

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
The cytokine network in rheumatoid arthritis (RA) is a complex field, with a lot of cytokines showing pleiotropic actions and many different targets. To keep it simple, the network can be divided in two groups, the pro-inflammatory and anti-inflammatory cytokines.Controling the balance between these two groups is considered as an important therapeutic goal.
Two key pro-inflammatory cytokines in RA are IL-1 and TNFα. Regulation of these cytokines is of crucial importance in the RA disease. First data of clinical trials showed efficacy, however, revealed also that blockade of these cytokines did not fully control the arthritis in all patients. Recent discoveries of novel cytokines in the pathology of arthritis, such as IL-17, IL-18 and RANK ligand (RANKL) will help us to get a better understanding of the pathogenesis of chronic arthritis and may contribute to improvement of current therapies. IL-4 and IL-10 are pleiotropic cytokines, and are considered as promising modulators in the control of RA.
Rheumatoid arthritis (RA) is a chronic systemic disorder of unknown etiology. This disease affects about 1% of the population worldwide, most commonly middle-aged women. It is characterized by chronic inflammation of the synovium, particularly of small joints, which often leads to destruction of articular cartilage and juxtaarticular bone.1 The clinical and laboratory features are suggestive of an autoimmune disease. However, the autoantigen is still unknown, hampering specific immunomodulation as a straightforward therapeutic approach.
The pathogenesis of RA is not identified and seems to be multifactorial. A major research goal in the field of arthritis is to unravel the pathogenesis of chronic arthritis and the concomitant joint destruction. During the last 20 years, the understanding of the basic biology of RA has increased enormously. This will help to define targeted therapies, selectively inhibiting the progression of destructive arthritis, yet leaving host-defence mechanisms virtually intact. Targeting the cytokine disbalance might represent a solid way to control this disease.2
Pathways in the Pathogenesis of RA
The current concept is that inflammation and tissue destruction results from complex cell-cell interactions in the rheumatoid synovium.3,4 The major cytokines and cellular pathways currently implicated in the pathogenesis of RA are presented in Figure 1. These events can be amplified or initiated by an interaction between antigen presenting cells (APC) and CD4+ T cells; APC display complexes of class II major histocompatibility complex (MHC) molecules and peptide antigen(s) that bind to specific receptors on the T cells. Macrophage activation occurs, with abundant secretion of proinflammatory cytokines such as IL-1 and NFα. These cytokines stimulate synovial fibroblasts and chondrocytes in the nearby articular cartilage to secrete enzymes that degrade proteoglycans and collagen, leading to tissue destruction.
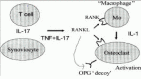
Figure 1
Schematic overview of cytokines in RA.
Whether this process of destruction is driven by T cells or reflects mainly macrophage and synovial fibroblast activation is still a matter of debate. It has been shown that RA synovial fibroblasts are capable of mediating progressive joint destruction in the absence of T cells or other inflammatory cells,5 suggesting T cell independent pathways in joint destruction.6 Detailed analysis of mediators production in the inflamed synovial tissue reveals a relative lack of T cell factors and an abundance of cytokines and growth factors, produced by macrophages and synovial fibroblasts.7
Proinflammatory Cytokines IL-1 and TNF
It is well established that TNF and IL-1 are key cytokines in the process of chronic joint inflammation and the concomitant erosive changes in cartilage and bone. Animal model studies have greatly contributed to this identification. The initial studies analysed the arthritogenic potential of recombinant cytokines when directly injected into the knee joints of rabbits and rodents. This provided the first suggestive evidence that TNFα was an inflammatory mediator, whereas IL-1 was a crucial cytokine in both arthritis and cartilage destruction. TNFα alone was hardly destructive, but it could enhance in a synergistic way the destructive behaviour of IL-1.8,9 Follow-up studies in TNFα transgenics further underlined the fact that TNFα over-expression, in the absence of functional T and B cells, was arthritogenic.10 Recent observations clarified that there is no requirement for soluble TNFα but that the full expression of arthritis can occur even with a membrane-bound form of TNFα (mTNFα).11 The consequences of this is that therapies focused on TNF blockade should preferably make use of antibodies or scavenging soluble receptors that have excellent access to cell surfaces. The development of arthritis in TNF transgenic mice could be prevented with antibodies to TNFα, which seems obvious. More interestingly, pathology could also be fully blocked with antibodies against the IL-1 receptor.12 This strongly indicates that 1) IL-1 is the secondary mediator responsible for the arthritic changes, and 2) TNFα alone is neither arthritogenic nor destructive towards joints.
