

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
Readers and authors of this book alike are the allies of scientists and scientists-to-be in the fight against one of our most common and mightiest enemies, Alzheimer's disease (AD), which deprives individuals of their basic human dignity after decades of (generally) respectful lives with families and friends. As a professional scientist, I am personally grateful to be able to live in the present time when scientists from different ethnic groups, some of which fought against each other in a brutal manner in the past, can now work together to fight the real common enemy through friendly collaborations or in open competition under hopefully true democracies.
However, as authors, we would like readers to know that we do not always share entirely identical ideas or hypotheses until such times as everyone reaches a relevant consensus concerning different aspects of science. We actually need to critically evaluate each other's work for the advancement of science while at the same time maintaining good human relationships (see, for instance, the open debate between Berislav Zlokovic, one of the authors who kindly contributed to this book, and myself).1 In this respect, not all the chapters here are necessarily consistent with each other, reflecting the differences in opinions. The AD research community is still in the process of proposing different potentially beneficial strategies for the development of preventive and therapeutic measures to combat AD. The ultimate proof of the relevance of any hypothesis or of any experimental results will be real clinical success in a practical manner. Therefore, some of the seemingly relevant strategies are undergoing or will undergo a form of natural selection, such that those, in which the merits outweigh the demerits, will eventually remain. The rest of this book will demonstrate that we are getting closer and closer to clinical success in an accelerated manner, particularly since around the end of the 1980s. The discoveries of gene mutations that cause familial AD (FAD) in the amyloid precursor protein (APP) gene and presenilin genes were the most significant milestones in the 1990s (see section Etiology of AD in this chapter and Chapters 2 to 5 for details). (For the nonspecialists, the major primary key abbreviations often used in this book are listed in Table 1.1).
Table 1
Major primary abbreviations used in this book.
In any case, I certainly hope that this “scientific globalization,” in a positive sense, sharing scientific achievements as cultural common human properties, will be even more improved in the near future because the disease generally does not distinguish between different ethnic groups and because the number of patients in the world will keep growing. Of note is the fact that aging is the strongest risk factor for AD2 (see section Towards the Scientific Control of Brain Aging).
For instance, mainland China, with a population of more than one billion people, does not seem to have as high an incidence of AD patients as might be expected from such a large population because the average life span there has been much shorter than in what politicians call more developed countries. However, if China maintains its current rate of industrial and economical growth, this country with its “one-child-per-family” policy, will have not only the largest number but also the highest population ratio of patients with AD and other aging-associated disorders within decades. Unless we do something substantial for the prevention and therapy of these diseases, this scenario would not only result in millions of tragic situations within families but could even induce political destabilization and threaten the peace in some areas, particularly in East Asia, as a result of serious recessions that would be caused by the unexpectedly heavy economic burdens of caring for such people. BUT, if our research efforts can contribute to helping people over the age of 60 stay healthier both physically and intellectually, then society will profit from more active participation of the elderly. This will have benefits for younger generations by reducing the inherent burdens on social welfare systems and on individual care giving, leading to more stable political environments.
The same logic could also apply to other regions of the world. In fact, it is generally established among economists that one major factor which caused recessions in the US and Western Europe in the 1970s to 80s and in Japan in 90s was the change in the ratio of those who needed to be supported economically (and medically) to those who had to support them. (Japan is still struggling with the transition processes even in the 21st century.) Therefore, all of us are responsible not only for the advancement of science but also for the future of human kind. Personally, I wish I could also do something about the other neurodegenerative disorder, Parkinson's disease, too, because my American mother, Dorothy J. Comings, with whom I stayed for one year as a high school exchange student from 1976 to 1977, is suffering from this disorder.
Etiology of AD
AD is the major cause of senile dementia in the present world. The estimated number of patients is approximately 20 million worldwide and is expected to keep growing as the world population ages. Now that Mild cognitive impairment3,4 (MCI), a condition characterized by a significantly reduced memory with cognition being within a normal range, is considered as a prodromal form of dementing disorders primarily represented by AD, the actual number of people being affected by AD pathology is probably much greater than 20 million. Our general understanding is that at the age of 85 one out of every two people is affected either by AD or MCI. In fact, if we consider the temporal distance of decades between the cause and effect in AD pathogenesis, it is possible that some aspects of what has been considered as being part of “normal” brain aging may be prodromal to MCI-associated conditions. (Note that even the autosomal dominantly inherited gene mutations that cause aggressive early-onset forms of AD (see the later part of this section) require at least 30-60 years before definitive clinical diagnosis of the onset of the disease can be made.)
