

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2004-2013.

| Chemical name: | [125I]2-Iodo-N-[(S)-{(S)-1-methylpiperidin-2-yl}(phenyl)methyl]3-trifluoromethyl-benzamide |
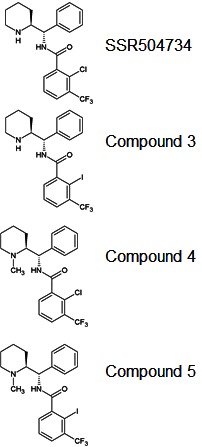
|
| Abbreviated name: | [125I]5 | |
| Synonym: | ||
| Agent Category: | Compounds | |
| Target: | Glycine transporter 1 (GlyT1) | |
| Target Category: | Transporters | |
| Method of detection: | Single-photon emission computed tomography (SPECT), planar imaging | |
| Source of signal / contrast: | 125I | |
| Activation: | No | |
| Studies: |
| Structures of relevant compounds (1). |
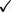 In vitro
In vitro
Background
[PubMed]
[125I]2-Iodo-N-[(S)-{(S)-1-methylpiperidin-2-yl}(phenyl)methyl]3-trifluoromethyl-benzamide, abbreviated as [125I]5, is a 2-iodo-substituted derivative of SSR504734, which was synthesized by Fuchigami et al. for imaging neuropsychiatric disorders by targeting glycine transporter 1 (GlyT1) (1).
Glycine is involved in excitatory and inhibitory neurotransmission in the mammalian central nervous system. Glycine metabolism is controlled by GlyT1 and glycine transporter 2 (GlyT2) (2). GlyT1 is widely distributed in the forebrain areas such as the cortex, hippocampus, and thalamus, as well as glycine-innervated regions such as the spinal cord, brainstem, and cerebellum (1, 3). This expression pattern of GlyT1, especially in the neocortex, is closely related to the distribution of glutamatergic N-methyl-d-aspartate receptors (NMDAR). NMDAR dysfunction has been shown to be involved in various disorders such as schizophrenia, stroke, Parkinson’s disease, and Alzheimer’s disease, while glycine modulates excitatory neurotransmission by acting as a necessary co-agonist for NMDAR in the frontal brain areas (4-6). In this process, GlyT1 is considered to play an important role by regulating glycine concentration (5, 7, 8).
Because of these findings, pharmacological manipulation of glycine-mediated neurotransmission with GlyT1 inhibitors has become an active field for the development of novel treatments for neuropsychiatric disorders, and evidence has demonstrated the beneficial effect of the inhibitors on the negative and cognitive symptoms of schizophrenia (3, 9). Meaningful progress has also been made in the development of imaging probes for GlyT1 to obtain the disease information and the occupancy of GlyT1 inhibitors in vivo (1, 10).
Depoortere et al. synthesized the piperidine benzamide derivative 2-chloro-N-[(S)-phenyl[(2S)-piperidin-2-yl] methyl]-3-trifluoromethyl benzamide (SSR504734) and determined that SSR504734 is a selective and reversible inhibitor of human, rat, and mouse GlyT1 (half-maximal inhibitory concentration (IC50) = 18, 15, and 38 nM, respectively) (9). SSR504734 reversibly blocks the cortical uptake of glycine, thus increasing the extracellular levels of glycine in prefrontal cortex, and it exhibits therapeutic activity in schizophrenia, anxiety, and depression models in rats (9). Recently, 11C-labeled SSR504734 and its derivatives have been shown to exhibit high brain uptake and accumulation in the monkey brain, consistent with the GlyT1 distribution (10). Fuchigami et al. labeled the SSR504734 derivatives for imaging purposes by introducing a radioiodine atom into their 2-position (1). These compounds, especially [125I]5, show high blood–brain barrier permeability and specific brain distribution in GlyT1-rich regions (1). This chapter summarizes the data obtained with [125I]5.
Related Resource Links:
The protein sequence of Gly T1
PubChem Bioassay data of Gly T1
Gly T1 related clinical trials in ClinicalTrials.gov
Synthesis
[PubMed]
Fuchigami et al. described the synthesis of [125I]5 in detail (1). The compound (S)-{(S)-1-allylpiperidin-2-yl}(phenyl)methanamine was first prepared and then coupled with 3-trifluoromethyl-2-iodobenzoic acid (71% yield). Subsequent Pd(0)-catalyzed deprotection of the allyl group resulted in compound 3 (2-iodo-N-[(S)-{(S)-1-piperidin-2-yl}(phenyl)methyl]3-trifluoromethyl-benzamide; 88% yield). Reductive amination of SSR504734 and compound 3 yielded 94% of compound 4 (2-chloro-N-[(S)-{(S)-1-methylpiperidin-2-yl}(phenyl)methyl]3-trifluoromethyl-benzamide) and 95% of compound 5, respectively. Compound 4 is the N-methyl derivative, and compounds 3 and 5 are the 2-iodo-substituted derivatives of SSR504734.
