

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Nelson HD, Fu R, Humphrey L, et al. Comparative Effectiveness of Medications To Reduce Risk of Primary Breast Cancer in Women [Internet]. Rockville (MD): Agency for Healthcare Research and Quality (US); 2009 Sep. (AHRQ Comparative Effectiveness Reviews, No. 17.)
This publication is provided for historical reference only and the information may be out of date.

Comparative Effectiveness of Medications To Reduce Risk of Primary Breast Cancer in Women [Internet].
Show detailsTopic Development
The topic for this comparative effectiveness review was nominated in a public process. With input from technical experts, the Scientific Resource Center (SRC) for the AHRQ Effective Health Care Program drafted the initial key questions and, after approval from AHRQ, posted them to a public web site. The public was invited to comment on these questions. After reviewing the public commentary, the SRC drafted final key questions and submitted them to AHRQ for approval.
The key questions went through three subsequent revisions. After discussions with a technical expert panel, the key questions were further refined to identify specific outcomes of interest for key questions 1, 2, and 3. These changes were submitted to AHRQ for approval before literature searches were conducted. The second change to the key questions occurred in September 2008 after publication of a new study of breast cancer risk reduction with the medication tibolone. After discussion with AHRQ, it was determined that the current report would be amended to include tibolone. New key questions including tibolone were then approved by AHRQ. The third change was in response to peer reviewers who suggested that the terms “chemotherapy” and “prevention” were misnomers. The term “medications to reduce risk” is a better representation of the intervention. Therefore, all references to “chemoprevention” were edited, including the key questions and report title.
We created an analytic framework incorporating the key questions to guide our examination of a chain of evidence about the effectiveness and potential adverse effects of medications to reduce risk of primary breast cancer (Figure 1). The analytic framework outlines the target population, interventions, and outcomes defined by the scope of this review. The target population includes women without pre-existing invasive or noninvasive breast cancer or precursor conditions, and who are not known carriers of breast cancer susceptibility mutations (BRCA1, BRCA2, or others). Outcomes are defined by the key questions and include a wide range of health outcomes as opposed to intermediate outcomes. Health outcomes are signs, symptoms, conditions, or events that individuals experience, such as myocardial infarction. Intermediate outcomes are health measures that individuals do not personally experience, such as laboratory test results.
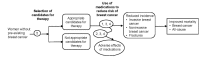
Figure 1
Analytic framework. Note: Numbers refer to key questions.
Search Strategy
We used the National Library of Medicine’s Medical Subject Headings (MeSH®) keyword nomenclature developed for MEDLINE® and adapted for use in other databases. With assistance from a research librarian, we searched OVID MEDLINE® (1950 to January Week 3, 2009), Cochrane Central Register of Controlled Trials (4th Quarter 2008), and Cochrane Database of Systematic Reviews (4th Quarter 2008) for relevant studies, systematic reviews, and meta-analyses. The searches were limited to papers published in English language. The texts of the major search strategies are provided in Appendix A1. We also searched clinical trial registries and conducted secondary referencing by manually reviewing reference lists of papers and reviewing citations indicated for key trials by Web of Science. ® After identifying several large trials meeting inclusion criteria for the review, we contacted the investigators to request additional unpublished data specifically addressing the subpopulations described in key question 3. No additional data have been received.
In addition, we received the following materials from the Scientific Resource Center:
- Searches of clinical trial registries: www.clinicaltrials.gov; www.controlled-trials.com; www.clinicalstudyresults.org; www.Drugs@FDA.gov; and the American Society of Clinical Oncology website: (http://www.asco.org/ASCO/Abstracts+%26+Virtual+Meeting/Abstracts).
- Scientific information packet from Eli Lilly for Evista® (raloxifene). Scientific information packets were requested from manufacturers of tamoxifen, however no information was provided. A scientific information packet was requested from the manufacturers of tibolone by the Scientific Resource Center in November 2008. As of publication of this draft, no information has been received.
The searches identified a total of 4,842 unique citations. Some citations were relevant to multiple key questions. Investigators reviewed 4,230 citations for key questions 1, 2, and 3; 1,644 citations for key question 4; and 1,364 citations for key question 5. All citations were imported into an electronic database (EndNote X1).
Study Selection
Prior to our review of abstracts and articles, we developed inclusion and exclusion criteria for studies based on the patient populations, interventions, outcome measures, and types of evidence specified in each key question (Appendix A2). We applied these criteria to the abstracts and articles identified by our searches. After an initial review of citations and abstracts, we retrieved full-text articles of potentially relevant material and conducted a second review to determine inclusion. A second reviewer confirmed results of the initial reviewer. Articles with questionable eligibility were reviewed and discussed by the investigator team before determining their inclusion. Results published only in abstract form were not included in our review because adequate information was not available to assess the validity of the data. Excluded studies and their main reasons for exclusion are listed in Appendix B.
