.: Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 65 (1): 104-121, 2017. [PubMed: 27775819]
- Carrillo-Reixach J, Torrens L, Simon-Coma M, et al.: Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol 73 (2): 328-341, 2020. [PubMed: 32240714]
- Gröbner SN, Worst BC, Weischenfeldt J, et al.: The landscape of genomic alterations across childhood cancers. Nature 555 (7696): 321-327, 2018. [PubMed: 29489754]
- Sekiguchi M, Seki M, Kawai T, et al.: Integrated multiomics analysis of hepatoblastoma unravels its heterogeneity and provides novel druggable targets. NPJ Precis Oncol 4: 20, 2020. [PMC free article: PMC7341754] [PubMed: 32656360]
- Cairo S, Armengol C, Maibach R, et al.: A combined clinical and biological risk classification improves prediction of outcome in hepatoblastoma patients. Eur J Cancer 141: 30-39, 2020. [PubMed: 33125945]
- Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu, Cancer Genome Atlas Research Network: Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169 (7): 1327-1341.e23, 2017. [PMC free article: PMC5680778] [PubMed: 28622513]
- Albrecht S, von Schweinitz D, Waha A, et al.: Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 54 (19): 5041-4, 1994. [PubMed: 7923113]
- Cairo S, Armengol C, De Reyniès A, et al.: Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 14 (6): 471-84, 2008. [PubMed: 19061838]
- Yoon JM, Burns RC, Malogolowkin MH, et al.: Treatment of infantile choriocarcinoma of the liver. Pediatr Blood Cancer 49 (1): 99-102, 2007. [PubMed: 16206190]
- Boman F, Bossard C, Fabre M, et al.: Mesenchymal hamartomas of the liver may be associated with increased serum alpha foetoprotein concentrations and mimic hepatoblastomas. Eur J Pediatr Surg 14 (1): 63-6, 2004. [PubMed: 15024683]
- Blohm ME, Vesterling-Hörner D, Calaminus G, et al.: Alpha 1-fetoprotein (AFP) reference values in infants up to 2 years of age. Pediatr Hematol Oncol 15 (2): 135-42, 1998 Mar-Apr. [PubMed: 9592840]
- Wu JT, Book L, Sudar K: Serum alpha fetoprotein (AFP) levels in normal infants. Pediatr Res 15 (1): 50-2, 1981. [PubMed: 6163129]
- Schneider DT, Calaminus G, Göbel U: Diagnostic value of alpha 1-fetoprotein and beta-human chorionic gonadotropin in infancy and childhood. Pediatr Hematol Oncol 18 (1): 11-26, 2001 Jan-Feb. [PubMed: 11205836]
- Nakagawara A, Ikeda K, Tsuneyoshi M, et al.: Hepatoblastoma producing both alpha-fetoprotein and human chorionic gonadotropin. Clinicopathologic analysis of four cases and a review of the literature. Cancer 56 (7): 1636-42, 1985. [PubMed: 2411379]
- Perilongo G, Malogolowkin M, Feusner J: Hepatoblastoma clinical research: lessons learned and future challenges. Pediatr Blood Cancer 59 (5): 818-21, 2012. [PubMed: 22678761]
- Trobaugh-Lotrario AD, Katzenstein HM: Chemotherapeutic approaches for newly diagnosed hepatoblastoma: past, present, and future strategies. Pediatr Blood Cancer 59 (5): 809-12, 2012. [PubMed: 22648979]
- Trobaugh-Lotrario AD, Chaiyachati BH, Meyers RL, et al.: Outcomes for patients with congenital hepatoblastoma. Pediatr Blood Cancer 60 (11): 1817-25, 2013. [PubMed: 23798361]
- Czauderna P, Haeberle B, Hiyama E, et al.: The Children's Hepatic tumors International Collaboration (CHIC): Novel global rare tumor database yields new prognostic factors in hepatoblastoma and becomes a research model. Eur J Cancer 52: 92-101, 2016. [PMC free article: PMC5141607] [PubMed: 26655560]
- Otte JB, Pritchard J, Aronson DC, et al.: Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer 42 (1): 74-83, 2004. [PubMed: 14752798]
- Meyers RL, Rowland JR, Krailo M, et al.: Predictive power of pretreatment prognostic factors in children with hepatoblastoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 53 (6): 1016-22, 2009. [PMC free article: PMC4408767] [PubMed: 19588519]
- Ortega JA, Douglass EC, Feusner JH, et al.: Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children's Cancer Group and the Pediatric Oncology Group. J Clin Oncol 18 (14): 2665-75, 2000. [PubMed: 10894865]
- Malogolowkin MH, Katzenstein HM, Meyers RL, et al.: Complete surgical resection is curative for children with hepatoblastoma with pure fetal histology: a report from the Children's Oncology Group. J Clin Oncol 29 (24): 3301-6, 2011. [PMC free article: PMC3158601] [PubMed: 21768450]
- De Ioris M, Brugieres L, Zimmermann A, et al.: Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: the SIOPEL group experience. Eur J Cancer 44 (4): 545-50, 2008. [PubMed: 18166449]
- Pritchard J, Brown J, Shafford E, et al.: Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma: a successful approach--results of the first prospective study of the International Society of Pediatric Oncology. J Clin Oncol 18 (22): 3819-28, 2000. [PubMed: 11078495]
- Perilongo G, Brown J, Shafford E, et al.: Hepatoblastoma presenting with lung metastases: treatment results of the first cooperative, prospective study of the International Society of Paediatric Oncology on childhood liver tumors. Cancer 89 (8): 1845-53, 2000. [PubMed: 11042582]
- Perilongo G, Shafford E, Maibach R, et al.: Risk-adapted treatment for childhood hepatoblastoma. final report of the second study of the International Society of Paediatric Oncology--SIOPEL 2. Eur J Cancer 40 (3): 411-21, 2004. [PubMed: 14746860]
- Zsíros J, Maibach R, Shafford E, et al.: Successful treatment of childhood high-risk hepatoblastoma with dose-intensive multiagent chemotherapy and surgery: final results of the SIOPEL-3HR study. J Clin Oncol 28 (15): 2584-90, 2010. [PubMed: 20406943]
- Katzenstein HM, Furman WL, Malogolowkin MH, et al.: Upfront window vincristine/irinotecan treatment of high-risk hepatoblastoma: A report from the Children's Oncology Group AHEP0731 study committee. Cancer 123 (12): 2360-2367, 2017. [PMC free article: PMC5665173] [PubMed: 28211941]
- O'Neill AF, Towbin AJ, Krailo MD, et al.: Characterization of Pulmonary Metastases in Children With Hepatoblastoma Treated on Children's Oncology Group Protocol AHEP0731 (The Treatment of Children With All Stages of Hepatoblastoma): A Report From the Children's Oncology Group. J Clin Oncol 35 (30): 3465-3473, 2017. [PMC free article: PMC5648177] [PubMed: 28892430]
- Fuchs J, Rydzynski J, Von Schweinitz D, et al.: Pretreatment prognostic factors and treatment results in children with hepatoblastoma: a report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer 95 (1): 172-82, 2002. [PubMed: 12115331]
- Aronson DC, Schnater JM, Staalman CR, et al.: Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol 23 (6): 1245-52, 2005. [PubMed: 15718322]
- Meyers RL, Maibach R, Hiyama E, et al.: Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. Lancet Oncol 18 (1): 122-131, 2017. [PMC free article: PMC5650231] [PubMed: 27884679]
- Haeberle B, Rangaswami A, Krailo M, et al.: The importance of age as prognostic factor for the outcome of patients with hepatoblastoma: Analysis from the Children's Hepatic tumors International Collaboration (CHIC) database. Pediatr Blood Cancer 67 (8): e28350, 2020. [PubMed: 32383794]
- Dall'Igna P, Brugieres L, Christin AS, et al.: Hepatoblastoma in children aged less than six months at diagnosis: A report from the SIOPEL group. Pediatr Blood Cancer 65 (1): , 2018. [PubMed: 28921839]
- Ortega JA, Krailo MD, Haas JE, et al.: Effective treatment of unresectable or metastatic hepatoblastoma with cisplatin and continuous infusion doxorubicin chemotherapy: a report from the Childrens Cancer Study Group. J Clin Oncol 9 (12): 2167-76, 1991. [PubMed: 1720452]
- Douglass EC, Reynolds M, Finegold M, et al.: Cisplatin, vincristine, and fluorouracil therapy for hepatoblastoma: a Pediatric Oncology Group study. J Clin Oncol 11 (1): 96-9, 1993. [PubMed: 8380296]
- Meyers RL, Czauderna P, Otte JB: Surgical treatment of hepatoblastoma. Pediatr Blood Cancer 59 (5): 800-8, 2012. [PubMed: 22887704]
- Hiyama E, Hishiki T, Watanabe K, et al.: Resectability and tumor response after preoperative chemotherapy in hepatoblastoma treated by the Japanese Study Group for Pediatric Liver Tumor (JPLT)-2 protocol. J Pediatr Surg 51 (12): 2053-2057, 2016. [PubMed: 27712887]
- Becker K, Furch C, Schmid I, et al.: Impact of postoperative complications on overall survival of patients with hepatoblastoma. Pediatr Blood Cancer 62 (1): 24-8, 2015. [PubMed: 25251521]