The synthesis of [125I]5 from its tributyltin precursor was initially carried out; however, radioiodination of the tributyltin precursor did not proceed, probably due to steric hindrance of the precursor (data not shown). Therefore, Fuchigami et al. first prepared the trimethyltin precursor of compound 5via an iodo-to-trimethyltin exchange reaction (67% yield), and then generated the [125I]5 by reaction of the trimethyltin precursor and [125I]NaI (carrier-free) in the presence of chloramine-T and HCl at 60°C for 40 min (1). The radiochemical yield was 13%–19%, and the radiochemical purity was >98%. The lipophilicity (logD7.4) value of [125I]5 was 2.86 (detailed data not shown). The specific activity was not reported.
In Vitro Studies: Testing in Cells and Tissues
[PubMed]
Fuchigami et al. evaluated and compared the inhibitory activities on GlyT1 among five compounds (SSR504734, 3, 4, 5, and ALX5407) using a [3H]glycine uptake-JAR cell system (1). JAR is a human choriocarcinoma cell line that expresses GlyT1. ALX5407 is a first-generation GlyT1 inhibitor with high inhibitory potency, but its safety profile and pharmacokinetics in vivo are unfavorable (11). The inhibitory effect of each compound on [3H]glycine uptake was expressed as IC50 value (Table 1).
SSR504734 showed modest inhibitory activity, but its N-methyl derivative 4 showed 11-fold higher activity. Introduction of an iodine atom into the 2-position of SSR504734 resulted in the improvement of inhibitory activity (compounds 3 and 5). Compound 5 exhibited the best inhibitory activity, which was 18-fold and 3-fold higher than 3 and 4, respectively. These results indicate the importance of the methyl group for GlyT1 inhibition, and the iodine atom is preferred over the chloride atom for inhibitory activity. The inhibitory activity of ALX5407 was 6-fold higher than that of 5.
Table 1: Inhibition on [3H]glycine uptake of JAR cells.
| Compound | SSR504734 | 3 | 4 | 5 | ALX5047 |
| IC50 (nM) | 80.1 ± 7.55 | 43.0 ± 4.12 | 7.51 ± 1.91 | 2.35 ± 0.35 | 0.39 ± 0.05 |
The binding property of [125I]5 was investigated with rat cortical brain homogenates and saturation assay (1). The binding of [125I]5 was calculated to be a well-fitted one binding site model (R2 = 0.985) with high affinity (Kd = 1.58 ± 0.34 nM). The maximal binding value was estimated to be 3.40 ± 0.18 pmol/mg protein.
The in vitro brain distribution of [125I]5 was characterized with rat brain sections and autoradiography (1). The brain sections were incubated with [125I]5 (100 kBq (2.70 µCi)) (0.03–0.04 nM) for 30 min at 25°C. [125I]5 showed high accumulation in the corpus callosum, thalamus, midbrain, medullary, and cerebellar white matter, and low uptake in the cerebral cortex, hippocampus, striatum, and cerebellar gray matter. The accumulation pattern of [125I]5 was similar to the distribution of GlyT1 immunoreactivity in the rat brain. Nonspecific binding was determined in the presence of nonradioactive 5 (10 μM), which resulted in a significant reduction of radioactivity accumulation in the whole brain. Regional brain differences for [125I]5 binding disappeared. These results indicate that the binding of [125I]5 in the rat brain slices correlates with GlyT1 expression level.
The specificity of [125I]5 (100 kBq (2.70 µCi) (0.03–0.04 nM) binding to GlyT1 was examined with four GlyT1 inhibitors (SSR504734, compound 4, ALX5047, and HPCP ((+)-N-[cis-1-(2-hydroxy-2-phenyl-cyclohexyl)-piperidin-4-yl]-4-methoxy-N-phenyl-benzenesulfonamide), 10 μM/agent), one GlyT1 substrate (glycine, 1 mM or 10 mM), one GlyT2 inhibitor (ORG25543, 10 μM), and one NMDAR glycine site antagonist (L-701,324, 10 μM) (1). SSR504734 derivatives are competitive, and ALX5407 is a noncompetitive inhibitor of the GlyT1. HPCP has a different structure from [125I]5 (1).