Meanwhile, studies with neutralizing antibodies against TNFα have been instrumental in the elucidation of TNF as a major target in more natural arthritis models, with a T cell-driven pathogenetic pathway, compared with the plain over-expression of a single mediator.
Ample studies have been performed in the generally accepted murine autoimmune model of collagen-induced arthritis (CIA). CIA is based on T cell and antibody-mediated autoimmune reactivity against collagen type II, the major component of cartilage. The model is characterized by severe and rapid cartilage and bone erosion. Suppression of collagen arthritis was achieved both with neutralizing antibodies against TNFα and with soluble TNF receptors.13,14 Intriguingly, it was found that TNFα was crucial at the onset of the arthritis but appeared less dominant in the later stages.15 In fact, studies in TNF receptor knockout mice demonstrated that the incidence and severity of arthritis were less in such mice; once the joints became affected, however, full progression to erosive damage was noted in an apparently TNF-independent fashion.16
As state above, IL-1 is a potent cytokine in the induction of cartilage destruction8,9 and a pivotal secondary mediator in arthritis and tissue destruction in TNF transgenic over-expression models.12 In addition, it has been found that IL-1 is not necessarily a dominant cytokine in the acute, inflammatory stages of most arthritis models, but plays a crucial role in the propagation of joint inflammation and concomitant cartilage and bone erosion in collagen arthritis. Transgenic over-expression of IL-1 produced erosive arthritis.17,18
In CIA, it was shown that treatment with a set of neutralizing antibodies against both IL-1a and IL-1β was still highly effective in established arthritis, reducing both inflammation and the progression of cartilage destruction. Studies with antibodies to seperate IL-1 isoforms revealed that IL-1β is more crucial.15,19 This is in line with the clear efficacy in this model of ICE (IL-1β-converting enzyme) inhibitors and the observation of reduced CIA in ICE-deficient mice.20 Similarly, the local overexpression of IL-1ra by retroviral gene transfer in inflamed knee joints was effective at the site.21 In line with the identification of TNFα and IL-1β as separate targets in animal models of arthritis, it has been convincingly demonstrated that combination therapy with both TNF and IL-1 blockers provides optimal protection.22
Role of T Cell Cytokines in Pathology of RA
Rheumatoid arthritis is considered as an Th1-associated disease.23 However, the factors that initiate and sustain Th1 responses in RA synovium are still not identified. The discovery of new cytokines such as IL-15, IL-17 and RANKL have reconsidered the importance of T cells in the pathology of RA.
IL-15
IL-15 shares many biologic activities with the T cell cytokine IL-2. IL-15 is produced in substantial amounts by macrophages and fibroblasts in the rheumatoid synovial membrane.24 It may recruit and activate synovial T cells in the relative absence of IL-2.25 IL-15 induces T cell proliferation, B cell maturation and isotype switching, and may protect T cells from apoptosis.25,26 In addition, IL-15 has novel activity to stimulate the differentiation of osteoclast progenitors into preosteoclasts.27 Blocking endogenous IL-15 by a soluble IL-15 receptor a-chain prevents murine collagen-induced arthritis, indicating a role of IL-15 in development of antigen-induced immunopathology.28 IL-15 recruits and activates CD45RO+ memory T cell subset in the synovial membrane and induces TNFα production in RA.25,26 Interestingly, these T cell subsets are IL-17 producer cells after stimulation and it has been shown that IL-15 triggers IL-17 production in vitro.29
IL-17
IL-17 is a recently discovered cytokine that is secreted by a restricted set of cells, whereas its receptor is ubiquitously expressed on many cell types.30–32 IL-17 production has been demonstrated in RA synovial tissue33 and it enhances IL-1 mediated IL-6 production in vitro.34 The CD4+CD45RO are the major source of IL-17. Th1/Th0, but not Th2 subsets of CD4+ T cell clones isolated from rheumatoid synovium produced IL-17.35 It is not clear whether IL-17 operates downstream of IL-15 and whether IL-17 has a direct role in T cell activation. The contribution of IL-17 in destructive arthritis was suggested by the fact that the cellular responses induced by IL-17 look similar to that of IL-1. Synergistic effects together with IL-1 and TNFα have been shown.36 Recently, adenoviral vector-mediated overexpression of IL-17 in the knee joint of type II collagen immunized mice was shown to promote destructive collagen arthritis (Fig. 2). It induces relatively high levels of IL-1β. Of extreme interest, part of the destructive effect of local overexpression of IL-17 in the knee joints of mice with collagen-induced arthritis seems independent of IL-1.37 Furthermore, amelioration of destructive collagen arthritis was noted after blocking endogenous IL-17 using soluble IL-17 receptor.37 IL-17 could therefore be a novel target for the treatment of destructive arthritis and this may have implications for tissue destruction in other autoimmune diseases as well.