Thus, to understand the etiology and mechanism of AD is important not only in conquering this cruel disease but also in realizing the historical dream of human beings to control brain aging (see section Towards the Scientific Control of Brain Aging). Figure 1.1 shows a simplified history of AD research since its initial scientific description by Alois Alzheimer in Germany in 1906 (published in 1907).5 Pathological and patho-biochemical studies mostly up to 1990 established the chronology of the major pathological events at least in neocortex (Fig. 1.2) and identified the molecules comprising the pathological structures. Senile plaques, present extracellularly, consist mainly of amyloid β (Αβ) peptide (Fig. 1.3) while neurofibrillary tangles (NFTs), present intracellularly, contain primarily tau protein. The research community seems to be reaching a consensus that these pathological structures as they appear may not be the direct causes of the symptoms, but rather, that the processes, not fully identified yet, that lead to the pathology may be essential in the pathogenesis.
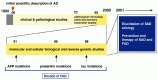


In any case, as mentioned above, the temporal distance of decades between the cause and effect has been the most challenging factor in AD research. The chronology of pathological events alone does not establish any cause-and-effect relationship. AD research resembles that of archaeology in that researchers need to collect a large body of consistent circumstantial evidence to form a consensus. The observations of pathological structures such as senile plaques and neurofibrillary tangles in AD brain, for instance, have always engendered the arguments as to whether these structures represent pathologically essential and significant pathways or just by-products or consequences of something else essential.
In this respect, identification of the FAD- and tauopathy (FTDP-17: fronto-temporal dementia associated with chromosome 17) causing gene mutations and analyses of their phenotypes in the 1990s have played a major indispensable role in resolving the etiology of AD.6-8 Consensus has now been reached in that the decades-long cascade leading to dementia is initiated by the deposition of amyloid-β peptide (Αβ) in the brain and that tauopathy is likely to play a major role in the neurodegenerative processes. Thus, the 1990s are called the ‘decade of FAD.’ I surely hope that the first 10 years of the 2000s will be a decade of SAD, which represents the vast majority of all AD cases.
More than 100 mutations that cause FAD6,7 have been identified in the three genes encoding the proteins, APP, presenilin 1, and presenilin 2, involved in Αβ generation as shown in Figure 1.4. (See Chapters 2–5 for more details.) A number of papers (probably several hundred) in the 1990s described the phenotype caused by these mutations. Some papers examined the effects of the mutations on Αβ production and others examined effects on cytoskeletal abnormalities involving tau protein, cell death or apoptosis, endoplasmic reticulum (ER)-stresses, etc. These studies have established a consensus that the only phenotype shared by essentially all the mutations in vitro (in cell culture), in vivo (in transgenic and knock-in (KI) mice), and in patients is the increased production of a specific species of Αβ, Αβ1-42, which is much more hydrophobic and fibrillogenic than the other major species, Αβ1-40. These results have strongly suggested that Αβ1-42 is the primary pathogenic agent in the AD cascade. Figure 1.4 shows the processing of APP. Typically, most of the presenilin 1 mutations, accounting for more than 70% of all FAD mutations thus far identified, cause very aggressive presenile Αβ amyoidosis in humans by increasing the steady-state Αβ1-42 level approximately 1.5-fold, as detected in the brains of mutant presenilin 1 transgenic or KI mice.9,10 (See also section on Towards the Scientific Control of Brain Aging).
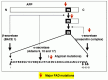
Atypical mutations in the intra-Αβ sequences of APP, such as the Dutch, Flemish, Italian, and Arctic mutations,6 have also been identified. Most of these mutations result in hemorrhages or strokes caused by unusually severe cerebral Αβ amyloid angiopathy (CAA) that also accompanies the presenile parenchymal Αβ deposition. A number of test-tube experiments have demonstrated that these mutations promote Αβ aggregation by altering the peptide conformation.11-13 Therefore, these rather atypical mutations are generally believed to cause Αβ accumulation through augmenting aggregation11,12 or protofibril formation.13 We have recently found that these mutations also cause Αβ to be more resistant to degradation by a physiologically relevant peptidase, neprilysin described in Chapter 6 (Tsubuki S, Takaki Y, Saido TC, submitted for publication). Therefore, these mutations may exert dual pathogenic effects associated not only with aggregation/protofibril formation but also with one major aspect of metabolism, proteolytic degradation.
