Brain MRI Findings
Common findings
Cerebellar hypoplasia and varying degrees of cerebellar atrophy (more severe in PCH4).
Ventral pontine atrophy, present in the majority of cases (more severe in PCH4).
Cerebellar hemispheres more affected than cerebellar vermis
Cerebral cortical atrophy, progressive with age
Pericerebral CSF accumulation and delayed neocortical maturation in PCH4
Less common findings (not present in all persons) (see )
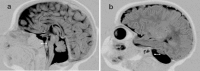
MRI of the brain of an infant age two months with PCH2 a. Midsagittal image showing hypoplastic vermis and flat ventral pons (arrow)
Striatal hypoplasia or atrophy
Delayed myelination of the brain in the first years; no demyelination; gliosis in PCH4
Exceptional: cerebellar hemispheric cysts in PCH2
Family history consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
Diagnosis of TSEN54-PCH is
established in a proband with suggestive findings and biallelic TSEN54 pathogenic (or likely pathogenic) variants identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic TSEN54 variants of uncertain significance (or of one known TSEN54 pathogenic variant and one TSEN54 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing or multigene panel) and comprehensive
genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive brain imaging findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of TSEN54-PCH has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of TSEN54 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. Typically, if only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications; however, to date such variants have not been identified as a cause of this disorder.
Note: Targeted analysis for the common c.919G>T variant can be performed first, since this is by far the most frequent pathogenic variant in TSEN54.
A cerebellar hypoplasia
multigene panel that includes TSEN54 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Of note, given the rarity of TSEN54 pontocerebellar hypoplasia, some panels for cerebellar hypoplasia may not include this gene. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis; however, to date such variants have not been identified as a cause of TSEN54 pontocerebellar hypoplasia.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in TSEN54 Pontocerebellar Hypoplasia
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
|
TSEN54
| Sequence analysis 3 | 100% 4 |
| Gene-targeted deletion/duplication analysis 5 | Unknown 6 |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
Data derived from the subscription-based professional view of the Human Gene Mutation Database [Stenson et al 2020]
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
No data on detection rate of gene-targeted deletion/duplication analysis are available.