Figure 2
Local IL-17 gene transfer promotes collagen arthritis.
RANKL
T cell IL-17 may be a crucial cytokine for osteoclastic bone resorption in vitro via RANKL expression.38–40 Osteoclasts are potent bone-resorbing cells and RANKL has been shown to be a key regulator of osteoclastogenesis.39 RANKL binds to its receptor, RANK (receptor activator of nuclear factor κB) inducing NFκB activation via TRAF 6.41 The decoy receptor OPG binds with the soluble and cell-bound forms of RANKL and thus prevents their interaction with, and stimulation of, RANK (Fig. 3).42–45 The RANKL/RANK/OPG balance seems of crucial importance in osteoclastogenesis and the bone erosion process during RA.46 Immunohistochemical and in situ hybridization studies have localized RANKL expression to T cells within lymphoid aggregates of inflamed synovial tissues in patients with RA.47–49 RANKL mRNA and protein were also detected in synovial fibroblasts from RA patients,48,49 and these fibroblasts promoted osteoclastogenesis when stimulated with 1,25-dihydroxyvitamin D3. This was mediated by increase in RANKL and a decrease in OPG production and could be abrogated by administration of OPG.49 In CIA, RANKL expression was found in synovial infiltrating mononuclear cells, fibroblast-like cells and chondrocytes.50,51 In vivo it was demonstrated that neutralization of RANKL by daily injections of recombinant OPG completely prevents bone and joint abnormalities in rat adjuvant arthritis, without interfering with the inflammatory process.39 However, recently a RANKL-independent role of TNF in osteoclastogenesis in vitro has been reported. Further studies in vivo are needed to evaluate the relation between the proinflammatory cytokines IL-17, IL-1, TNF and the RANKL/RANK/OPG pathway.
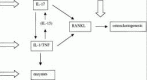
Figure 3
Schematic overview of mediators involved in osteoclastogenesis and bone erosion.
IL-12/IL-18
The production of the pro-inflammatory cytokines IL-1 and TNF is influenced by other cytokines. Disease promoting mediators can on the one hand induce or sustain direct production of IL-1 and TNF or on the other hand propagate arthritis via Th1 immune-stimulatory activity. IL-12 and the novel cytokine IL-18 (and IL-15 see above) have been shown to be potent Th1-driving cytokines, but can also induce the production of TNF and IL-1 in a T cell independent way. Administration of IL-12 during the early onset of collagen-induced arthritis accelerated onset and enhanced severity.52 Blocking endogenous IL-12 during onset using specific antibodies inhibited the onset of CIA, indicating that IL-12 is a pivotal mediator in the expression of CIA. However, continued treatment did not suppress established arthritis. Instead, these mice showed marked exacerbation of arthritis shortly after cessation of anti-IL-12 treatment, implying impairment of endogenous control. Enhanced expression of IL-1β and TNFα was noted in the synovium. Treating established CIA with recombinant mIL-12 suppresses the arthritis. Elevated levels of IL-10 seems responsible for this effect, since the anti-inflammatory effects of IL-12 is reversed by anti-IL-10 treatment. This dual role of IL-12 in early and late stages of CIA needs subtle tuning of IL-12-directed therapy in human arthritis.