SSR504734 and its N-methyl derivative 4 partially decreased the accumulation of [125I]5 in the GlyT1-rich regions, such as the corpus callosum, thalamus, midbrain, medullary, and cerebellum (21%−27% and 20%−29% compared with control, respectively; P < 0.05). Reduction of radioactivity was also observed in the GlyT1-poor regions, such as the cerebral cortex, hippocampus, and striatum (32%−45% and 32%−46% for SSR504734 and 4, respectively). HPCP showed partial inhibition of the [125I]5 binding to GlyT1-rich regions (11%−23%) and moderate inhibition to GlyT1-poor regions (31%−56%). ALX5407 decreased [125I]5 binding in all brain regions, though to a lower extent than other GlyT1 inhibitors, which is thought to be caused by allosteric inhibition of ALX5047 to the [125I]5 binding for GlyT1. Glycine had almost no effect on [125I]5 binding at 1 mM, and when increased to 10 mM glycine reduced the accumulated radioactivity in all brain regions. These results suggest that compound 5 is a competitive inhibitor of the glycine binding site of GlyT1 and that the binding affinity of glycine to the [125I]5 binding site is much lower than that of the GlyT1 inhibitors. ORG25543 and L-701,324 did not affect the [125I]5 binding in any brain regions.
Animal Studies
Rodents
[PubMed]
The biodistribution of [125I]5 was studied in normal ddY mice (n = 5–6/time point) after tail vein injection of 11.1 kBq (0.3 µCi) [125I]5, and its accumulation in organs was expressed as percentage of injected dose per gram tissue (% ID/g) (1). High pulmonary uptake of [125I]5 was observed at all time points (9.34%−17.19% ID/g), which could be attributed to the piperidine moiety of this tracer. Initial brain uptake of [125I]5 was 0.51% ID/g (at 0.5 min), then increased over time and peaked at 2.42% ID/g at 30 min, suggesting good blood–brain barrier permeability of [125I]5. The brain uptake of [125I]5 gradually decreased after 30 min, but the brain–blood ratio remained constant for up to 60 min after injection (2.46 and 2.32 at 30 min and 60 min, respectively). High brain uptake of [125I]5 can be explained partially by its ideal lipophilicity. Radiotracers with moderate lipophilicity (logD7.4 values, 2.0–3.5) usually have optimal passive brain entry under in vivo condition.
Metabolism of [125I]5 in mouse brain was analyzed with brain homogenates obtained at different times after injection of [125I]5 (185 kBq (5 µCi)) and 92%, 90%, and 89% of [125I]5 remained unchanged at 5, 30, and 60 min after injection, respectively (data not shown), suggesting that metabolism should have little or no influence on the in vivo brain distribution pattern of [125I]5 (1).
The specificity of regional brain binding of [125I]5 (33.3 kBq (0.9 µCi)) for GlyT1 was analyzed with pre-administration of nonradioactive 5, compound 4, SSR504734, and ALX5407 (2 mg/kg per blocking agent), respectively, 15 min before injection of [125I]5 (n = 5–6 mice/time point per agent) (1). Because the brain/blood ratio of [125I]5 peaked at 30 min and 60 min in biodistribution studies, regional brain distribution studies were therefore conducted at 30 min and 60 min after the radiotracer injection. The blocking effect on [125I]5 accumulation was expressed as a percentage of the control (no administration of blocking agents) at 30 min and 60 min, respectively. The results showed that pre-injection of nonradioactive compound 5 resulted in partial reduction of [125I]5 accumulation in the GlyT1-rich regions, such as the thalamus (45% and 58%; P < 0.05), midbrain (48% and 51%; P < 0.05), and medulla (53% and 52%; P < 0.05). In contrast, the extent of [125I]5 uptake reduction in the GlyT1-poor regions, such as the hippocampus and striatum, was relatively lower than the GlyT1-rich regions. Pre-administration of compound 4 also led to a partial decrease in regional radioactivity uptake in the thalamus (56% and 56%; P < 0.05), midbrain (55% and 48%; P < 0.05), and medulla (59% and 47%; P < 0.05), but also resulted in little displacement of radioactivity from the hippocampus and striatum. The estimated brain region/blood ratios were also significantly decreased by nonradioactive compound 5 and compound 4 in the GlyT1-rich regions. The radioactivity level of [125I]5 in all brain regions became homogenous with the use of these blockers. These results indicate that in vivo binding of [125I]5 is specifically mediated by GlyT1. SSR504734 did not significantly block the uptake of [125I]5 in any brain regions (P > 0.05), which might be due to its much lower inhibitory activity and binding affinity for GlyT1 than its N-methyl analogs (compounds 4 and 5). Pretreatment with ALX5407 showed no effect on the brain uptake of [125I]5 at 30 min and weak inhibitory effect at 60 min (P > 0.05).