- McAteer JP, Goldin AB, Healey PJ, et al.: Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant 17 (8): 744-50, 2013. [PubMed: 23992390]
- Schnater JM, Aronson DC, Plaschkes J, et al.: Surgical view of the treatment of patients with hepatoblastoma: results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer 94 (4): 1111-20, 2002. [PubMed: 11920482]
- Zsiros J, Brugieres L, Brock P, et al.: Dose-dense cisplatin-based chemotherapy and surgery for children with high-risk hepatoblastoma (SIOPEL-4): a prospective, single-arm, feasibility study. Lancet Oncol 14 (9): 834-42, 2013. [PMC free article: PMC3730732] [PubMed: 23831416]
- Van Tornout JM, Buckley JD, Quinn JJ, et al.: Timing and magnitude of decline in alpha-fetoprotein levels in treated children with unresectable or metastatic hepatoblastoma are predictors of outcome: a report from the Children's Cancer Group. J Clin Oncol 15 (3): 1190-7, 1997. [PubMed: 9060563]
- Nguyen R, McCarville MB, Sykes A, et al.: Rapid decrease of serum alpha-fetoprotein and tumor volume predicts outcome in children with hepatoblastoma treated with neoadjuvant chemotherapy. Int J Clin Oncol 23 (5): 900-907, 2018. [PubMed: 29744604]
- Brown J, Perilongo G, Shafford E, et al.: Pretreatment prognostic factors for children with hepatoblastoma-- results from the International Society of Paediatric Oncology (SIOP) study SIOPEL 1. Eur J Cancer 36 (11): 1418-25, 2000. [PubMed: 10899656]
- D'Antiga L, Vallortigara F, Cillo U, et al.: Features predicting unresectability in hepatoblastoma. Cancer 110 (5): 1050-8, 2007. [PubMed: 17661341]
- Czauderna P, Lopez-Terrada D, Hiyama E, et al.: Hepatoblastoma state of the art: pathology, genetics, risk stratification, and chemotherapy. Curr Opin Pediatr 26 (1): 19-28, 2014. [PubMed: 24322718]
- López-Terrada D, Alaggio R, de Dávila MT, et al.: Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 27 (3): 472-91, 2014. [PubMed: 24008558]
- Weinberg AG, Finegold MJ: Primary hepatic tumors of childhood. Hum Pathol 14 (6): 512-37, 1983. [PubMed: 6303939]
- Haas JE, Muczynski KA, Krailo M, et al.: Histopathology and prognosis in childhood hepatoblastoma and hepatocarcinoma. Cancer 64 (5): 1082-95, 1989. [PubMed: 2547506]
- Trobaugh-Lotrario AD, Tomlinson GE, Finegold MJ, et al.: Small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood Cancer 52 (3): 328-34, 2009. [PMC free article: PMC2946187] [PubMed: 18985717]
- Rowland JM: Hepatoblastoma: assessment of criteria for histologic classification. Med Pediatr Oncol 39 (5): 478-83, 2002. [PubMed: 12228903]
- Conran RM, Hitchcock CL, Waclawiw MA, et al.: Hepatoblastoma: the prognostic significance of histologic type. Pediatr Pathol 12 (2): 167-83, 1992 Mar-Apr. [PubMed: 1315024]
- Gunawan B, Schäfer KL, Sattler B, et al.: Undifferentiated small cell hepatoblastoma with a chromosomal translocation t(22;22)(q11;q13). Histopathology 40 (5): 485-7, 2002. [PubMed: 12010372]
- Zhou S, Gomulia E, Mascarenhas L, et al.: Is INI1-retained small cell undifferentiated histology in hepatoblastoma unfavorable? Hum Pathol 46 (4): 620-4, 2015. [PubMed: 25649007]
- Vokuhl C, Oyen F, Häberle B, et al.: Small cell undifferentiated (SCUD) hepatoblastomas: All malignant rhabdoid tumors? Genes Chromosomes Cancer 55 (12): 925-931, 2016. [PubMed: 27356182]
- Cornet M, De Lambert G, Pariente D, et al.: Rhabdoid tumor of the liver: Report of 6 pediatric cases treated at a single institute. J Pediatr Surg 53 (3): 567-571, 2018. [PubMed: 28966010]
- Haas JE, Feusner JH, Finegold MJ: Small cell undifferentiated histology in hepatoblastoma may be unfavorable. Cancer 92 (12): 3130-4, 2001. [PubMed: 11753992]
- Meyers RL, Tiao G, de Ville de Goyet J, et al.: Hepatoblastoma state of the art: pre-treatment extent of disease, surgical resection guidelines and the role of liver transplantation. Curr Opin Pediatr 26 (1): 29-36, 2014. [PubMed: 24362406]
- Katzenstein HM, Langham MR, Malogolowkin MH, et al.: Minimal adjuvant chemotherapy for children with hepatoblastoma resected at diagnosis (AHEP0731): a Children's Oncology Group, multicentre, phase 3 trial. Lancet Oncol 20 (5): 719-727, 2019. [PMC free article: PMC6499702] [PubMed: 30975630]
- Hiyama E, Hishiki T, Watanabe K, et al.: Outcome and Late Complications of Hepatoblastomas Treated Using the Japanese Study Group for Pediatric Liver Tumor 2 Protocol. J Clin Oncol 38 (22): 2488-2498, 2020. [PubMed: 32421442]
- Aronson DC, Weeda VB, Maibach R, et al.: Microscopically positive resection margin after hepatoblastoma resection: what is the impact on prognosis? A Childhood Liver Tumours Strategy Group (SIOPEL) report. Eur J Cancer 106: 126-132, 2019. [PubMed: 30528797]
- Perilongo G, Maibach R, Shafford E, et al.: Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. N Engl J Med 361 (17): 1662-70, 2009. [PubMed: 19846851]
- Reyes JD, Carr B, Dvorchik I, et al.: Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr 136 (6): 795-804, 2000. [PubMed: 10839879]
- Molmenti EP, Wilkinson K, Molmenti H, et al.: Treatment of unresectable hepatoblastoma with liver transplantation in the pediatric population. Am J Transplant 2 (6): 535-8, 2002. [PubMed: 12118897]
- Czauderna P, Otte JB, Aronson DC, et al.: Guidelines for surgical treatment of hepatoblastoma in the modern era--recommendations from the Childhood Liver Tumour Strategy Group of the International Society of Paediatric Oncology (SIOPEL). Eur J Cancer 41 (7): 1031-6, 2005. [PubMed: 15862752]
- Austin MT, Leys CM, Feurer ID, et al.: Liver transplantation for childhood hepatic malignancy: a review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg 41 (1): 182-6, 2006. [PubMed: 16410130]
- Pham TA, Gallo AM, Concepcion W, et al.: Effect of Liver Transplant on Long-term Disease-Free Survival in Children With Hepatoblastoma and Hepatocellular Cancer. JAMA Surg 150 (12): 1150-8, 2015. [PubMed: 26308249]
- Khan AS, Brecklin B, Vachharajani N, et al.: Liver Transplantation for Malignant Primary Pediatric Hepatic Tumors. J Am Coll Surg 225 (1): 103-113, 2017. [PubMed: 28232059]
- Xianliang H, Jianhong L, Xuewu J, et al.: Cure of hepatoblastoma with transcatheter arterial chemoembolization. J Pediatr Hematol Oncol 26 (1): 60-3, 2004. [PubMed: 14707717]
- Malogolowkin MH, Stanley P, Steele DA, et al.: Feasibility and toxicity of chemoembolization for children with liver tumors. J Clin Oncol 18 (6): 1279-84, 2000. [PubMed: 10715298]
- Aguado A, Dunn SP, Averill LW, et al.: Successful use of transarterial radioembolization with yttrium-90 (TARE-Y90) in two children with hepatoblastoma. Pediatr Blood Cancer 67 (9): e28421, 2020. [PubMed: 32603027]
- Madanur MA, Battula N, Davenport M, et al.: Staged resection for a ruptured hepatoblastoma: a 6-year follow-up. Pediatr Surg Int 23 (6): 609-11, 2007. [PubMed: 17066271]
- Aronson DC, Meyers RL: Malignant tumors of the liver in children. Semin Pediatr Surg 25 (5): 265-275, 2016. [PubMed: 27955729]
- Venkatramani R, Stein JE, Sapra A, et al.: Effect of neoadjuvant chemotherapy on resectability of stage III and IV hepatoblastoma. Br J Surg 102 (1): 108-13, 2015. [PubMed: 25349947]
- von Schweinitz D, Hecker H, Harms D, et al.: Complete resection before development of drug resistance is essential for survival from advanced hepatoblastoma--a report from the German Cooperative Pediatric Liver Tumor Study HB-89. J Pediatr Surg 30 (6): 845-52, 1995. [PubMed: 7545228]
- Fuchs J, Cavdar S, Blumenstock G, et al.: POST-TEXT III and IV Hepatoblastoma: Extended Hepatic Resection Avoids Liver Transplantation in Selected Cases. Ann Surg 266 (2): 318-323, 2017. [PubMed: 27501172]
- Hemming AW, Reed AI, Fujita S, et al.: Role for extending hepatic resection using an aggressive approach to liver surgery. J Am Coll Surg 206 (5): 870-5; discussion 875-8, 2008. [PubMed: 18471713]
- Fonseca A, Gupta A, Shaikh F, et al.: Extreme hepatic resections for the treatment of advanced hepatoblastoma: Are planned close margins an acceptable approach? Pediatr Blood Cancer 65 (2): , 2018. [PubMed: 28921939]
- de Freitas Paganoti G, Tannuri ACA, Dantas Marques AC, et al.: Extensive Hepatectomy as an Alternative to Liver Transplant in Advanced Hepatoblastoma: A New Protocol Used in a Pediatric Liver Transplantation Center. Transplant Proc 51 (5): 1605-1610, 2019. [PubMed: 31155201]