Αβ versus Tau
Until some years ago, there used to be arguments between “Baptists” who believed that Αβ was more important and “Tauists” who believed that tau was more important in terms of their contributions to the AD pathogenesis. However, those familiar with the major publications on AD and related disorders in the late 1990s (see the previous section and also Chapters 2–4) do not participate in this kind of discussion any more. I believe that most AD researchers would agree with my view that both are likely to be equally important, particularly in clinical terms. In the AD pathological cascade, it now seems to be just that Αβ is more closely associated with the primary cause while tau is closer to the consequences, such as neurodegeneration. The importance of tau in AD pathogenesis is also apparent from the fact that the quantities of tau accumulated in AD brains are much larger than those in the brains of other neurodegenerative disorders accompanying tauopathy (Taniguchi and Hasegawa, personal communication). Therefore, tau may be a better target for improving the symptoms of patients in a clinically pragmatic manner. Not knowledgeable enough in this specific subject, I will leave this issue to such well-known specialists as J.P. Brion, P. Davies, A. Delacourte, M.. Goedert, M. Hasegawa, Y. Ihara, K. Iqbal, V. Lee, Eva & Eckhard Mandelkow, M. Morishima-Kawashima, A. Takashima, R. Terry, J. Trojanowski, C.M. Wischik, the late H.M. Wisniewski (names in alphabetical order), and others (just net-surf for the reviews and articles under these names), and instead just present my current personal views as follows.
The primary question that is yet to be answered in the domain of tau research is the role of phosphorylation; the tau proteins accumulated in NFT are heavily phosphorylated.14,15 Although this is an issue beyond the scope of this book on Αβ metabolism, the observations demonstrating that the PHF tau proteins in transgenic mice overexpressing FTDP-17 mutation-carrying tau are also highly phosphorylated16-18 provide some insights. They seem to logically imply to me that phosphorylation is more likely to be a consequence, possibly of neurons struggling to protect themselves, rather than a cause of NFT formation, unless the mutations exert their effects through altering the phosphorylation/dephosphorylation status of tau, which has never been demonstrated to my knowledge.
Αβ Metabolism: Three Major Targets
Another very important finding in the 1990s is that Αβ is a physiological peptide secreted from neurons under normal conditions both in vitro and in vivo.19-21 Besides, Αβ does not appear to play a major physiological role; the apparent role of APP processing by the α- and β-secretases is the release of soluble forms of APP, APPs, known for its neuroprotective and neurotrophic functions.2224
Besides, splice variants containing the insert sequences corresponding to the Kunitz-type protease inhibitor (KPI) domain have been identified as protease nexin II, an endogenous inhibitor against a group of serine proteases including thrombin.25 The other possible function may be the release of a cytoplasmic fragment, generated by γ-secretase, which may translocate to the nucleus and play a regulatory role in transcription in a manner similar to the cleavage of Notch by the β-secretase activity (See Chapter 5).26,27 Thus far, several substrates for γ-secretase have been identified. In any of the known activities, the fragment that corresponds to Αβ in APP does not seem to play any major role. Therefore, at present it is most likely that Αβ is simply an unwanted by-product of APP processing.
In any case, APP processing occurs constitutively in the brains of both young and old, and, at least in the brains of young and healthy individuals, no Αβ deposition takes place. Taken all together, these observations clearly indicate that Αβ is constantly anabolized and rapidly catalyzed before being deposited under normal conditions. This catabolism can take place inside the brain or in the circulatory system after transport out of the brain. The kinetic relationships between these three metabolic processes are schematized in Figure 1.5. (Αβ40 is left out for the sake of simplicity.)
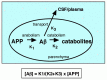
K1, K2, and, K3 are the rate constants for production, in-parenchyma degradation, and out-of-brain transport of Αβ, respectively. Under the assumptions that the kinetics of the reactions can essentially be analyzed linearly, that these rate constants are independent of each other, and that these processes exist in steady-state equilibrium (see one of the previous reviews for details regarding these assumptions),28 the relationship between the amounts of Αβ42 and APP, represented as [Αβ42] and [APP], respectively, can be expressed by the following equation.
[Αβ42] = K1/(K2 + K3) × [APP] (Formula 1.1)
This is based on the following differential equation (See again ref. 31 for details).
d[Αβ42]/dt = K1 × [APP] [K2 + K3] × [Αβ42] = 0 (Formula 1.2)
A measure of time is expressed as “t” in Formula 1.2. Formula 1.1 is consistent with the phenotypes of almost all the FAD mutations in APP and presenilin 1 genes; K1 is approximately 1.5-fold greater than that in normal controls, meaning that [Αβ42] also becomes 1.5-fold greater. It also is consistent with one of the phenotypes of Down's syndrome caused by trisomy of chromosome 21 carrying the APP gene; [APP] is 1.5-fold greater than in normal controls and [Αβ42] also becomes 1.5-fold greater.