Another pivotal cytokine for the development of Th1 responses is the recently discovered proinflammatory cytokine IL-18.53 IL-18 is a member of the IL-1 family of proteins and has been demonstrated in RA synovium.53 Synergistic activity was noted with IL-12 and IL-15 in sustaining both Th1 responses (IFNg) and monokine production in RA.53 Both articular chondrocytes and osteoblasts express IL-18. Mice lacking IL-18 revealed reduced incidence and severity of collagen-induced arthritis.54 This was accompanied by reduced Ag-specific proliferation and pro-inflammatory cytokine (IFNg, TNFα, IL-6 and IL-12) production by spleen and lymph node cells in response to bovine type II collagen in vitro, paralleled in vivo by a significant reduction in serum anti-CII IgG2a Ab level. Interestingly, blockade of endogenous IL-18 in murine streptococcal cell wall-induced arthritis revealed an IFN-γ-independent role of IL-18.55 Significant suppression of local TNFI and IL-1 was found under these conditions, indicating regulation of these proinflammatory cytokines by IL-18. Blocking IL-18 could therefore represent a new therapeutic approach that warrants further testing in the clinic.
Regulation by IL-4/IL-10
Apart from direct interference with TNF and IL-1, regulation of arthritis can also be exerted at the level of modulatory cytokines, such as interleukin-4 (IL-4) and interleukin-10 (IL-10). These regulatory mediators can inhibit Th1 cell activity by suppressing IFN-γ expression. In addition, they may have a direct inhibitory effect on the macrophage activity in the synovium.
Both actions will lead to less IL-1 and TNFα production in the synovium. Moreover, IL-4 and IL-10 may up-regulate natural inhibitors of IL-1 and TNFα, such as IL-1 receptor antagonist (IL-1Ra), soluble TNFα receptor (sTNFαR), and tissue inhibitor of metalloproteinase (TIMP), suggesting surplus value to anti-IL-1/TNFα treatment.
Elevated levels of IL-10 has been shown in the synovial fluid of RA patients. No IL-4 has been found in the synovial fluid of RA patients. In vitro studies have shown that IL-4 and IL-10 regulated the production of IL-1 and TNFα by RA synovial tissue.56–61
IL-10 is a dominant suppressive cytokine in the CIA model.56,62–68 Blocking both IL-4 and IL-10, however, resulted in the best acceleration of CIA onset. Treatment with IL-10 was only marginally effective, with variation probably linked to variable involvement of endogenous IL-10. Low dose of IL-4 alone did not provoke any effect. Pronounced protection against cartilage destruction was only achieved with combination treatment of IL-4 and IL-10. This cooperatieve effect was noted after early treatment but also occurred when treatment was started during full-blown arthritis. The mechanism of protection is linked to suppressed generation of TNFα and IL-1 and up-regulation of the IL-1Ra/IL-1β balance in the synovium and, in particular, in the arthritic cartilage.62 Initial trials with IL-10 were disappointing, and it is expected that in the treatment of RA patients too, IL-10 and IL-4 have to be combined.
IL-4 could not be detected in synovial fluid, synovial supernatants, or synovium of RA patients.23 This lack of IL-4 is likely to contribute to the uneven Th1/Th2 balance and to the chronic nature of RA. Local IL-4 overexpression in the knee joint of type II collagen immunized mice has been shown to enhance the onset and aggravated the synovial inflammation. However, impressive prevention of chondrocyte death and cartilage erosion was noted.69 Chondrocyte proteoglycan synthesis was enhanced in the articular cartilage by local IL-4. Reduction of cartilage erosion was substantiated by lack of expression of the MMP-dependent cartilage proteoglycan breakdown neoepitope VDIPEN in the local IL-4-treated knee joints. The protective effect was associated with a reduction of PMN's in the synovial joint space, decreased NO synthesis, down-regulation of IL-1β and a reduction of the MMP-3/TIMP disbalance in the synovium. Furthermore, IL-4 gene therapy reduced IL-17 and RANKL expression in the synovium and prevents bone erosion.51 This protective effect was associated with decreased formation of osteoclast-like cells and reduced mRNA levels of cysteine proteinase cathepsin K. Interestingly, IL-4 prevented collagen type I breakdown, but enhanced the formation of type I procollagen in bone samples from RA patients, suggesting promotion of tissue repair. These data suggest that therapeutic strategies that enhance local IL-4 production may protect against cartilage and bone destruction in RA (Fig. 4).