- Katzenstein HM, Malogolowkin MH, Krailo MD, et al.: Doxorubicin in combination with cisplatin, 5-flourouracil, and vincristine is feasible and effective in unresectable hepatoblastoma: A Children's Oncology Group study. Cancer 128 (5): 1057-1065, 2022. [PMC free article: PMC9066555] [PubMed: 34762296]
- Wang S, Yang C, Zhang J, et al.: First experience of high-intensity focused ultrasound combined with transcatheter arterial embolization as local control for hepatoblastoma. Hepatology 59 (1): 170-7, 2014. [PubMed: 23813416]
- Meyers RL, Katzenstein HM, Krailo M, et al.: Surgical resection of pulmonary metastatic lesions in children with hepatoblastoma. J Pediatr Surg 42 (12): 2050-6, 2007. [PubMed: 18082706]
- Karski EE, Dvorak CC, Leung W, et al.: Treatment of hepatoblastoma with high-dose chemotherapy and stem cell rescue: the pediatric blood and marrow transplant consortium experience and review of the literature. J Pediatr Hematol Oncol 36 (5): 362-8, 2014. [PubMed: 24577552]
- Partrick DA, Bensard DD, Teitelbaum DH, et al.: Successful thoracoscopic lung biopsy in children utilizing preoperative CT-guided localization. J Pediatr Surg 37 (7): 970-3; discussion 970-3, 2002. [PubMed: 12077751]
- Katzenstein HM, Rigsby C, Shaw PH, et al.: Novel therapeutic approaches in the treatment of children with hepatoblastoma. J Pediatr Hematol Oncol 24 (9): 751-5, 2002. [PubMed: 12468918]
- Palmer RD, Williams DM: Dramatic response of multiply relapsed hepatoblastoma to irinotecan (CPT-11). Med Pediatr Oncol 41 (1): 78-80, 2003. [PubMed: 12764754]
- Qayed M, Powell C, Morgan ER, et al.: Irinotecan as maintenance therapy in high-risk hepatoblastoma. Pediatr Blood Cancer 54 (5): 761-3, 2010. [PubMed: 20063426]
- Zsíros J, Brugières L, Brock P, et al.: Efficacy of irinotecan single drug treatment in children with refractory or recurrent hepatoblastoma--a phase II trial of the childhood liver tumour strategy group (SIOPEL). Eur J Cancer 48 (18): 3456-64, 2012. [PubMed: 22835780]
- Sue K, Ikeda K, Nakagawara A, et al.: Intrahepatic arterial injections of cisplatin-phosphatidylcholine-Lipiodol suspension in two unresectable hepatoblastoma cases. Med Pediatr Oncol 17 (6): 496-500, 1989. [PubMed: 2555659]
- Habrand JL, Nehme D, Kalifa C, et al.: Is there a place for radiation therapy in the management of hepatoblastomas and hepatocellular carcinomas in children? Int J Radiat Oncol Biol Phys 23 (3): 525-31, 1992. [PubMed: 1319426]
- Semeraro M, Branchereau S, Maibach R, et al.: Relapses in hepatoblastoma patients: clinical characteristics and outcome--experience of the International Childhood Liver Tumour Strategy Group (SIOPEL). Eur J Cancer 49 (4): 915-22, 2013. [PubMed: 23146961]
- Shi Y, Geller JI, Ma IT, et al.: Relapsed hepatoblastoma confined to the lung is effectively treated with pulmonary metastasectomy. J Pediatr Surg 51 (4): 525-9, 2016. [PubMed: 26607968]
- Matsunaga T, Sasaki F, Ohira M, et al.: Analysis of treatment outcome for children with recurrent or metastatic hepatoblastoma. Pediatr Surg Int 19 (3): 142-6, 2003. [PubMed: 12768314]
- Malogolowkin MH, Katzenstein HM, Krailo M, et al.: Redefining the role of doxorubicin for the treatment of children with hepatoblastoma. J Clin Oncol 26 (14): 2379-83, 2008. [PMC free article: PMC4558624] [PubMed: 18467729]
- Trobaugh-Lotrario AD, Meyers RL, Feusner JH: Outcomes of Patients With Relapsed Hepatoblastoma Enrolled on Children's Oncology Group (COG) Phase I and II Studies. J Pediatr Hematol Oncol 38 (3): 187-90, 2016. [PubMed: 26583620]
- Yevich S, Calandri M, Gravel G, et al.: Reiterative Radiofrequency Ablation in the Management of Pediatric Patients with Hepatoblastoma Metastases to the Lung, Liver, or Bone. Cardiovasc Intervent Radiol 42 (1): 41-47, 2019. [PubMed: 30350173]
- Liu B, Zhou L, Huang G, et al.: First Experience of Ultrasound-guided Percutaneous Ablation for Recurrent Hepatoblastoma after Liver Resection in Children. Sci Rep 5: 16805, 2015. [PMC free article: PMC4649467] [PubMed: 26578035]
Hepatocellular Carcinoma
Incidence
The annual incidence of hepatocellular carcinoma in the United States is 0.4 cases per 1 million children between the ages of 0 and 14 years and 1.4 cases per 1 million adolescents aged 15 to 19 years.[1] Although the incidence of hepatocellular carcinoma in adults in the United States has steadily increased since the 1970s, possibly because of the increased frequency of chronic hepatitis C infection,[2] the incidence in children has not increased. In several Asian countries, the incidence of hepatocellular carcinoma in children is 10 times higher than in North America. The high incidence appears to be related to the incidence of perinatally acquired hepatitis B, which can be prevented in most cases by vaccination and administration of hepatitis B immune globulin to the newborn child.[3]
Fibrolamellar hepatocellular carcinoma, a subtype of hepatocellular carcinoma that is unrelated to cirrhosis, hepatitis B virus (HBV), or hepatitis C virus (HCV) infection, generally occurs in adolescents and young adults, but has been reported in infants.[4]
Risk Factors
Conditions associated with hepatocellular carcinoma are described in Table 7.
Table 7. Conditions Associated With Hepatocellular Carcinoma
| Associated Disorder | Clinical Findings |
|---|---|
| Alagille syndrome [5] | Broad prominent forehead, deep-set eyes, and small prominent chin. Abnormality of bile ducts leads to intrahepatic scarring. For more information, see the Alagille syndrome section. |
| Glycogen storage diseases I–IV [6] | Symptoms vary by individual disorder. |
| Hepatitis B and C [7-9] | For more information, see the Hepatitis B and hepatitis C infection section. |
| Progressive familial intrahepatic cholestasis [10,11] | Symptoms of jaundice, pruritus, and failure to thrive begin in infancy and progress to portal hypertension and liver failure. |
| Tyrosinemia [12] | First few months of life: failure to thrive, vomiting, jaundice. |
Alagille syndrome
Alagille syndrome is an autosomal dominant genetic syndrome that is usually caused by a mutation in or deletion of the JAG1 gene. It involves the bile ducts of the liver, as well as the heart and blood vessels in the brain and kidney. Patients develop a characteristic facies.[5]
Hepatitis B and hepatitis C infection
In children, hepatocellular carcinoma is associated with perinatally acquired HBV. In adults, it is associated with chronic HBV and HCV infection.[7-9] Widespread hepatitis B immunization has decreased the incidence of hepatocellular carcinoma in Asia.[3] Compared with adults, the incubation period from hepatitis virus infection to the genesis of hepatocellular carcinoma is extremely short in a small subset of children with perinatally acquired virus. Mutations in the MET gene could be one mechanism that results in a shortened incubation period.[13]
Hepatitis C infection is associated with development of cirrhosis and hepatocellular carcinoma that takes decades to develop and is generally not seen in children.[9] Unlike in adults, cirrhosis in children is much less commonly involved in the development of hepatocellular carcinoma and is found in only 20% to 35% of children with hepatocellular carcinoma tumors.
Nonviral liver injury
Specific types of nonviral liver injury and cirrhosis that are associated with hepatocellular carcinoma in children include the following:
- Tyrosinemia. Patients with tyrosinemia are regularly screened for hepatocellular carcinoma, even if they are treated with nitisinone.[12] Nitisinone can prevent cirrhosis and decrease the incidence of hepatocellular carcinoma, especially when administered during infancy, after neonatal screening is used to diagnose tyrosinemia.[14] As of 2014, only a minority of state screening programs had adopted a highly recommended, new, more predictive newborn screen that is much more effective in newborn children aged 24 to 48 hours.[15]In an Iranian study, 36 children underwent liver transplant for tyrosinemia.[16] Twenty-two children had liver nodules greater than 10 cm, and in 20 children, the nodules were cirrhotic. Median age at transplant was 3.9 years. Five of 19 children older than 2 years had hepatocellular carcinoma, and no children younger than 2 years had hepatocellular carcinoma in the resected liver.