Therefore, an increase of K1 (production) or decreases in K2 (in-parenchyma degradation) and K3 (out-of-brain transport) can elevate [Αβ42] and thus be causal of pathological Αβ deposition. This logic also indicates that down-regulation of K1 (production) or up-regulation of K2 (in-parenchyma-degradation) and K3 (out-of-brain transport) can decelerate Αβ deposition in the brain and thus will be effective in the prevention and therapy of AD if indeed Αβ plays a primary pathogenic role. Note that the activation of α-secretase(s) would also contribute to reducing K1 (production) (Chapter 3). The current status of these strategies to achieve the goal of preventing Αβ accumulation is schematized in Figure 1.6. Within the scope of this book, Chapters 1-5 thus focus on the production, 6 and 7 on the inparenchyma degradation, and 10-12 on the out-of-brain transport. Chapters 9 and 10 describe the possible role of the lipid raft in parenchymal Αβ metabolism and also in pathological Αβ accumulation. Chapter 12 refers to the cellular mechanism of Αβ clearance and to the inflammatory processes associated with Αβ vaccination.
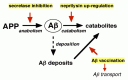
Incidentally, there have been some discussions regarding the relative importance of K2 (in-parenchyma degradation) versus K3 (out-of-brain transport) in Αβ clearance.1 If K2 is excessively greater than K3, Formula 1.1 would practically simplify to [Αβ42]3 K1/K2 × [APP], whereas, if K2 is excessively smaller than K3, it would be [Αβ42]3 K1/K3 × [APP].
I predict a bright future for the control of Αβ levels in the brain through the pursuit of these pathways. Actual approaches and future approaches, based on these strategies, which will have to survive ‘natural selection’ in a clinical sense (i.e., successes in clinical trials) will be optimally combined so that we will be able to control the Αβ levels in our brains in a manner similar to that of the "cocktail therapy" employed for the treatment of acquired immunodeficiency syndrome (AIDS).29 In this latter treatment protocol used worldwide, a cocktail of three different antihuman immunodeficiency virus (HIV) strategies suppresses disease development, whereas use of one or two of the three agents generally is ineffective. Moreover, a combination of these anti-Αβ strategies with other strategies such as that targeted at tauopathy will make future prevention and therapy even more promising.
Questions Regarding the Mechanisms of the Cascade of Αβ-Initiated Pathology
One currently unresolved issue is the elucidation of the precise mechanism(s), by which Αβ deposition causes subsequent pathological processes, i.e., tauopathy, dysfunction and degeneration of neurons. This will allow possible opportunities for therapeutic interventions at various time points between Αβ deposition and neurodegeneration. The accelerating effect of excessive amounts of Αβ on tauopathy demonstrated in mouse models30,31 would indicate the presence of something unknown that relays pathological signals from the former to the latter.
However, perhaps the most important and fundamental question yet to be answered is why Αβ is deposited in SAD which accounts for 99% or more of all AD cases32 (See Table 1.2). It should be noted that the number of SAD patients will grow as the average life span increases, whereas the number of FAD patients simply remains proportional to the total population. Because the up-regulation of Αβ production (i.e., increases in [APP] or K1 in formula 1.1.) is rarely observed prior to the pathological Αβ deposition upon aging, a decrease in the in-parenchyma degradation (K2) or in the out-of-brain transport (K3) (or both) is a logical candidate for the primary cause of the majority SAD cases, the expanding burden of aging populations.
Table 2
Estimated numbers of FAD and SAD patients in Japan.
Animal Models of AD: The Issues to Be Further Addressed
I would like to point out some precautions that we need to bear in mind in relying on research results stemming from the presently available animal models for AD. All the presently available, widely accepted Αβ amyloidosis mouse models are transgenic mice that highly overexpress human APP. Although they do reconstitute some of the pathological features of AD (other than Αβ amyloidosis) including synaptic dysfunction and degeneration, dystrophic neurites, inflammatory responses involving activated astrocytes and microglia, and behavioral abnormalities, none of them show the major pathological hallmarks that are actually essential in defining AD: tauopathy and neurodegeneration. The accelerated tauopathy described in the published literature concerning the double APP/tau-transgenic mice31 is in a sense a matter of course and may simply depend on the high-level expression; even co-overexpression of other proteins such as bovine serum albumin (BSA) may also exert a similar effect if the expressed amount is extremely large. In addition, while the synaptic dysfunction or cognitive deficit has been shown to precede Αβ deposition in the mouse models,33-35 the AD symptoms become apparent many years after the initial Αβ deposition in humans. Before this deposition, there is little or no apparent sign of synaptic dysfunction in human brains even in those carrying FAD mutations. If it is the “soluble” Αβ oligomers, rather than deposits, that are causing the dysfunction in mice,36,37 they should be detectable in a well-defined and measurable form that correlates with synaptic dysfunction. To date, this has not been demonstrated to the best of my knowledge. There can be a number of possible reasons accounting for the lack of tauopathy and neurodegeneration in mice as follows.
- It may be just a matter of time. Mice only live up to three years of age at most, whereas even the most aggressive form of FAD takes at least a couple of decades to present. However, the assumption is not consistent with the fact that APP-transgenic mice accumulate as much Αβ as AD patients do,37 if Αβ plays the major role in AD pathogenesis.