Fuchigami et al. further investigated the in vivo binding properties of [125I]5 to GlyT1 with ex vivo autoradiography (1). Mice (n = 5) were injected intravenously with [125I]5 (200 kBq (5.41 µCi)) and euthanized at 60 min after administration. The results showed high uptake of [125I]5 in the GlyT1-rich regions (thalamus, midbrain, cerebellum, and medullary) and low uptake in the GlyT1-poor regions (cerebral cortex, hippocampus, and striatum). The low radioactivity of [125I]5 in the corpus callosum was inconsistent with that obtained in the in vitro autoradiography studies, which might be due to the influence of lower regional blood flow in the white matter, such as the corpus callosum, than in other brain regions. To further confirm the in vivo specificity of [125I]5 for GlyT1, blocking studies were performed by pretreatment with nonradioactive compound 5 or compound 4 (2 mg/kg body weight) 15 min before [125I]5 injection (n = 5 mice/group). Both compound 5 and compound 4 abolished the heterogenous distribution of [125I]5 in the brain regions. Accumulation of [125I]5 was significantly reduced by these compounds only in the GlyT1-rich regions (P < 0.05).
References
- 1.
- Fuchigami T., Haratake M., Magata Y., Haradahira T., Nakayama M. Synthesis and characterization of [(1)(2)I]2-iodo N-[(S)-{(S)-1-methylpiperidin-2-yl}(phenyl)methyl]3-trifluoromethyl-benzamide as novel imaging probe for glycine transporter 1. Bioorg Med Chem. 2011;19(21):6245–53. [PubMed: 21975065]
- 2.
- Zafra F., Gimenez C. Glycine transporters and synaptic function. IUBMB Life. 2008;60(12):810–7. [PubMed: 18798526]
- 3.
- Wolkenberg S.E., Sur C. Recent progress in the discovery of non-sarcosine based GlyT1 inhibitors. Curr Top Med Chem. 2010;10(2):170–86. [PubMed: 20166956]
- 4.
- Sur C., Kinney G.G. Glycine transporter 1 inhibitors and modulation of NMDA receptor-mediated excitatory neurotransmission. Curr Drug Targets. 2007;8(5):643–9. [PubMed: 17504107]
- 5.
- Gomeza, J.,Armsen, W.,Betz, H., and Eulenburg, V. Lessons from the knocked-out glycine transporters. Handb Exp Pharmacol. 2006(175): 457-83. [PubMed: 16722246]
- 6.
- Eulenburg V., Gomeza J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res Rev. 2010;63(1-2):103–12. [PubMed: 20097227]
- 7.
- Betz H., Laube B. Glycine receptors: recent insights into their structural organization and functional diversity. J Neurochem. 2006;97(6):1600–10. [PubMed: 16805771]
- 8.
- Betz H., Gomeza J., Armsen W., Scholze P., Eulenburg V. Glycine transporters: essential regulators of synaptic transmission. Biochem Soc Trans. 2006;34(Pt 1):55–8. [PubMed: 16417482]
- 9.
- Depoortere R., Dargazanli G., Estenne-Bouhtou G., Coste A., Lanneau C., Desvignes C., Poncelet M., Heaulme M., Santucci V., Decobert M., Cudennec A., Voltz C., Boulay D., Terranova J.P., Stemmelin J., Roger P., Marabout B., Sevrin M., Vige X., Biton B., Steinberg R., Francon D., Alonso R., Avenet P., Oury-Donat F., Perrault G., Griebel G., George P., Soubrie P., Scatton B. Neurochemical, electrophysiological and pharmacological profiles of the selective inhibitor of the glycine transporter-1 SSR504734, a potential new type of antipsychotic. Neuropsychopharmacology. 2005;30(11):1963–85. [PubMed: 15956994]
- 10.
- Toyohara J., Ishiwata K., Sakata M., Wu J., Nishiyama S., Tsukada H., Hashimoto K. In vivo evaluation of carbon-11-labelled non-sarcosine-based glycine transporter 1 inhibitors in mice and conscious monkeys. Nucl Med Biol. 2011;38(4):517–27. [PubMed: 21531289]
- 11.