- Aggressive familial intrahepatic cholestasis. Hepatocellular carcinoma may also arise in very young children with mutations in the ABCB11 gene (encodes bile salt export pump protein), which causes progressive familial intrahepatic cholestasis.[10]
Genomics of Hepatocellular Carcinoma
Molecular features of hepatocellular carcinoma
Genomic findings related to hepatocellular carcinoma include the following:
- One case of pediatric hepatocellular carcinoma was analyzed by whole-exome sequencing, which showed a higher mutation rate (53 variants) and the coexistence of CTNNB1 and NFE2L2 mutations.[17]
- One study investigated pediatric nonfibrolamellar hepatocellular carcinoma tumors (N = 15) using multiple analytic tools. These tumors were found to frequently carry alterations in a subset of genes that are commonly mutated in adult hepatocellular carcinoma, including CTNNB1 and TERT, but the molecular mechanisms of the mutations are different. The TP53 mutation was rare in this pediatric hepatocellular carcinoma cohort. Pediatric hepatocellular carcinoma that arose in the background of underlying metabolic disease had fewer mutations and a distinct molecular profile. Typical driver mutations were lacking in this group of patients.[18]
- Fibrolamellar hepatocellular carcinoma is a rare subtype of hepatocellular carcinoma observed in older children and young adults. It is characterized by an approximately 400 kB deletion on chromosome 19 that results in production of a chimeric RNA coding for a protein containing the amino-terminal domain of DNAJB1, a homolog of the molecular chaperone DNAJ, fused in frame with PRKACA, the catalytic domain of protein kinase A.[19]
- A rare, more aggressive subtype of childhood liver cancer (hepatocellular neoplasm, not otherwise specified, also termed transitional liver cell tumor) occurs in older children. It has clinical and histopathological findings of both hepatoblastoma and hepatocellular carcinoma.
To date, these genetic mutations have not been used to select therapeutic agents for investigation in clinical trials.
Diagnosis
For more information, see the Diagnosis section in the Hepatoblastoma section.
Prognosis and Prognostic Factors
Prognosis
The 5-year overall survival (OS) rate is 42% for children and adolescents with hepatocellular carcinoma.[22] The 5-year survival for patients with hepatocellular carcinoma may depend on the stage of the disease. In an intergroup chemotherapy study conducted in the 1990s, seven of eight stage I patients survived, and less than 10% of stage III and IV patients survived.[22,23] An analysis of Surveillance, Epidemiology, and End Results (SEER) Program data found a 5-year OS rate of 24%, a 10-year rate of 23%, and a 20-year rate of 8% in patients aged 19 years and younger, suggesting improved outcome related to more recent treatment. In a multivariate analysis of the SEER data, surgical resection, localized tumor, and non-Hispanic ethnicity were all associated with improved outcome. Patients who had a complete surgical resection had an OS rate of 60%, compared with an OS rate of 0% for patients who had an incomplete resection.[24][Level of evidence C1]
The 5-year OS rates by PRE-Treatment EXTent of disease (PRETEXT) group for patients with hepatocellular carcinoma in the SIOPEL-1 trial were found to be the following:[25]
- 44% for patients with PRETEXT I group tumors.
- 44% for patients with PRETEXT II group tumors.
- 22% for patients with PRETEXT III group tumors.
- 8% for patients with PRETEXT IV group tumors.
For more information about PRETEXT grouping, see the PRETEXT and POSTTEXT Group Definitions section.
Hepatocellular carcinoma prognosis by Evans surgical stage. For more information, see the Evans Surgical Staging for Childhood Liver Cancer (Historical) section.
- Stage I.Children with stage I hepatocellular carcinoma have a good outcome.[26]
- Stage II.Stage II is too rarely seen to predict outcome.
- Stages III and IV.
Prognostic factors
Factors affecting prognosis include the following:
- Treatment-related factors: Cure of hepatocellular carcinoma requires gross tumor resection. However, hepatocellular carcinoma is often extensively invasive or multicentric, and less than 30% of tumors are resectable. Orthotopic liver transplant has been successful in selected children with hepatocellular carcinoma.[27,28]
- PRETEXT group: PRETEXT group (resectability) is also a prognostic factor. For more information, see the Tumor Stratification by Imaging and Evans Surgical Staging for Childhood Liver Cancer section.
- Tumor histology: For more information, see the Histology section.
Histology
The cells of hepatocellular carcinoma are epithelial in appearance. Hepatocellular carcinoma commonly arises in the right lobe of the liver.
Fibrolamellar carcinoma
A distinctive histological variant of hepatocellular carcinoma, termed fibrolamellar carcinoma, has been described in the livers of older children, young adults, and, rarely, infants.[4,29] This histology is characterized by a fusion transcript created by deletion of a 400 kb section of chromosome 19, which was found in 15 of 15 tumors that were tested.[19]
Unlike nonfibrolamellar hepatocellular carcinoma in adults, fibrolamellar hepatocellular carcinoma in older children and adults is not clearly increasing in incidence over time.[2,29] Fibrolamellar carcinoma is not associated with cirrhosis and was previously thought to be associated with an improved prognosis.[2,29,30] The improved outcomes of patients with fibrolamellar carcinoma in older studies may be related to a higher proportion of tumors being less invasive and more resectable in the absence of cirrhosis. However, the outcomes of patients with fibrolamellar carcinoma in recent prospective studies, when compared stage to stage and PRETEXT group to PRETEXT group, are the same as the outcomes of patients with conventional hepatocellular carcinomas.[25,31]; [32][Level of evidence C1]
Hepatocellular neoplasm, not otherwise specified (NOS)
Hepatocellular neoplasm, NOS, is also known as transitional liver cell tumor. This tumor, with characteristics of both hepatoblastoma and hepatocellular carcinoma, is a rare neoplasm found in older children and adolescents. It has a putative intermediate position between hepatoblasts and more mature hepatocyte-like tumor cells. The tumor cells may vary in regions of the tumor between classical hepatoblastoma and obvious hepatocellular carcinoma. In the international consensus classification, these tumors are referred to as hepatocellular neoplasm, NOS.[33] The tumors are usually unifocal and may have central necrosis at presentation. Response to chemotherapy has not been rigorously studied, but it is thought to be similar to that of hepatocellular carcinoma.[34]
Treatment of Hepatocellular Carcinoma
Treatment options for newly diagnosed hepatocellular carcinoma depend on the following:
- Whether the cancer is resectable at diagnosis.
- How the cancer responds to chemotherapy.
- Whether the cancer has metastasized.
- Whether the cancer is HBV related.
Treatment options for hepatocellular carcinoma that is resectable at diagnosis
Treatment options for hepatocellular carcinoma that is resectable at diagnosis include the following:
- Complete surgical resection of the primary tumor followed by chemotherapy.
- Chemotherapy followed by complete surgical resection of the primary tumor.[25]
- Complete surgical resection without chemotherapy.
Surgical resection and chemotherapy are the mainstays of treatment for resectable hepatocellular carcinoma.
Evidence (complete surgical resection followed by chemotherapy):
- Seven of eight patients with stage I hepatocellular carcinoma who received adjuvant cisplatin-based chemotherapy survived disease free.[23]
- In a survey of childhood liver tumors treated before the consistent use of chemotherapy, only 12 of 33 patients with hepatocellular carcinoma who had complete excision of the tumor survived.[35] This suggests that treatment with adjuvant chemotherapy may benefit children with completely resected hepatocellular carcinoma.
- In an analysis of SEER data for children and adolescents younger than 20 years who were diagnosed between 1976 and 2009, patients who underwent a complete resection had a 5-year OS rate of 60%, and patients who did not have a complete resection had a 5-year OS rate of 0%.[24][Level of evidence C1]
Cisplatin and doxorubicin may be administered as adjuvant therapy because these agents may have activity in the treatment of hepatocellular carcinoma.[25]
Evidence (complete surgical resection without chemotherapy):
- In a single-institution retrospective report, 12 patients with stage I hepatocellular carcinoma were treated with surgery. Ten patients received no chemotherapy and two patients received a short course of chemotherapy based on oncologist preference.[36][Level of evidence C1]
- All 12 patients were alive without evidence of disease at a median of 54 months.
Despite improvements in surgical techniques, chemotherapy delivery, and patient supportive care in the past 20 years, clinical trials of chemotherapy have not shown improved survival rates for pediatric patients with hepatocellular carcinoma.[25] The International Childhood Liver Tumors Strategy Group (SIOPEL) studies in Europe have observed no improvement in 5-year OS since 1990. The only long-term survivors were patients whose tumors were resectable at diagnosis, which was less than 30% of children entered in the study.[37] However, some liver transplant studies (complete resection with transplant with or without neoadjuvant chemotherapy) have shown OS rates that are superior to the SIOPEL studies.[28,38-41]
Treatment options for nonmetastatic hepatocellular carcinoma that is not resectable at diagnosis
Treatment options for nonmetastatic hepatocellular carcinoma that is not resectable at diagnosis include the following:
- Chemotherapy followed by reassessment of surgical resectability. If the primary tumor is resectable, complete surgical resection.
- Chemotherapy with or without transarterial radioembolization (TARE) followed by reassessment of surgical resectability. If the primary tumor remains unresectable:
- Orthotopic liver transplant.
- Temporizing transarterial chemoembolization (TACE) or TARE followed by complete resection or liver transplant.
- TACE or TARE alone.
The use of neoadjuvant chemotherapy or TACE to enhance resectability or liver transplant, which may result in complete resection of tumor, is necessary for a cure.