For key question 1 and any outcomes relating to risk reduction benefits for key question 3, we included only randomized controlled trials (RCT) of tamoxifen, raloxifene, or tibolone for primary prevention of breast cancer enrolling women without breast cancer. We included trials that were designed and powered to demonstrate invasive breast cancer incidence as a primary or secondary outcome. The technical expert panel advised including only RCTs for several reasons. These include lack of observational studies of tamoxifen and raloxifene with breast cancer outcomes in women without breast cancer, and concerns for bias among users in observational studies. For example, women using tibolone to treat menopausal hot flashes are more likely to have a hysterectomy/oophorectomy than nonusers, reducing their breast cancer risk.
For key question 2 and outcomes relating to harms for key question 3, we defined our inclusion criteria more broadly. We included RCTs and observational studies of tamoxifen, raloxifene, or tibolone in women without breast cancer that were designed for multiple types of outcomes. However, studies must have had a nonuser comparison group, or direct comparisons between tamoxifen, raloxifene, or tibolone to be included. We included studies with treatment durations of 3 months or more that enrolled 100 or more participants to assure adequate drug exposure and power to support results.
For key question 4, RCTs, observational studies, and descriptive studies evaluating benefits or harms and treatment adherence, persistence, concordance, or treatment choice with tamoxifen, raloxifene, or tibolone in women without breast cancer were included.
For key question 5, we included studies of risk stratification models that could be used in a primary care setting to identify women at higher than average risk for breast cancer. Only studies reporting discriminatory accuracy of the models were included. We did not include models designed to evaluate family history in order to determine risk for deleterious BRCA mutations because women with these mutations are outside the target population for this review. We also excluded studies of single risk factors or laboratory tests.
Data Extraction
For the included RCTs and observational studies, we abstracted the following data: study design; setting; participant characteristics (including age, ethnicity, diagnosis); enrollment criteria; interventions (dose and duration); numbers enrolled and lost to follow-up; methods of outcome ascertainment; and results for each outcome. For descriptive studies of treatment choice, we abstracted: study design; intervention; setting; population characteristics; eligibility and exclusion criteria; response rates; procedure for data collection; and results for each outcome. For studies of risk stratification models, we abstracted: study design; population characteristics; eligibility and exclusion criteria; breast cancer incidence rates; risk factors included in the models; and performance measures of the models. A second reviewer confirmed the accuracy of abstracted information.
Quality Assessment
We used predefined criteria developed by the U.S. Preventive Services Task Force to assess the quality of studies of benefits and harms of medications (Appendix C-1).13 To determine quality of risk assessment instruments, we adapted the U.S. Preventive Services Task Force criteria for diagnostic accuracy studies (Appendix C-1).13 We did not evaluate descriptive studies for quality because specific criteria are not available for these study designs. Two investigators independently rated the quality of each eligible study (good, fair, poor) and final ratings were determined by consensus.
Applicability
We assessed applicability of studies by following the population, intervention, comparator, outcomes, timing of outcomes measurement, and setting (PICOTS) format (Appendix C-1).14 When possible, we highlighted effectiveness studies conducted in settings relevant to clinical practice, with subjects selected with less stringent eligibility criteria, assess health outcomes, and have longer follow-up periods than most efficacy studies. The results of effectiveness studies are more applicable to the spectrum of patients that will use a medication, have a test, or undergo a procedure than results from highly selected populations in efficacy studies. Two investigators independently rated the quality of each eligible study (good, fair, poor) and final ratings were determined by consensus.
Rating the Body of Evidence
We assessed the overall strength of the body of evidence through group consensus using the EPC GRADE (Grading of Recommendations Assessment, Development and Evaluation) approach.15 This approach uses a two step process for each key outcome. First, we assessed risk of bias, consistency of effect, directness, and precision for each outcome. We also determined the magnitude of effect for key outcomes using results of meta-analyses of trials as described below. Additional optional domains in the EPC GRADE table were not included in the table because they are not relevant to this review. Definitions and criteria for scoring these domains are described in Appendixes C-2, C-3, and C-4. Second, we determined overall grades based on qualitative combinations of the ratings for each domain. The EPC GRADE classifications for the overall strength of the body of evidence are: high, moderate, low, and insufficient (Appendix C-3). A grade of high indicates high confidence that the evidence reflects the true effect and further research is very unlikely to change our confidence in the estimate of effect. A grade of moderate indicates moderate confidence that the evidence reflects the true effect and that further research may change our confidence in the estimate of effect and may change the estimate. A grade of low indicates low confidence that the evidence reflects the true effect and that further research is likely to change our confidence in the estimate of effect and is likely to change the estimate. A grade of insufficient is given when the evidence either is unavailable or does not permit estimation of an effect.
Data Synthesis
Statistical Analysis
We combined results of eligible placebo-controlled trials in several meta-analyses to obtain more precise estimates of major health outcomes for the target population (key questions 1 and 2), and explore whether the combined estimates differ among subpopulations (key question 3). To determine the appropriateness of meta-analysis, we considered clinical and methodological diversity and assessed statistical heterogeneity.