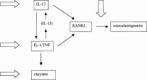
Figure 4
Potential targets of IL-4.
References
- 1.
- Harris E D Jr. Rheumatoid arthritis: Pathophysiology and implications for therapy. N Engl J Med. 1990;322:1277–1289. [PubMed: 2271017]
- 2.
- Miossec P, Chomarat P, Dechanet J. Bypassing the antigen to control rheumatoid arthritis. Immunol Today. 1996;17:170–173. [PubMed: 8871348]
- 3.
- Arend WP. The pathophysiology and treatment of rheumatoid arthritis. Arthritis Rheum. 1997;40:595–597. [PubMed: 9125239]
- 4.
- Moreland LW, Heck L W Jr, Koopman WJ. Biologic agents for treating rheumatoid arthritis. Concepts and progress. Arthritis Rheum. 1997;40:397–409. [PubMed: 9082924]
- 5.
- Müller-Ladner U, Kriegsmann J, Franklin BN. et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;49:1607–1615. [PMC free article: PMC1865262] [PubMed: 8909250]
- 6.
- Franz JK, Pap T, Müller-Ladner U. T cell-independent joint destruction. In: Miossec P, Van den Berg WB, Firestein GS, editors. T Cells in Arthritis. Basel; Birkhäuser Verlag: 1998.
- 7.
- Firestein GS, Alvaro-Garcia JS, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990;144:3347–3353. [PubMed: 2109776]
- 8.
- VandeLoo A A J, VandenBerg WB. Effects of murine recombinant IL-1 on synovial joints in mice: Measurements of patellar cartilage metabolism and joint inflammation. Ann Rheum Dis. 1990;49:238–245. [PMC free article: PMC1004046] [PubMed: 2339905]
- 9.
- Henderson B, Pettipher ER. Arthritogenic actions of recombinant IL-1 and TNF in the rabbit: Evidence for synergistic interactions between cytokines in vivo. Clin Exp Immunol. 1989;75:306–310. [PMC free article: PMC1542106] [PubMed: 2784740]
- 10.
- Keffer J, Probert L, Cazlaris H. et al. Transgenic mice expressing human tumor necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991:4025–4031. [PMC free article: PMC453150] [PubMed: 1721867]
- 11.
- Georgopoulos S, Plows D, Kollias G. Transmembrane TNF is sufficient to induce localized tissue toxicity and chronic inflammatory arthritis in transgenic mice. J Inflamm. 1996;46:86–97. [PubMed: 8734789]
- 12.
- Probert L, Plows D, Kontogeorgos G. et al. The type I IL-1 receptor acts in serie with TNFα to induce arthritis in TNFα transgenic mice. Eur J Immunol. 1995;25:1794–1797. [PubMed: 7615010]
- 13.
- Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–9788. [PMC free article: PMC50217] [PubMed: 1409699]
- 14.
- Wooley PH, Dutcher J, Widmer MB. et al. Influence of a recombinant human soluble tumor necrosis factor receptor FC fusion protein on type II collagen-induced arthritis in mice. J Immunol. 1993;151:6602–6607. [PubMed: 8245488]
- 15.
- Joosten L A B, Helsen M M A, Van de Loo F A J. et al. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice: A comparative study using anti-TNFα, anti-IL-1a/b, and IL-1Ra. Arthritis Rheum. 1996;39:797–809. [PubMed: 8639177]
- 16.
- Mori L, Iselin S, de Libero G. et al. Attenuation of collagen-induced arthritis in 55-kDa TNF receptor type I (TNFRI)-IgGI-treated and TNFRI-deficient mice. J Immunol. 1996;157:3178–3182. [PubMed: 8816431]
- 17.