- It may be a matter concerning the primary structure of Ab. In human AD cases, in which the majority (approximately 90%) of both soluble and insoluble Αβ is N-truncated, the most abundant species being Αβ3(pE: pyroglutamate)42.37-39 In contrast, the majority of Αβ accumulated in the mice overexpressing APP is full-length Αβ1-40/42.37 The former species differs from the latter by lacking one positive and two negative charges, thus making it more hydrophobic at the N-terminus than the latter, and thus could be more pathogenic, in a manner analogous to Αβ42 versus Αβ40.38,40 Although overexpression of an APP mutant that produces Αβ3(pE: pyroglutamate)-42 in primary neurons does not seem to have any significant neurodegenerative effect,41 I believe that the effect must be examined under relevant in vivo conditions.
- The extent of APP overexpression is unphysiologically high, being several-fold to several tens of times greater than nontransgenic controls. What could one expect if other secretase substrates such as Notch are overexpressed? Most would expect to observe enhanced Notch signaling with an increased release of Notch intracellular domain (NICD); very few would expect any physiological or pathological effect of the overproduced fragment that corresponds to Αβ in APP. There is no reason to reject the same logic from being applied to the APP overexpression paradigm. Indeed, the NICD counterpart fragment of APP, APP-ICD or AICD, has been shown to play a similar transcription-associated role,26,27 which could be unphysiologically enhanced by APP overexpression. Besides, the primary role of APP processing has been well known for a long time, i.e., release of soluble forms of APP, APPs, which plays neurotrophic and neuroprotective roles22,23 and also inhibits proteinases such as matrix metalloproteinase (MMP) and a group of serine proteinases including thrombin.25,42 The presence of too much APPs may account for the absence of neurodegeneration in these mice.
- The exaggerated overexpression of APP may also affect the way that APP and other proteins are metabolized. For instance, proteins including Αβ, which are axonally transported to presynapses under normal conditions, may instead undergo endosomal-lysosomal metabolism mainly in the soma. Consistently, the relative increases in the Αβ levels (ratios to the endogenous Αβ) are much smaller than those in APP levels in all of the transgenic mice in my knowledge.
- Depending on the promoter to drive the expression of transgenic APP, the effects may rather be overly artificial. Neurons are primarily categorized into two major groups, excitatory neurons and inhibitory neurons, and each group is further categorized into a number of subgroups. APP mRNA is not evenly expressed in every neuron and those expressing platelet-derived growth factor (PDGF), prion protein, and thy1 antigen mRNAs do not probably express the transgene-derived APP in a manner identical to that of the endogenous APP.
- Humans and mice differ from each other in many ways. A good example is the complement system.43 Indeed, T. Wyss-Coray demonstrated that inhibition of the complement system led to increased Αβ deposition and occurrence of neurodegeneration.44 We may thus need to humanize the mice in logically and physiologically relevant manners.
These and other possible reasons and the lack of NFT and apparent neurodegeneration strongly indicate that the animal models that we have been employing are far from the real model of AD. Therefore, it probably is safe to point out the intrinsic limitation of the use of the present mouse models. Such a careful statement would be even more highly evaluated in the future if, for instance, all vaccination-related data unfortunately turned out to be artifacts that can be observed only in the APP-transgenic mice but not in humans. We need to come up with much more improved models that demonstrate all the major pathological features of AD with minimum artificial manipulation(s). Besides, behind the scenes of the most transgenic studies, one has to make tens, sometimes nearly a hundred, of different lines of mice and choose the ones that show the pathological features seemingly worth publishing. Presence of such processes does not seem to be as scientific as standard science should be because involvement of researchers' wishful thoughts is difficult to fully exclude.
From time to time, the cutting-edge techniques in science keep arising and being refined. Now may be the time when AD researchers fully recognize the consensus of basic cell biologists that overexpression paradigms are often unphysiological and can produce artifacts that may go unnoticed unless there are definitive physiologically relevant controls. Without very careful and logical interpretation and proper controls, the entire research community, led by a relatively small number of influential scientists, could head off in the wrong direction. To perform good science is to discover or to create something new that human beings have never known, not just to put the trendy key words of the era together.
In fact, the term “neurodegeneration” itself is to be outdated for the frontrunners of neuroscience, being too general without specifying which of so many types of neurons undergo neurodegeneration, in what chronology, and through what mechanisms it is preceded by neuronal dysfunction. There already are invisible competitions among frontrunners of neuroscience to be the first to establish each interneuron type-targeted manipulation of specific gene expression for understanding the role of each gene product in both chronological and spatial terms in a relevant in vivo manner.