Evidence (chemotherapy followed by surgery):
- In a prospective study of 41 patients who received preoperative cisplatin/doxorubicin chemotherapy, the following was observed:[25]
- Treatment resulted in a decrease in tumor size, with a decrease in alpha-fetoprotein (AFP) levels in about 50% of patients.
- The responders had a superior tumor resectability and survival, although the OS rate was 28% and only those undergoing complete resection survived.
Evidence (chemotherapy, TARE, or TACE followed by reassessment of surgical resectability; treatment options, including liver transplant, for unresectable primary tumor after chemotherapy, TARE, or TACE):
- A review of SEER data for hepatocellular carcinoma treatment in patients younger than 20 years revealed that 75% of patients underwent resection and 25% underwent liver transplant.[45]
- The 5-year OS rate was 53.4% with resection and 85.3% with transplant, suggesting that the criteria for transplantation in hepatocellular carcinoma might be liberalized for overall patient benefit. This data has not been verified in a prospective clinical trial.
- TACE followed by complete surgical resection of the primary tumor may be an alternative to the use of chemotherapy followed by surgical resection.
- Studies in adults in China suggest that repeated hepatic TACE before surgery may improve the outcome of subsequent hepatectomy.[46]
- A meta-analysis found seven randomized trials that compared resection alone with TACE followed by resection. There was no difference in the 3-year event-free survival (EFS) and OS between the two groups, but the 5-year EFS and OS favored TACE followed by resection.[47]
If the primary tumor is not resectable after chemotherapy and the patient is not a transplant candidate, alternative treatment approaches used in adults include the following:
- Sorafenib.
- TACE or TARE.
- Cryosurgery.
- Intratumoral injection of alcohol.
- Radiation therapy.
There are limited data on the use of these alternative treatment approaches in children.
Limited data from a European pilot study suggest that sorafenib was well tolerated in 12 children and adolescents with newly diagnosed advanced hepatocellular carcinoma when given in combination with standard chemotherapy of cisplatin and doxorubicin.[50] Additional study is needed to define its role in the treatment of children with hepatocellular carcinoma.
Cryosurgery, intratumoral injection of alcohol, and radiofrequency ablation can successfully treat small (<5 cm) tumors in adults with cirrhotic livers.[46,51,52] Some local approaches such as cryosurgery, radiofrequency ablation, and TACE, which suppress hepatocellular carcinoma tumor progression, are used as bridging therapy in adults to delay tumor growth while on a waiting list for cadaveric liver transplant.[53] In a pediatric study of eight patients with hepatocellular carcinoma, two patients died of progressive disease without transplant. Treatment with TACE stabilized disease in six patients, for a mean of 141 days to reach transplant.[54][Level of evidence C1] Five patients were alive at the end of the observation period, and one patient died of disease. For more information, see Adult Primary Liver Cancer Treatment.
Treatment options for hepatocellular carcinoma with metastases at diagnosis
No specific treatment has proven effective for metastatic hepatocellular carcinoma in the pediatric age group.
In two prospective trials, cisplatin plus either vincristine/fluorouracil or continuous-infusion doxorubicin was ineffective in adequately treating 25 patients with metastatic hepatocellular carcinoma.[23,25] Occasional patients may transiently benefit from treatment with cisplatin/doxorubicin therapy, especially if the localized hepatic tumor shrinks adequately enough to allow resection of disease and the metastatic disease disappears or becomes resectable.
Treatment options for HBV-related hepatocellular carcinoma
Although HBV-related hepatocellular carcinoma is not common in children in the United States, nucleotide/nucleoside analog HBV inhibitor treatment improves postoperative prognosis in children and adults treated in China.[55]
Treatment options for HBV-related hepatocellular carcinoma include the following:
- Antiviral therapy.
Evidence (antiviral therapy):
- In a randomized controlled trial, 163 patients post–radical hepatectomy were evaluated for response to one of three antiviral treatments.[55]
- Antiviral treatment significantly decreased hepatocellular carcinoma recurrence, with a hazard ratio (HR) of 0.48 (95% confidence interval [CI], 0.32–0.70), and hepatocellular carcinoma–related death, with an HR of 0.26 (95% CI, 0.14–0.50), in multivariate Cox analyses.
- Patients who received antiviral treatment had significantly decreased early recurrence (HR, 0.41; 95% CI, 0.27–0.62) and improved liver function 6 months after surgery than did the control patients (P < .001).
Treatment of Progressive or Recurrent Hepatocellular Carcinoma
The prognosis for a patient with recurrent or progressive hepatocellular carcinoma is extremely poor.[56]
Treatment options for progressive or recurrent hepatocellular carcinoma include the following:
- Phase I and phase II clinical trials may be appropriate and should be considered.
Treatment with sorafenib has resulted in improved progression-free survival in adults with advanced hepatocellular carcinoma. For adult patients who received sorafenib, the median survival and time to radiologic progression were about 3 months longer than for patients who received a placebo.[58]
Treatment Options Under Clinical Evaluation for Hepatocellular Carcinoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- AHEP1531 (NCT03533582) (Cisplatin and Combination Chemotherapy in Treating Children and Young Adults with Hepatoblastoma or Liver Cancer After Surgery):This is the COG's participation in a large international trial (Pediatric Hepatic Malignancy International Therapeutic Trial [PHITT]) of treatment of all stages of hepatoblastoma and hepatocellular carcinoma in children.
- Hepatocellular carcinoma that is potentially resectable is treated with complete resection without any chemotherapy if the mass appears to derive from underlying liver disease. If the hepatocellular carcinoma appears to be de novo without underlying disease, it is treated with resection followed by four cycles of cisplatin/doxorubicin.
- Patients with hepatocellular carcinoma that is metastatic or appears unresectable at diagnosis undergo consultation with interventional radiology and liver transplant services. Patients are then randomly assigned to receive either cisplatin/doxorubicin/sorafenib for three 21-day cycles or cisplatin/doxorubicin/sorafenib alternating with gemcitabine/oxaliplatin/sorafenib for four 14-day cycles. Responding patients continue chemotherapy for the same number of cycles (three or four). They receive the same chemotherapy regimen to which they were originally assigned.
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified in a patient's tumor (refractory or recurrent). Children and adolescents aged 1 to 21 years are eligible for the trial.Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the NCI website and ClinicalTrials.gov website.
References
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 6, 2022.
- El-Serag HB, Davila JA, Petersen NJ, et al.: The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med 139 (10): 817-23, 2003. [PubMed: 14623619]
- Chang MH, Chen TH, Hsu HM, et al.: Prevention of hepatocellular carcinoma by universal vaccination against hepatitis B virus: the effect and problems. Clin Cancer Res 11 (21): 7953-7, 2005. [PubMed: 16278421]
- Cruz O, Laguna A, Vancells M, et al.: Fibrolamellar hepatocellular carcinoma in an infant and literature review. J Pediatr Hematol Oncol 30 (12): 968-71, 2008. [PubMed: 19131794]
- Keeffe EB, Pinson CW, Ragsdale J, et al.: Hepatocellular carcinoma in arteriohepatic dysplasia. Am J Gastroenterol 88 (9): 1446-9, 1993. [PubMed: 8395765]
- Siciliano M, De Candia E, Ballarin S, et al.: Hepatocellular carcinoma complicating liver cirrhosis in type IIIa glycogen storage disease. J Clin Gastroenterol 31 (1): 80-2, 2000. [PubMed: 10914784]
- Ni YH, Chang MH, Hsu HY, et al.: Hepatocellular carcinoma in childhood. Clinical manifestations and prognosis. Cancer 68 (8): 1737-41, 1991. [PubMed: 1655224]
- Tsukuma H, Hiyama T, Tanaka S, et al.: Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 328 (25): 1797-801, 1993. [PubMed: 7684822]
- González-Peralta RP, Langham MR, Andres JM, et al.: Hepatocellular carcinoma in 2 young adolescents with chronic hepatitis C. J Pediatr Gastroenterol Nutr 48 (5): 630-5, 2009. [PubMed: 19412012]
- Knisely AS, Strautnieks SS, Meier Y, et al.: Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 44 (2): 478-86, 2006. [PubMed: 16871584]
- Alonso EM, Snover DC, Montag A, et al.: Histologic pathology of the liver in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr 18 (2): 128-33, 1994. [PubMed: 8014759]
- van Spronsen FJ, Bijleveld CM, van Maldegem BT, et al.: Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro- methylbenzoyl)-1, 3-cyclohexanedione treatment. J Pediatr Gastroenterol Nutr 40 (1): 90-3, 2005. [PubMed: 15625434]
- Park WS, Dong SM, Kim SY, et al.: Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res 59 (2): 307-10, 1999. [PubMed: 9927037]
- de Laet C, Dionisi-Vici C, Leonard JV, et al.: Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 8: 8, 2013. [PMC free article: PMC3558375] [PubMed: 23311542]
- De Jesús VR, Adam BW, Mandel D, et al.: Succinylacetone as primary marker to detect tyrosinemia type I in newborns and its measurement by newborn screening programs. Mol Genet Metab 113 (1-2): 67-75, 2014 Sep-Oct. [PMC free article: PMC4533100] [PubMed: 25066104]
- Bahador A, Dehghani SM, Geramizadeh B, et al.: Liver Transplant for Children With Hepatocellular Carcinoma and Hereditary Tyrosinemia Type 1. Exp Clin Transplant 13 (4): 329-32, 2015. [PubMed: 24679101]
- Vilarinho S, Erson-Omay EZ, Harmanci AS, et al.: Paediatric hepatocellular carcinoma due to somatic CTNNB1 and NFE2L2 mutations in the setting of inherited bi-allelic ABCB11 mutations. J Hepatol 61 (5): 1178-83, 2014. [PubMed: 25016225]
- Haines K, Sarabia SF, Alvarez KR, et al.: Characterization of pediatric hepatocellular carcinoma reveals genomic heterogeneity and diverse signaling pathway activation. Pediatr Blood Cancer 66 (7): e27745, 2019. [PubMed: 30977242]
- Honeyman JN, Simon EP, Robine N, et al.: Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343 (6174): 1010-4, 2014. [PMC free article: PMC4286414] [PubMed: 24578576]
- Eichenmüller M, Trippel F, Kreuder M, et al.: The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 61 (6): 1312-20, 2014. [PubMed: 25135868]
- Nault JC, Mallet M, Pilati C, et al.: High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 4: 2218, 2013. [PMC free article: PMC3731665] [PubMed: 23887712]
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012, Section 29. Also available online. Last accessed August 11, 2022.