- Ghivizzani SC, Kang R, Georgescu HI. et al. Constitutive intra-articular expression of human IL-1β following gene transfer to rabbit synovium produces all major pathologies of human rheumatoid arthritis. J Immunol. 1997;159:3604–3612. [PubMed: 9317160]
- 18.
- Niki Y, Yadmada H, Kikuchi T. et al. Membrane associated IL-1 contributes to chronic synovitis in human IL-1a transgenic mice. Arthritis Rheum. 1998;41:S212. [PubMed: 14688369]
- 19.
- Vanden Berg WB, Joosten L A B, Helsen M M A. et al. Amelioration of established murine collagen induced arthritis with anti-IL-1 treatment. Clin Exp Immunol. 1994;95:237–243. [PMC free article: PMC1534928] [PubMed: 8306498]
- 20.
- Ku G, Faust T, Laufer LL. et al. IL-1β converting enzyme inhibition blocks progression of type II collagen induced arthritis in mice. Cytokine. 1996;8:377–386. [PubMed: 8726666]
- 21.
- Bakker AC, Joosten L A B, Arntz OJ. et al. Prevention of murine collagen-induced arthritis in the knee and ipsilateral paw by local expression of human IL-1Ra protein in the knee. Arthritis Rheum. 1997;40:893–900. [PubMed: 9153551]
- 22.
- Vanden Berg WB. Anti-cytokine therapy in chronic destructive arthritis. Arthritis Res. 2001;3:18–26. [PMC free article: PMC128880] [PubMed: 11178124]
- 23.
- Miossec P, van den Berg WB. Th1/Th2 cytokine balance in arthritis. Arthritis Rheum. 1997;40:2105–2115. [PubMed: 9416846]
- 24.
- McInnes IB, Leung BP, Stuock RD. et al. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-a production in rheumatoidrthritis. Nature Med. 1997;3:189–195. [PubMed: 9018238]
- 25.
- McInnes IB, Al-Mughales J, Field M. et al. The role of interleukin-15 in T-cell migration and activation in rheumatoid arthritis. Nat Med. 1996;2:175–182. [PubMed: 8574962]
- 26.
- McInnes IB, Liew FY. Interleukin-15: A proinflammatory role in rheumatoid arthritis synovitis. Immunol Today. 1998;19:75–79. [PubMed: 9509762]
- 27.
- Ogata Y, Kukita A, Kukita T. et al. A novel role of IL-15 in the development of osteoclasts: Inability to replace its activity with IL-2. J Immunol. 1999;162:2754–2760. [PubMed: 10072521]
- 28.
- Ruchatz H, Leung BP, Wei X. et al. Soluble IL-15 receptor a-chain administration prevents murine collagen-induced arthritis: A role for IL-15 in development of antigen-induced immunopathology. J Immunol. 1998;160:5654–5660. [PubMed: 9605172]
- 29.
- Ziolkowska M, Koch A, Luszczykiewics G. et al. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–2838. [PubMed: 10679127]
- 30.
- Fossiez F, Djossou O, Chomarat P. et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. [PMC free article: PMC2192621] [PubMed: 8676080]
- 31.
- Yao Z, Painter SL, Fanslow WC. et al. Human IL-17: A novel cytokine derived from T cells. J Immunol. 1995;155:5483–5486. [PubMed: 7499828]
- 32.
- Yao Z, Fanslow WC, Seldin MF. et al. Herpesvirus Saimiri encodes a new cytokine IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811. [PubMed: 8777726]
- 33.
- Chabaud M, Durand JM, Buchs N. et al. Human interleukin-17.A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963. [PubMed: 10323452]
- 34.
- Chabaud M, Fossiez F, Taupin JL. et al. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409. [PubMed: 9647250]
- 35.
- Aarvak T, Chabaud M, Miossec P. et al. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246. [PubMed: 9973376]
- 36.
- Chabaud M, Lubberts E, Joosten L. et al. IL-17 derived from juxta-articular bone and synovium ontributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001;3:168–177. [PMC free article: PMC30709] [PubMed: 11299057]
- 37.
- Lubberts E, Joosten L A B, Oppers B. et al. IL-1 independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–1013. [PubMed: 11441109]
- 38.