Moreover, I predict that we will need to introduce the concepts, facts, and methods of Systems Neuroscience to fully understand how mainstream pathological events in the AD cascade lead to the actual symptoms, i.e., loss of memory followed by abnormal reduction of cognition often accompanying psychiatric symptoms such as unusual aggressiveness. I also predict that human genetics to search for genetic risk factors will need to be further developed by establishing more mathematically refined methodology (see Chapter 6.7. for the actual proposal) although I am no specialist in this specific subject.
Towards the Scientific Control of Brain Aging
Recent studies indicate that virtually all humans start to accumulate Αβ in the brain upon aging.4750 This suggests that Αβ deposition is an important factor that could determine the life span of our brain and that an AD cascade is initiated by the normal aging process; humans whose “brain life span” is shorter than their “body life span” will suffer from AD in the later stages of their lives. AD has even been described as an “ultimate form of brain aging.” This idea provides us with the hope that a large portion of AD cases can be prevented if we can make the brain life span longer than the body life span by reducing Αβ deposition. As outlined in Figure 1.7, most humans start to accumulate Αβ between the ages of 40 and 80.50–52 This accumulation is initially slow but, after a certain point, deposition begins to accelerate exponentially. Typically, the amounts of Αβ42 in the brains of AD patients are 1,000 to 10,000 times higher than in young normal controls. The catastrophic increase implies the presence of multiple positive-feedback vicious cycles originally caused by a slight increase of the steady-state Αβ levels in the brain. Therefore, the profiles of Αβ accumulation in FAD and Down's syndrome cases being presenile can be explained by considering the original curve for SAD is shifted to the left as shown in Figure 1.8.

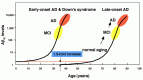
I am optimistic in a sense that our research efforts will eventually make it possible to control the Αβ levels in our brains. If we can do so in the early stages of AD development, before the massive neurodegeneration takes place, it will serve as a postsymptomatic therapy (Figure 1.7). If it becomes possible to prediagnose the MCI prodromal to AD, we will be able to initiate a presymptomatic intervention. Because the conversion of what has been interpreted as “normal aging” to AD via MCI appears as a continuous process primarily caused by the gradually accelerating accumulation of Ab, we may even become able to partially control some very significant aspects of brain aging by maintaining low Αβ levels throughout our lives.
Final Comments
Before closing, I need to inform the readers that I did not succeed in obtaining chapters from some outstanding scientists whose views may differ from some of those included in this book. However, I and other authors have tried to acknowledge their papers and perspectives so that the book covers all the major aspects of Αβ metabolism and adequately represents divergent views of respected researchers. The authors would be more than happy if this book contributes to Alzheimer research by imparting not only the accepted facts, but some of the cutting edge excitement of scientific controversy and inspiring new investigators, particularly young people, to further achievement.
I would like thank all the authors, the very top (relatively young and very active) scientists in the world, and Ronald G. Landes, Landes Bioscience, who led me to edit this book, helped me in many ways. I am very much grateful for Gregory Cole, UCLA, John Trojanowski, University of Pennsylvania, Cynthia Lemere, Harvard Medical School, David Holtzman, Washington University School of Medicine, Maho Morishima-Kawashima, University of Tokyo, and Yasuo Ihara, University of Tokyo, for critical reading of the chapter. I also thank Paul Robbins, University of Pittsburgh, for productive comments on the Introduction. (For the acknowledgements for my colleagues, collaborators, etc., see Chapter 6.) I myself have learned a lot through all the editorial procedures, although this book will probably be the last book that I will be editing for many years. Finally, responsibility for all the statements made in this chapter rests with TCS, not with the other authors of this book.
References
- 1.
- Zlokovic BV, Yamada S, Holtzman D. et al. Clearance of amyloid β-peptide from brain: transport or metabolism? Nat Med 20006(7):718: Iwata N, Tsubuki S, Hama Eet al. Reply to: ‘Clearance of amyloid β-peptide from brain: transport or metabolism?’ Nat Med 2000; 6(7): 718-719. [PubMed: 10888892]
- 2.
- Bick KL, Katzman R. Alzheimer's disease: The road to 2000. Neurosci News. 2000;3(4):6–7.
- 3.
- Flicker C, Ferris SH, Reisberg B. Mild cognitive impairment in the elderly: predictors of dementia. Neurology. 1991;41(7):1006–1009. [PubMed: 2067629]
- 4.
- Golomb J, Kluger A, Ferris SH. Mild cognitive impairment: identifying and treating the earliest stages of Alzheimer's disease. Neurosci News. 2000;3(4):46–53.
- 5.
- Alzheimer A. Allgem Z PsychiatrGerich Med 190764146–148.. Translated version: Schwab C. Neurosci News 2000; 3(4):8-13.