- Katzenstein HM, Krailo MD, Malogolowkin MH, et al.: Hepatocellular carcinoma in children and adolescents: results from the Pediatric Oncology Group and the Children's Cancer Group intergroup study. J Clin Oncol 20 (12): 2789-97, 2002. [PubMed: 12065555]
- Allan BJ, Wang B, Davis JS, et al.: A review of 218 pediatric cases of hepatocellular carcinoma. J Pediatr Surg 49 (1): 166-71; discussion 171, 2014. [PubMed: 24439603]
- Czauderna P, Mackinlay G, Perilongo G, et al.: Hepatocellular carcinoma in children: results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol 20 (12): 2798-804, 2002. [PubMed: 12065556]
- Douglass E, Ortega J, Feusner J, et al.: Hepatocellular carcinoma (HCA) in children and adolescents: results from the Pediatric Intergroup Hepatoma Study (CCG 8881/POG 8945). [Abstract] Proceedings of the American Society of Clinical Oncology 13: A-1439, 420, 1994.
- Austin MT, Leys CM, Feurer ID, et al.: Liver transplantation for childhood hepatic malignancy: a review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg 41 (1): 182-6, 2006. [PubMed: 16410130]
- Vinayak R, Cruz RJ, Ranganathan S, et al.: Pediatric liver transplantation for hepatocellular cancer and rare liver malignancies: US multicenter and single-center experience (1981-2015). Liver Transpl 23 (12): 1577-1588, 2017. [PMC free article: PMC5725660] [PubMed: 28834194]
- Eggert T, McGlynn KA, Duffy A, et al.: Fibrolamellar hepatocellular carcinoma in the USA, 2000-2010: A detailed report on frequency, treatment and outcome based on the Surveillance, Epidemiology, and End Results database. United European Gastroenterol J 1 (5): 351-7, 2013. [PMC free article: PMC4040774] [PubMed: 24917983]
- Mayo SC, Mavros MN, Nathan H, et al.: Treatment and prognosis of patients with fibrolamellar hepatocellular carcinoma: a national perspective. J Am Coll Surg 218 (2): 196-205, 2014. [PMC free article: PMC4596238] [PubMed: 24315886]
- Katzenstein HM, Krailo MD, Malogolowkin MH, et al.: Fibrolamellar hepatocellular carcinoma in children and adolescents. Cancer 97 (8): 2006-12, 2003. [PubMed: 12673731]
- Weeda VB, Murawski M, McCabe AJ, et al.: Fibrolamellar variant of hepatocellular carcinoma does not have a better survival than conventional hepatocellular carcinoma--results and treatment recommendations from the Childhood Liver Tumour Strategy Group (SIOPEL) experience. Eur J Cancer 49 (12): 2698-704, 2013. [PubMed: 23683550]
- López-Terrada D, Alaggio R, de Dávila MT, et al.: Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 27 (3): 472-91, 2014. [PubMed: 24008558]
- Prokurat A, Kluge P, Kościesza A, et al.: Transitional liver cell tumors (TLCT) in older children and adolescents: a novel group of aggressive hepatic tumors expressing beta-catenin. Med Pediatr Oncol 39 (5): 510-8, 2002. [PubMed: 12228909]
- Exelby PR, Filler RM, Grosfeld JL: Liver tumors in children in the particular reference to hepatoblastoma and hepatocellular carcinoma: American Academy of Pediatrics Surgical Section Survey--1974. J Pediatr Surg 10 (3): 329-37, 1975. [PubMed: 49416]
- D'Souza AM, Shah R, Gupta A, et al.: Surgical management of children and adolescents with upfront completely resected hepatocellular carcinoma. Pediatr Blood Cancer 65 (11): e27293, 2018. [PubMed: 29968976]
- Murawski M, Weeda VB, Maibach R, et al.: Hepatocellular Carcinoma in Children: Does Modified Platinum- and Doxorubicin-Based Chemotherapy Increase Tumor Resectability and Change Outcome? Lessons Learned From the SIOPEL 2 and 3 Studies. J Clin Oncol 34 (10): 1050-6, 2016. [PubMed: 26811523]
- Kelly D, Sharif K, Brown RM, et al.: Hepatocellular carcinoma in children. Clin Liver Dis 19 (2): 433-47, 2015. [PubMed: 25921672]
- Malek MM, Shah SR, Atri P, et al.: Review of outcomes of primary liver cancers in children: our institutional experience with resection and transplantation. Surgery 148 (4): 778-82; discussion 782-4, 2010. [PubMed: 20728194]
- Ismail H, Broniszczak D, Kaliciński P, et al.: Liver transplantation in children with hepatocellular carcinoma. Do Milan criteria apply to pediatric patients? Pediatr Transplant 13 (6): 682-92, 2009. [PubMed: 19496985]
- Pham TA, Gallo AM, Concepcion W, et al.: Effect of Liver Transplant on Long-term Disease-Free Survival in Children With Hepatoblastoma and Hepatocellular Cancer. JAMA Surg 150 (12): 1150-8, 2015. [PubMed: 26308249]
- Reyes JD, Carr B, Dvorchik I, et al.: Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr 136 (6): 795-804, 2000. [PubMed: 10839879]
- Bilik R, Superina R: Transplantation for unresectable liver tumors in children. Transplant Proc 29 (7): 2834-5, 1997. [PubMed: 9365581]
- Romano F, Stroppa P, Bravi M, et al.: Favorable outcome of primary liver transplantation in children with cirrhosis and hepatocellular carcinoma. Pediatr Transplant 15 (6): 573-9, 2011. [PubMed: 21797955]
- McAteer JP, Goldin AB, Healey PJ, et al.: Surgical treatment of primary liver tumors in children: outcomes analysis of resection and transplantation in the SEER database. Pediatr Transplant 17 (8): 744-50, 2013. [PubMed: 23992390]
- Zhang Z, Liu Q, He J, et al.: The effect of preoperative transcatheter hepatic arterial chemoembolization on disease-free survival after hepatectomy for hepatocellular carcinoma. Cancer 89 (12): 2606-12, 2000. [PubMed: 11135222]
- Yu T, Xu X, Chen B: TACE combined with liver resection versus liver resection alone in the treatment of resectable HCC: a meta-analysis. Chinese-German J Clin Oncol 12 (11): 532-6, 2013.
- Tohme S, Bou Samra P, Kaltenmeier C, et al.: Radioembolization for Hepatocellular Carcinoma: A Nationwide 10-Year Experience. J Vasc Interv Radiol 29 (7): 912-919.e2, 2018. [PubMed: 29843996]
- Aguado A, Ristagno R, Towbin AJ, et al.: Transarterial radioembolization with yttrium-90 of unresectable primary hepatic malignancy in children. Pediatr Blood Cancer 66 (7): e27510, 2019. [PubMed: 30406959]
- Schmid I, Häberle B, Albert MH, et al.: Sorafenib and cisplatin/doxorubicin (PLADO) in pediatric hepatocellular carcinoma. Pediatr Blood Cancer 58 (4): 539-44, 2012. [PubMed: 21922643]
- Zhou XD, Tang ZY: Cryotherapy for primary liver cancer. Semin Surg Oncol 14 (2): 171-4, 1998. [PubMed: 9492887]
- Lencioni RA, Allgaier HP, Cioni D, et al.: Small hepatocellular carcinoma in cirrhosis: randomized comparison of radio-frequency thermal ablation versus percutaneous ethanol injection. Radiology 228 (1): 235-40, 2003. [PubMed: 12759473]
- Lubienski A: Hepatocellular carcinoma: interventional bridging to liver transplantation. Transplantation 80 (1 Suppl): S113-9, 2005. [PubMed: 16286887]
- Weiss KE, Sze DY, Rangaswami AA, et al.: Transarterial chemoembolization in children to treat unresectable hepatocellular carcinoma. Pediatr Transplant 22 (4): e13187, 2018. [PubMed: 29707868]
- Yin J, Li N, Han Y, et al.: Effect of antiviral treatment with nucleotide/nucleoside analogs on postoperative prognosis of hepatitis B virus-related hepatocellular carcinoma: a two-stage longitudinal clinical study. J Clin Oncol 31 (29): 3647-55, 2013. [PubMed: 24002499]
- Malogolowkin MH, Stanley P, Steele DA, et al.: Feasibility and toxicity of chemoembolization for children with liver tumors. J Clin Oncol 18 (6): 1279-84, 2000. [PubMed: 10715298]
- Otte JB, Pritchard J, Aronson DC, et al.: Liver transplantation for hepatoblastoma: results from the International Society of Pediatric Oncology (SIOP) study SIOPEL-1 and review of the world experience. Pediatr Blood Cancer 42 (1): 74-83, 2004. [PubMed: 14752798]
- Llovet JM, Ricci S, Mazzaferro V, et al.: Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359 (4): 378-90, 2008. [PubMed: 18650514]
Undifferentiated Embryonal Sarcoma of the Liver
Incidence
Undifferentiated embryonal sarcoma of the liver (UESL) is a distinct clinical and pathological entity and accounts for 2% to 15% of pediatric hepatic malignancies.[1]
Diagnosis
UESL presents as an abdominal mass, often with pain or malaise, usually between the ages of 5 and 10 years. Widespread infiltration throughout the liver and pulmonary metastasis is common. It may appear solid or cystic on imaging, frequently with central necrosis. Undifferentiated sarcomas, like small cell undifferentiated hepatoblastomas, should be examined for loss of SMARCB1 expression by immunohistochemistry to help rule out rhabdoid tumor of the liver.