- Kotake S, Udagawa N, Takahashi N. et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345. [PMC free article: PMC408356] [PubMed: 10225978]
- 39.
- Kong YY, Feige U, Sarosi I. et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304. [PubMed: 10580503]
- 40.
- van Bezooijen RL, Farih-Sips H C M, Papapoulos SE. et al. Interleukin-17: A new bone acting cytokine in vitro. J Bone Miner Res. 1999;14:1513–1521. [PubMed: 10469279]
- 41.
- Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin-17 signal transduction. J Exp Med. 2000;191:1233. [PMC free article: PMC2193168] [PubMed: 10748240]
- 42.
- Quinn J M W, Elliott J, Gillespie MT. et al. A combination of osteoclast differentiation factor and macrophage-colony stimulating factor is sufficient for both human and mouse osteoclat formation in vitro. Endocrinology. 1998;139:4424–4427. [PubMed: 9751528]
- 43.
- Fuller K, Wong B, Fox S. et al. TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med. 1998;188:997–1001. [PMC free article: PMC2213394] [PubMed: 9730902]
- 44.
- Burgess TL, Qian Y, Kaufman S. et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J Cell Biol. 1999;145:527–538. [PMC free article: PMC2185088] [PubMed: 10225954]
- 45.
- Jimi E, Akiyama S, Tsurukai T. et al. Osteoclast differentiation factor acts as a multifunctional regulator in murine osteoclast differentiation and function. J Immunol. 1999;163:434–442. [PubMed: 10384146]
- 46.
- Hofbauer LC, Heufelder AE. The role of osteoprotegerin and receptor activator of nuclear factor κB ligand in the pathogenesisand treatment of rheumatoid arthritis. Arthritis Rheum. 2001;44:253–259. [PubMed: 11229454]
- 47.
- Horwood NJ, Kartsogiannis V, Quinn J M W. et al. Activated T cells support osteoclast formation in vitro. Biochem Biophys Res Commun. 1999;265:144–150. [PubMed: 10548505]
- 48.
- Gravallese EM, Manning C, Tsay A. et al. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250–258. [PubMed: 10693863]
- 49.
- Takayanagi H, Iizuka H, Juji T. et al. Involvement of receptor activator of nuclear factor κB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000;43:259–269. [PubMed: 10693864]
- 50.
- Romas E, Bakharevski O, Hards DK. et al. Expression of osteoclast differentiation factor at sites of bone erosion in collagen-induced arthritis. Arthritis Rheum. 2000;43:821–826. [PubMed: 10765926]
- 51.
- Lubberts E, Joosten L A B, Chabaud M. et al. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000;105:1697–1710. [PMC free article: PMC378501] [PubMed: 10862785]
- 52.
- Joosten L A B, Lubberts E, Helsen M M A. et al. Dual role of IL-12 in early and late stages of murine collagen type II arthritis. J Immunol. 1997;159:4094–4102. [PubMed: 9379000]
- 53.
- Gracie AJ, Forsey RJ, Chan WL. et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest. 1999;104:1393. [PMC free article: PMC409841] [PubMed: 10562301]
- 54.
- Wei XQ, Leung BP, Arthur H M L. et al. Reduced incidence and severity of collagen-induced arthritis in mice lacking IL-18. J Immunol. 2001;166:517–521. [PubMed: 11123331]
- 55.
- Joosten L A B, Van de Loo F A J, Lubberts E. et al. An IFN-g-independent proinflammatory role of IL-18 in murine streptococcal cell wall arthritis. J Immunol. 2000;165:6553–6558. [PubMed: 11086098]
- 56.
- Katsikis PD, Chu C -Q, Brennan FM. et al. Immuno regulatory role of interleukin-10 (IL-10) in rheumatoid arthritis. J Exp Med. 1994;179:1517–1527. [PMC free article: PMC2191503] [PubMed: 8163935]
- 57.
- Llorente L, Richaud-Patin Y, Kior R. et al. In vivo production of interleukin-10 by non-T cells in rheumatoid arthritis, Sjögren's syndrome, and systemic lupus erythematosus. A potential mechanism of B lymphocyte hyperactivity and autoimmunity. Arthritis Rheum. 1994;37:1647–1655. [PubMed: 7980676]
- 58.