- 6.
- Hardy J. APP Mutations Directory:http://www.alzforum.org/members/resources/app_mutations/index.html.
- 7.
- Hardy J. Presenilin Mutations Directoryhttp://www.alzforum.org/members/resources/pres_mutations/index.html.
- 8.
- Kwon J. Tau Mutations Directory:http://www.alzforum.org/res/com/mut/tau/default.asp.
- 9.
- Duff K, Eckman C, Zehr C. et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383(6602):710–713. [PubMed: 8878479]
- 10.
- Nakano Y, Kondoh G, Kudo T. et al. Accumulation of murine amyloidβ42 in a gene-dosage-dependent manner in PS1 ‘knock-in’ mice. Eur J Neurosci. 1999;11(7):2577–2581. [PubMed: 10383647]
- 11.
- Murakami K, Irie K, Morimoto A. et al. Synthesis, aggregation, neurotoxicity, and secondary structure of various Αβ 1-42 mutants of familial Alzheimer's disease at positions 21-23. Biochem Biophys Res Commun. 2002;294(1):5–10. [PubMed: 12054732]
- 12.
- Demeester N, Mertens C, Caster H. et al. Comparison of the aggregation properties, secondary structure and apoptotic effects of wild-type, Flemish and Dutch N-terminally truncated amyloid β peptides. Eur J Neurosci. 2001;13(11):2015–2024. [PubMed: 11422442]
- 13.
- Nilsberth C, Westlind-Danielsson A, Eckman CB. et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Αβ protofibril formation. Nat Neurosci. 2001;4(9):887–893. [PubMed: 11528419]
- 14.
- Ihara Y, Nukina N, Miura R. et al. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer's disease. J Biochem (Tokyo). 1986;99(6):1807–1810. [PubMed: 2427509]
- 15.
- Grundke-Iqbal I, Iqbal K, Tung YC. et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83(13):4913–4917. [PMC free article: PMC323854] [PubMed: 3088567]
- 16.
- Ishihara T, Hong M, Zhang B. et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24(3):751–762. [PubMed: 10595524]
- 17.
- Lewis J, McGowan E, Rockwood J. et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25(4):402–405. [PubMed: 10932182]
- 18.
- Tanemura K, Murayama M, Akagi T. et al. Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci. 2002;22(1):133–141. [PMC free article: PMC6757582] [PubMed: 11756496]
- 19.
- Shoji M, Golde TE, Ghiso J. et al. Production of the Alzheimer amyloid β protein by normal proteolytic processing. Science. 1992;258:126–129. [PubMed: 1439760]
- 20.
- Seubert P, Vigo-Pelfrey C, Esch F. et al. Isolation and quantification of soluble Alzheimer's β-peptide from biological fluids. Nature. 1992;359:325–327. [PubMed: 1406936]
- 21.
- van GoolWA, Schenk DB, Bolhuis PA. Concentrations of amyloid-β protein in cerebrospinal fluid increase with age in patients free from neurodegenerative disease. Neurosci Lett. 1994;172(12):122–124. [PubMed: 8084515]
- 22.
- Saitoh T, Sundsmo M, Roch JM. et al. Secreted form of amyloid β protein precursor is involved in the growth regulation of fibroblasts. Cell. 1989;58(4):615–622. [PubMed: 2475254]
- 23.
- Mattson MP, Cheng B, Culwell AR. et al. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron. 1993;10(2):243–254. [PubMed: 8094963]
- 24.
- Kang J, Lemaire HG, Unterbeck A. et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325(6106):733–736. [PubMed: 2881207]
- 25.
- Oltersdorf T, Fritz LC, Schenk DB. et al. The secreted form of the Alzheimer's amyloid precursor protein with the Kunitz domain is protease nexin-II. Nature. 1989;341(6238):144–147. [PubMed: 2506449]
- 26.
- Kimberly WT, Zheng JB, Guenette SY. et al. The intracellular domain of the β-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276(43):40288–40292. [PubMed: 11544248]
- 27.
- Leissring MA, Murphy MP, Mead TR. et al. A physiologic signaling role for the γ-secretase-derived intracellular fragment of APP. Proc Natl Acad Sci USA. 2002;99(7):4697–4702. [PMC free article: PMC123710] [PubMed: 11917117]
- 28.
- Saido TC. Alzheimer's disease as proteolytic disorders: anabolism and catabolism of β-amyloid. Neurobiol Aging. 1998;19(1 Suppl):S69–75. [PubMed: 9562472]
- 29.
- Flexner C. Dual protease inhibitor therapy in HIV-infected patients: pharmacologic rationale and clinical benefits. Annu Rev Pharmacol Toxicol. 2000;40:649–674. [PubMed: 10836150]
- 30.