We abstracted or calculated estimates of risk ratios (rate ratio, hazard ratio, or relative risk) and their standard errors from each trial and used them as the effect measures. For each outcome, we adopted the following steps to obtain the risk ratio and to account for the varying follow-up periods of the trials:
- If a study reported a rate ratio based on a Poisson model, where women-years of follow-up was incorporated in the estimates, or a hazard ratio from a Cox regression model, we used the reported estimate.
- If not, but the study reported the number of events and women-years of follow-up, or women-years of follow-up could be calculated from reported data, we calculated the rate ratio based on a Poisson distribution using the number of events and women-years of follow-up.
- If both 1) and 2) were not possible, we used the reported or calculated relative risk, which does not take into account the women-years of follow-up. However, the estimate of relative risk would be expected to be very close to the estimate of rate ratio since the mean or median follow-up time was similar between the treatment and control arms in the trials.
We assessed the presence of statistical heterogeneity among the studies using standard χ2 tests, and the magnitude of heterogeneity using the I2 statistic.16 We used a random effects model to account for variation among studies.17 When there is no variation among studies, the random effects model yields the same results as a fixed effects model. For all meta-analyses, we combined results separately for tamoxifen and raloxifene and provided 95% confidence intervals. We used STATA® 9.1 software for all these analyses (StataCorp, College Station, TX, 2006).
To explore whether combined estimates differ among subpopulations for key question 3, we performed subgroup analysis by age (≤50 yrs vs. > 50 yrs), family history of breast cancer (yes vs. no), use of menopausal hormone therapy (yes vs. no), menopausal status (pre vs. post), and body mass index (BMI) (≤25 vs. >25), when at least two studies reported results. We also performed subgroup analysis for tamoxifen trials stratified by active vs. post treatment periods when studies reported these data.
We also conducted an indirect comparison to compare the major benefits from trials of raloxifene with the one trial of tibolone using meta-regression. Since the raloxifene and tibolone trials recruited much older populations than the tamoxifen trials, we did not conduct indirect comparisons between the tamoxifen trials and raloxifene/tibolone trials.
Event Rates
To facilitate the evaluation of benefits and harms across trials, we abstracted or calculated event rates per 1000 women years for both treatment and placebo groups using steps similar to those obtaining risk ratios. When the event rates were not reported or calculable, we indicated them as such. To obtain the combined event rates, we conducted a meta-analysis of the placebo event rates by using a random effects Poisson model and raw data of number of events and women years of follow-up. We used PROC NLMIXED, SAS 9, 1.3. software for this analysis (SAS Institute Inc., Cary, NC, 2008).
Number Needed To Treat/Harm
To help interpret the clinical impact of the medications, we calculated the number of women needed to treat (NNT) to cause an outcome if each woman were to take the medication for 5 years. These numbers and the corresponding 95% confidence intervals were estimated using the combined risk ratios from the meta-analyses and the combined event rates from the placebo groups of included trials. To obtain the combined event rates, we conducted a meta-analysis of the placebo event rates as described above. We calculated the 95% confidence intervals for NNT by using a simulation method. We assumed that both logs of risk ratios and event rates have normal distributions, and we drew 10,000 random samples from them. The number needed to treat/harm and the number of events prevented/caused were then calculated from each sample, and the 95% confidence intervals were obtained by computing the 2.5% and 97.5% quantiles of the full sample.
Peer Review and Public Commentary
A draft of the report was sent to peer reviewers, anonymous reviewers identified by the United States Preventive Services Task Force, AHRQ representatives and the Scientific Resource Center. The draft report was also posted on the AHRQ Effective Health Care for a public comment period. Changes to the report were made based on comments received from peer and public reviewers. A summary of responses to comments will be publically available on the Effective Health Care website.
- Methods - Comparative Effectiveness of Medications To Reduce Risk of Primary Bre...Methods - Comparative Effectiveness of Medications To Reduce Risk of Primary Breast Cancer in Women
- Methods - Local Hepatic Therapies for Metastases to the Liver From Unresectable ...Methods - Local Hepatic Therapies for Metastases to the Liver From Unresectable Colorectal Cancer
- Results - PCA3 Testing for the Diagnosis and Management of Prostate CancerResults - PCA3 Testing for the Diagnosis and Management of Prostate Cancer
- Peer Reviewers - The Clinical Utility of Fractional Exhaled Nitric Oxide (FeNO) ...Peer Reviewers - The Clinical Utility of Fractional Exhaled Nitric Oxide (FeNO) in Asthma Management
- Peer Reviewers - The Role of Immunotherapy in the Treatment of AsthmaPeer Reviewers - The Role of Immunotherapy in the Treatment of Asthma
Your browsing activity is empty.
Activity recording is turned off.
See more...