- Cush JJ, Splawski JB, Thomas R. et al. Elevated interleukin-10 levels in patient with rheumatoid arthritis. Arthritis Rheum. 1995;38:96–104. [PubMed: 7818579]
- 59.
- Cohen S B A, Katsikis PD, Chu C -Q. et al. High level of interleukin-10 production by the activated T cell population within the rheumatoid synovial membrane. Arthritis Rheum. 1995;38:946–952. [PubMed: 7612044]
- 60.
- Chomarat P, Vannier E, Dechanet J. et al. Balance of IL-1 receptor antagonist/IL-1J in rheumatoid synovium and its regulation by IL-4 and IL-10. J Immunol. 1995;154:1432–1439. [PubMed: 7822808]
- 61.
- Isomäki P, Luukkainen R, Saario R. et al. Interleukin-10 functions as an anti-inflammatory cytokine in rheumatoid synovium. Arthritis Rheum. 1996;39:386–395. [PubMed: 8607887]
- 62.
- Joosten L A B, Lubberts E, Durez P. et al. Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum. 1997;40:249–260. [PubMed: 9041936]
- 63.
- Lubberts E, Joosten L A B, Helsen M M A. et al. Regulatory role of interleukin-10 in joint inflammation and cartilage dstruction in murine streptococcal cell wallarthritis.More therapeutic benefit with IL-4/IL-10 combination therapy than with IL-10 treatment alone. Cytokine. 1998;10:361–369. [PubMed: 9619374]
- 64.
- Lubberts E, Joosten L A B, Van den Bersselaar L. et al. Intra-articular IL-10 gene transfer regulates the expression of collagen-induced arthritis in the knee and ipsilateral paw. Clin Exp Immunol. 2000;120:375–383. [PMC free article: PMC1905648] [PubMed: 10792391]
- 65.
- Walmsley M, Katsikis PD, Abney E. et al. Interleukin-10 inhibition of the progression of established collagen-induced arthritis. Arthritis Rheum. 1996;39:495–503. [PubMed: 8607899]
- 66.
- Apparaily F, Verwaerde C, Jacquet C. et al. Adenovirus-mediated transfer of viral IL-10 gene inhibits murine collagen-induced arthritis. J Immunol. 1998;160:5213–5220. [PubMed: 9605116]
- 67.
- Ma Y, Thornton S, Duwel LE. et al. Inhibition of collagen-induced arthritis in mice by viral IL-10 gene transfer. J Immunol. 1998;161:1516–1524. [PubMed: 9686619]
- 68.
- Whalen JD, Lechman El, Carlos CA. et al. Adenoviral transfer of the viral IL-10 gene periarticularly to mouse paws suppresses development of collagen-induced arthritis in both injected and uninjected paws. J Immunol. 1999;162:3625–3632. [PubMed: 10092823]
- 69.
- Lubberts E, Joosten L A B, Van den Bersselaar L. et al. Adenoviral vector-mediated overexpression of IL-4 in the knee joints of mice with collagen-induced arthritis prevents cartilage destruction. J Immunol. 1999;163:4546–4556. [PubMed: 10510398]
- Cytokines in the Pathogenesis of Rheumatoid Arthritis and Collagen-Induced Arthr...Cytokines in the Pathogenesis of Rheumatoid Arthritis and Collagen-Induced Arthritis - Madame Curie Bioscience Database
- The Evolution of CDK-Activating Kinases - Madame Curie Bioscience DatabaseThe Evolution of CDK-Activating Kinases - Madame Curie Bioscience Database
- Cell Cycle Reactivation in Skeletal Muscle and Other Terminally Differentiated C...Cell Cycle Reactivation in Skeletal Muscle and Other Terminally Differentiated Cells - Madame Curie Bioscience Database
- A Conceptual Framework - Madame Curie Bioscience DatabaseA Conceptual Framework - Madame Curie Bioscience Database
- Biocompatibility of Polyurethanes - Madame Curie Bioscience DatabaseBiocompatibility of Polyurethanes - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...