It is important to make the diagnostic distinction between UESL and biliary tract rhabdomyosarcoma because they share some common clinical and pathological features, but treatment differs between the two, as shown in Table 8.[1] For more information, see Childhood Rhabdomyosarcoma Treatment.
Table 8. Diagnostic Differences Between Undifferentiated Embryonal Sarcoma of the Liver and Biliary Tract Rhabdomyosarcomaa
| Undifferentiated Embryonal Sarcoma of the Liver | Biliary Tract Rhabdomyosarcoma | |
|---|---|---|
| Age at Diagnosis | Median age, 10.5 y | Median age, 3.4 y |
| Tumor Location | Often arises in the right lobe of the liver | Often arises in the hilum of the liver |
| Biliary Obstruction | Unusual | Frequent; jaundice is a common presenting symptom |
| Treatment | Surgery and chemotherapy | Surgery (usually biopsy only), radiation therapy, and chemotherapy |
aAdapted from Nicol et al.[1]
Histology
Distinctive histological features of UESL include intracellular hyaline globules and marked anaplasia on a mesenchymal background.[2] Many UESL tumors contain diverse elements of mesenchymal cell maturation, such as smooth muscle and fat.
Strong clinical and histological evidence suggests that UESL can arise within preexisting mesenchymal hamartomas of the liver, which are large, benign, multicystic masses that present in the first 2 years of life.[1] In a report of 11 cases of UESL, 5 arose in association with mesenchymal hamartomas of the liver, and transition zones between the histologies were noted.[3] Many mesenchymal hamartomas of the liver have a characteristic translocation with a breakpoint at 19q13.4, and several UESLs have the same translocation.[4,5] Some UESLs arising from mesenchymal hamartomas of the liver may have complex karyotypes not involving 19q13.4.[4]
Prognosis and Prognostic Factors
The overall survival (OS) rates of children with UESL appears to be substantially higher than 50% when combining reports, although all series are small and may have selection bias.[6]; [7][Level of evidence C1]; [8-17][Level of evidence C1]
The Childhood Cancer Database, which does not provide central review of pathology or reliable details of nonsurgical treatment, reported on 103 children with UESL diagnosed between 1998 and 2012. The 5-year OS rate was 86% for all patients and 92% for those treated with combination surgery and chemotherapy. A multivariate analysis of the nonsurgical data revealed statistically significant poorer outcomes for patients with tumors larger than 15 cm. Seven of ten children who presented with metastases and ten of ten children who underwent orthotopic liver transplant survived at least 5 years, but details of their treatment were not presented.[18]
Treatment Options for UESL
UESL is rare. Only small series have been published regarding treatment.[19]
Treatment options for UESL include the following:
- Surgical resection and chemotherapy.
- Liver transplant for unresectable tumors.
The generally accepted approach is resection of the primary tumor mass in the liver when possible.[18] Use of aggressive chemotherapy regimens seems to have improved the OS of patients with UESL. Neoadjuvant chemotherapy can be effective in decreasing the size of an unresectable primary tumor mass, resulting in resectability.[8-11] Most patients are treated with chemotherapy regimens used for pediatric rhabdomyosarcoma or Ewing sarcoma without cisplatin.[6]; [7,20][Level of evidence C1]; [8-16][Level of evidence C1]
Evidence (surgical resection and chemotherapy):
- The only prospective series to treat patients with UESL came from the Italian and German Soft Tissue Sarcoma Cooperative Groups. Patients were treated with conservative surgery or biopsy followed by neoadjuvant chemotherapy consisting of varying combinations of vincristine, cyclophosphamide, dactinomycin, doxorubicin, and ifosfamide. Disease evaluation, usually after four cycles of chemotherapy, was followed by second-look surgery when appropriate to try to remove residual primary tumor followed by additional and/or adjuvant chemotherapy.[12]
- Ten of 17 patients survived in first complete remission, and one patient survived in third complete remission.
- In a single-center retrospective report, five patients with UESL were treated with surgery and adjuvant chemotherapy consisting of vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide. Four patients had stage I disease, and one patient had stage II disease. One patient received abdominal radiation for tumor rupture.[17][Level of evidence C1]
- All patients were alive (range, 5–19 years), with event-free survival and OS rates of 100%.
Liver transplant has occasionally been used to successfully treat an otherwise unresectable primary tumor.[14,16,18,21]
Treatment Options Under Clinical Evaluation for UESL
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified in a patient's tumor (refractory or recurrent). Children and adolescents aged 1 to 21 years are eligible for the trial.Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the NCI website and ClinicalTrials.gov website.
References
- Nicol K, Savell V, Moore J, et al.: Distinguishing undifferentiated embryonal sarcoma of the liver from biliary tract rhabdomyosarcoma: a Children's Oncology Group study. Pediatr Dev Pathol 10 (2): 89-97, 2007 Mar-Apr. [PubMed: 17378682]
- Stocker JT: Hepatic tumors in children. Clin Liver Dis 5 (1): 259-81, viii-ix, 2001. [PubMed: 11218918]
- Shehata BM, Gupta NA, Katzenstein HM, et al.: Undifferentiated embryonal sarcoma of the liver is associated with mesenchymal hamartoma and multiple chromosomal abnormalities: a review of eleven cases. Pediatr Dev Pathol 14 (2): 111-6, 2011 Mar-Apr. [PubMed: 20925497]
- Stringer MD, Alizai NK: Mesenchymal hamartoma of the liver: a systematic review. J Pediatr Surg 40 (11): 1681-90, 2005. [PubMed: 16291152]
- O'Sullivan MJ, Swanson PE, Knoll J, et al.: Undifferentiated embryonal sarcoma with unusual features arising within mesenchymal hamartoma of the liver: report of a case and review of the literature. Pediatr Dev Pathol 4 (5): 482-9, 2001 Sep-Oct. [PubMed: 11779051]
- Walther A, Geller J, Coots A, et al.: Multimodal therapy including liver transplantation for hepatic undifferentiated embryonal sarcoma. Liver Transpl 20 (2): 191-9, 2014. [PubMed: 24142883]
- Ismail H, Dembowska-Bagińska B, Broniszczak D, et al.: Treatment of undifferentiated embryonal sarcoma of the liver in children--single center experience. J Pediatr Surg 48 (11): 2202-6, 2013. [PubMed: 24210186]
- Chowdhary SK, Trehan A, Das A, et al.: Undifferentiated embryonal sarcoma in children: beware of the solitary liver cyst. J Pediatr Surg 39 (1): E9-12, 2004. [PubMed: 14694398]
- Baron PW, Majlessipour F, Bedros AA, et al.: Undifferentiated embryonal sarcoma of the liver successfully treated with chemotherapy and liver resection. J Gastrointest Surg 11 (1): 73-5, 2007. [PubMed: 17390190]
- Kim DY, Kim KH, Jung SE, et al.: Undifferentiated (embryonal) sarcoma of the liver: combination treatment by surgery and chemotherapy. J Pediatr Surg 37 (10): 1419-23, 2002. [PubMed: 12378446]
- Webber EM, Morrison KB, Pritchard SL, et al.: Undifferentiated embryonal sarcoma of the liver: results of clinical management in one center. J Pediatr Surg 34 (11): 1641-4, 1999. [PubMed: 10591560]
- Bisogno G, Pilz T, Perilongo G, et al.: Undifferentiated sarcoma of the liver in childhood: a curable disease. Cancer 94 (1): 252-7, 2002. [PubMed: 11815984]
- Urban CE, Mache CJ, Schwinger W, et al.: Undifferentiated (embryonal) sarcoma of the liver in childhood. Successful combined-modality therapy in four patients. Cancer 72 (8): 2511-6, 1993. [PubMed: 8402469]
- Okajima H, Ohya Y, Lee KJ, et al.: Management of undifferentiated sarcoma of the liver including living donor liver transplantation as a backup procedure. J Pediatr Surg 44 (2): e33-8, 2009. [PubMed: 19231519]
- Weitz J, Klimstra DS, Cymes K, et al.: Management of primary liver sarcomas. Cancer 109 (7): 1391-6, 2007. [PubMed: 17315167]
- Plant AS, Busuttil RW, Rana A, et al.: A single-institution retrospective cases series of childhood undifferentiated embryonal liver sarcoma (UELS): success of combined therapy and the use of orthotopic liver transplant. J Pediatr Hematol Oncol 35 (6): 451-5, 2013. [PubMed: 23138115]
- Mathias MD, Ambati SR, Chou AJ, et al.: A single-center experience with undifferentiated embryonal sarcoma of the liver. Pediatr Blood Cancer 63 (12): 2246-2248, 2016. [PMC free article: PMC5073002] [PubMed: 27427850]
- Shi Y, Rojas Y, Zhang W, et al.: Characteristics and outcomes in children with undifferentiated embryonal sarcoma of the liver: A report from the National Cancer Database. Pediatr Blood Cancer 64 (4): , 2017. [PMC free article: PMC5333454] [PubMed: 27781381]
- Techavichit P, Masand PM, Himes RW, et al.: Undifferentiated Embryonal Sarcoma of the Liver (UESL): A Single-Center Experience and Review of the Literature. J Pediatr Hematol Oncol 38 (4): 261-8, 2016. [PubMed: 26925712]
- Merli L, Mussini C, Gabor F, et al.: Pitfalls in the surgical management of undifferentiated sarcoma of the liver and benefits of preoperative chemotherapy. Eur J Pediatr Surg 25 (1): 132-7, 2015. [PubMed: 25259441]
- Kelly MJ, Martin L, Alonso M, et al.: Liver transplant for relapsed undifferentiated embryonal sarcoma in a young child. J Pediatr Surg 44 (12): e1-3, 2009. [PubMed: 20005995]
Infantile Choriocarcinoma of the Liver
Choriocarcinoma of the liver is a very rare tumor that appears to originate in the placenta during gestation. It presents with a liver mass in the first few months of life. Metastasis from the placenta to maternal tissues occurs in many cases, necessitating beta-human chorionic gonadotropin (beta-hCG) testing of the mother. Infants are often unstable at diagnosis because of hemorrhage of the tumor.