- Perry K.W., Falcone J.F., Fell M.J., Ryder J.W., Yu H., Love P.L., Katner J., Gordon K.D., Wade M.R., Man T., Nomikos G.G., Phebus L.A., Cauvin A.J., Johnson K.W., Jones C.K., Hoffmann B.J., Sandusky G.E., Walter M.W., Porter W.J., Yang L., Merchant K.M., Shannon H.E., Svensson K.A. Neurochemical and behavioral profiling of the selective GlyT1 inhibitors ALX5407 and LY2365109 indicate a preferential action in caudal vs. cortical brain areas. Neuropharmacology. 2008;55(5):743–54. [PubMed: 18602930]
- PubMedLinks to PubMed
- Synthesis and evaluation of 2-chloro N-[(S)-{(S)-1-[11 C]methylpiperidin-2-yl} (phenyl)methyl]3-trifluoromethyl-benzamide ([11 C]N-methyl-SSR504734) as a PET radioligand for glycine transporter 1.[EJNMMI Res. 2012]Synthesis and evaluation of 2-chloro N-[(S)-{(S)-1-[11 C]methylpiperidin-2-yl} (phenyl)methyl]3-trifluoromethyl-benzamide ([11 C]N-methyl-SSR504734) as a PET radioligand for glycine transporter 1.Fuchigami T, Takano A, Gulyás B, Jia Z, Finnema SJ, Andersson JD, Nakao R, Magata Y, Haratake M, Nakayama M, et al. EJNMMI Res. 2012 Jul 9; 2(1):37. Epub 2012 Jul 9.
- Inhibitors of GlyT1 affect glycine transport via discrete binding sites.[Mol Pharmacol. 2008]Inhibitors of GlyT1 affect glycine transport via discrete binding sites.Mezler M, Hornberger W, Mueller R, Schmidt M, Amberg W, Braje W, Ochse M, Schoemaker H, Behl B. Mol Pharmacol. 2008 Dec; 74(6):1705-15. Epub 2008 Sep 24.
- Synthesis and characterization of [¹²⁵I]2-iodo N-[(S)-{(S)-1-methylpiperidin-2-yl}(phenyl)methyl]3-trifluoromethyl-benzamide as novel imaging probe for glycine transporter 1.[Bioorg Med Chem. 2011]Synthesis and characterization of [¹²⁵I]2-iodo N-[(S)-{(S)-1-methylpiperidin-2-yl}(phenyl)methyl]3-trifluoromethyl-benzamide as novel imaging probe for glycine transporter 1.Fuchigami T, Haratake M, Magata Y, Haradahira T, Nakayama M. Bioorg Med Chem. 2011 Nov 1; 19(21):6245-53. Epub 2011 Sep 10.
- Review 2-chloro-N-((S)-((S)-1-[(11)C]methylpiperidine-2-yl)(thiophen-3-yl)methyl)-3-(trifluoromethyl)benzamide ([(11)C]SA1) and derivatives.[Molecular Imaging and Contrast...]Review 2-chloro-N-((S)-((S)-1-[(11)C]methylpiperidine-2-yl)(thiophen-3-yl)methyl)-3-(trifluoromethyl)benzamide ([(11)C]SA1) and derivatives.Chopra A. Molecular Imaging and Contrast Agent Database (MICAD). 2004
- Review Neurobiology of glycine transporters: From molecules to behavior.[Neurosci Biobehav Rev. 2020]Review Neurobiology of glycine transporters: From molecules to behavior.Marques BL, Oliveira-Lima OC, Carvalho GA, de Almeida Chiarelli R, Ribeiro RI, Parreira RC, da Madeira Freitas EM, Resende RR, Klempin F, Ulrich H, et al. Neurosci Biobehav Rev. 2020 Nov; 118:97-110. Epub 2020 Jul 24.
- [125I]2-Iodo-N-[(S)-{(S)-1-methylpiperidin-2-yl}(phenyl)methyl]3-trifluoromethyl...[125I]2-Iodo-N-[(S)-{(S)-1-methylpiperidin-2-yl}(phenyl)methyl]3-trifluoromethyl-benzamide - Molecular Imaging and Contrast Agent Database (MICAD)
- Gastric FistulaGastric FistulaAbnormal passage communicating with the STOMACH.<br/>MeSH
- Toll-Like Receptor 7Toll-Like Receptor 7A pattern recognition receptor that binds several forms of imidazo-quinoline including the antiviral compound Imiquimod.<br/>Year introduced: 2006(2005)MeSH
Your browsing activity is empty.
Activity recording is turned off.
See more...