- Götz J, Chen F, van DorpeJ. et al. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Αβ42 fibrils. Science. 2001;293:1491–1495. [PubMed: 11520988]
- 31.
- Lewis J, Dickson DW, Lin WL. et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. [PubMed: 11520987]
- 32.
- Campion D, Dumanchin C, Hannequin D. et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–670. [PMC free article: PMC1377972] [PubMed: 10441572]
- 33.
- Hsia AY, Masliah E, McConlogue L. et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96(6):3228–3233. [PMC free article: PMC15924] [PubMed: 10077666]
- 34.
- Moechars D, Dewachter I, Lorent K. et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. [PubMed: 10037741]
- 35.
- Mucke L, Masliah E, Yu GQ. et al. High-level neuronal expression of Αβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. [PMC free article: PMC6772621] [PubMed: 10818140]
- 36.
- Walsh DM, Klyubin I, Fadeeva JV. et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. [PubMed: 11932745]
- 37.
- Kawarabayashi T, Younkin LH, Saido TC. et al. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. [PMC free article: PMC6763819] [PubMed: 11160418]
- 38.
- Saido TC, Iwatsubo T, Mann DMA. et al. Dominant and differential deposition of distinct β-amyloid peptide species, ΑβN3(pE), in senile plaques. Neuron. 1995;14:457–466. [PubMed: 7857653]
- 39.
- Russo C, Schettini G, Saido TC. et al. Preferential deposition of truncated amyloid-β peptides in brain of presenilin 1 gene mutation carriers. Nature. 2000;405:531–532. [PubMed: 10850703]
- 40.
- Jarrett JT, Lansbury PT Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73(6):1055–8. [PubMed: 8513491]
- 41.
- Shirotani K, Tsubuki S, Lee HJ. et al. Generation of amyloid β peptide with pyroglutamate at position 3 in primary cortical neurons. Neurosci Lett. 2002;327(1):25–28. [PubMed: 12098492]
- 42.
- Miyazaki K, Hasegawa M, Funahashi K. et al. A metalloproteinase inhibitor domain in Alzheimer amyloid protein precursor. Nature. 1993;362(6423):839–841. [PubMed: 8479521]
- 43.
- Akiyama H, Barger S, Barnum S. et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21(3):383–421. [PMC free article: PMC3887148] [PubMed: 10858586]
- 44.
- Wyss-Coray T, Yan F, Lin AH. et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. 2002;99(16):10837–10842. [PMC free article: PMC125059] [PubMed: 12119423]
- 45.
- Behl C, Davis J, Cole GM. et al. Vitamin E protects nerve cells from amyloid β protein toxicity. Biochem Biophys Res Commun. 1992;186(2):944–950. [PubMed: 1497677]
- 46.
- Lim GP, Chu T, Yang F. et al. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21(21):8370–8377. [PMC free article: PMC6762797] [PubMed: 11606625]
- 47.
- Funato H, Yoshimura M, Kusui K. et al. Quantitation of amyloid β-protein (Αβ) in the cortex during aging and in Alzheimer's disease. Am J Pathol. 1998;152:1633–1640. [PMC free article: PMC1858449] [PubMed: 9626067]
- 48.
- Funato H, Enya M, Yoshimura M. et al. Presence of sodium dodecyl sulfate-stable amyloid β-protein dimers in the hippocampus CA1 not exhibiting neurofibrillary tangle formation. Am J Pathol. 1999;155(1):23–28. [PMC free article: PMC1866667] [PubMed: 10393832]
- 49.
- Morishima-Kawashima M, Oshima N, Ogata H. et al. Effect of apolipoprotein E allele ϵ4 on the initial phase of amyloid β-protein accumulation in the human brain. Am J Pathol. 2000;157:2093–2099. [PMC free article: PMC1885772] [PubMed: 11106581]
- 50.
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. [PubMed: 10415140]
- Overview Αβ Metabolism: From Alzheimer Research to Brain Aging Control - Madame ...Overview Αβ Metabolism: From Alzheimer Research to Brain Aging Control - Madame Curie Bioscience Database
- Metastatic Genitourinary Malignancies - Madame Curie Bioscience DatabaseMetastatic Genitourinary Malignancies - Madame Curie Bioscience Database
- Lipocalins in Arthropoda: Diversification and Functional Explorations - Madame C...Lipocalins in Arthropoda: Diversification and Functional Explorations - Madame Curie Bioscience Database
- Role of Superoxide in Post-Ischemic Liver Injury - Madame Curie Bioscience Datab...Role of Superoxide in Post-Ischemic Liver Injury - Madame Curie Bioscience Database
- Structure and Replication of Hepatitis Delta Virus RNA - Madame Curie Bioscience...Structure and Replication of Hepatitis Delta Virus RNA - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...