Diagnosis
Clinical diagnosis may be made without biopsy on the basis of tumor imaging of the liver associated with extremely high serum beta-hCG levels and normal alpha-fetoprotein (AFP) levels for age.[1]
Histology
Cytotrophoblasts and syncytiotrophoblasts are both present. The former are closely packed nests of medium-sized cells with clear cytoplasm, distinct cell margins, and vesicular nuclei. The latter are very large, multinucleated syncytia formed from the cytotrophoblasts.[2]
Prognosis
The prognosis of patients with infantile choriocarcinoma of the liver is often poor because of the instability at presentation from hemorrhage. A 2017 case report and literature review found 32 case reports, with 6 long-term survivors. The authors emphasized the opportunity for early diagnosis and treatment of this very chemosensitive tumor.[3]
Treatment Options for Infantile Choriocarcinoma of the Liver
Treatment options for infantile choriocarcinoma of the liver include the following:
- Surgical resection.[1]
- Chemotherapy followed by surgical resection.
- Chemotherapy followed by liver transplant.
Initial surgical removal of the tumor mass may be difficult because of its friability and hemorrhagic tendency. Surgical removal of the primary tumor is often performed after neoadjuvant chemotherapy.[1]
Maternal gestational trophoblastic tumors are exquisitely sensitive to methotrexate, and many women, including those with distant metastases, are cured with single-agent chemotherapy. Maternal and infantile choriocarcinoma both come from the same placental malignancy. The combination of cisplatin, etoposide, and bleomycin, as used in other pediatric germ cell tumors, has been effective in some patients and is followed by resection of the residual mass. Use of neoadjuvant methotrexate in infantile choriocarcinoma, although often resulting in a response, has not been uniformly successful.[1]
A case report of neoadjuvant chemotherapy followed by successful liver transplant highlights the opportunity for this therapy in children whose tumors remain unresectable after chemotherapy.[4]
Treatment Options Under Clinical Evaluation for Infantile Choriocarcinoma of the Liver
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified in a patient's tumor (refractory or recurrent). Children and adolescents aged 1 to 21 years are eligible for the trial.Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the NCI website and ClinicalTrials.gov website.
References
- Yoon JM, Burns RC, Malogolowkin MH, et al.: Treatment of infantile choriocarcinoma of the liver. Pediatr Blood Cancer 49 (1): 99-102, 2007. [PubMed: 16206190]
- Olson T, Schneider D, Perlman E: Germ cell tumors. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 6th ed. Lippincott Williams and Wilkins, 2011, pp 1045-1067.
- Alsharif S, Karsou A: Infantile choriocarcinoma of the liver: case report and review of the literature. Oncol Cancer Case Rep 3 (1): 2017.
- Hanson D, Walter AW, Dunn S, et al.: Infantile choriocarcinoma in a neonate with massive liver involvement cured with chemotherapy and liver transplant. J Pediatr Hematol Oncol 33 (6): e258-60, 2011. [PubMed: 21792032]
Vascular Liver Tumors
Careful attention to the clinical history, physical exam, laboratory evaluation, and radiological imaging is essential for an appropriate diagnosis of vascular liver tumors. If there is any doubt about the accuracy of the diagnosis, a biopsy should be performed.
The different diagnoses of vascular tumors of the liver include the following:
- Benign tumors.
- Focal congenital hemangiomas. For more information, see the Congenital Hemangiomas section in Childhood Vascular Tumors Treatment.
- Multiple or diffuse infantile hemangiomas. For more information, see the Infantile Hemangioma section in Childhood Vascular Tumors Treatment.
- Malignant tumors.
- Epithelioid hemangioendothelioma. For more information, see the Epithelioid Hemangioendothelioma section in Childhood Vascular Tumors Treatment.
- Angiosarcoma. For more information, see the Angiosarcoma of the Soft Tissue section in Childhood Vascular Tumors Treatment.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Changes to This Summary (01/19/2023)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Tumor Stratification by Imaging and Evans Surgical Staging for Childhood Liver Cancer
Added Figure 1 depicting the four sections and eight segments of the liver.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood liver cancer. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Liver Cancer Treatment are:
- Denise Adams, MD (Children's Hospital Boston)
- Andrea A. Hayes-Dixon, MD, FACS, FAAP (Howard University)
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Michael V. Ortiz, MD (Memorial Sloan Kettering Cancer Center)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Liver Cancer Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/liver/hp/child-liver-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389232]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
- General Information About Childhood Liver Cancer
- Cellular Classification of Childhood Liver Cancer
- Tumor Stratification by Imaging and Evans Surgical Staging for Childhood Liver Cancer
- Treatment Option Overview for Childhood Liver Cancer
- Special Considerations for the Treatment of Children With Cancer
- Hepatoblastoma
- Hepatocellular Carcinoma
- Undifferentiated Embryonal Sarcoma of the Liver
- Infantile Choriocarcinoma of the Liver
- Vascular Liver Tumors
- Current Clinical Trials
- Changes to This Summary (01/19/2023)
- About This PDQ Summary
- Review Childhood Vascular Tumors Treatment (PDQ®): Health Professional Version.[PDQ Cancer Information Summari...]Review Childhood Vascular Tumors Treatment (PDQ®): Health Professional Version.PDQ Pediatric Treatment Editorial Board. PDQ Cancer Information Summaries. 2002
- Review Childhood NUT Carcinoma Treatment (PDQ®): Health Professional Version.[PDQ Cancer Information Summari...]Review Childhood NUT Carcinoma Treatment (PDQ®): Health Professional Version.PDQ Pediatric Treatment Editorial Board. PDQ Cancer Information Summaries. 2002
- Review Osteosarcoma and Undifferentiated Pleomorphic Sarcoma of Bone Treatment (PDQ®): Health Professional Version.[PDQ Cancer Information Summari...]Review Osteosarcoma and Undifferentiated Pleomorphic Sarcoma of Bone Treatment (PDQ®): Health Professional Version.PDQ Pediatric Treatment Editorial Board. PDQ Cancer Information Summaries. 2002
- Review Childhood Rhabdomyosarcoma Treatment (PDQ®): Health Professional Version.[PDQ Cancer Information Summari...]Review Childhood Rhabdomyosarcoma Treatment (PDQ®): Health Professional Version.PDQ Pediatric Treatment Editorial Board. PDQ Cancer Information Summaries. 2002
- Review Childhood Craniopharyngioma Treatment (PDQ®): Health Professional Version.[PDQ Cancer Information Summari...]Review Childhood Craniopharyngioma Treatment (PDQ®): Health Professional Version.PDQ Pediatric Treatment Editorial Board. PDQ Cancer Information Summaries. 2002
- Childhood Liver Cancer Treatment (PDQ®) - PDQ Cancer Information SummariesChildhood Liver Cancer Treatment (PDQ®) - PDQ Cancer Information Summaries
- transcription factor ETV7 isoform X1 [Homo sapiens]transcription factor ETV7 isoform X1 [Homo sapiens]gi|767940157|ref|XP_011512961.1|Protein
- Structure Links for Protein (Select 19115207) (1)Structure
- Nucleotide Links for Protein (Select 19115207) (2)Nucleotide
- uncharacterized protein At2g38710-like isoform X1 [Cucurbita moschata]uncharacterized protein At2g38710-like isoform X1 [Cucurbita moschata]gi|1279757538|ref|XP_022956202.1|Protein
Your browsing activity is empty.
Activity recording is turned off.
See more...