

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Finding inhibitors of Αβ42 generation is a major goal of Alzheimer's disease drug development. Two target protease activities, β- and γ-secretases, were operationally defined more than 10 years ago, but progress in this area has been slow because the actual enzymes were not identified. Using an expression cloning strategy we have identified a novel membrane-bound aspartic protease, BACE1, as β-secretase. This finding has been confirmed, and BACE1 and its homologue BACE2 have been characterized in detail by many groups. Major progress has been made in two areas: First, the x-ray crystal structure, which is critical for rational inhibitor design, has been solved and shown to be similar to that of other pepsin family members. Second, knockout studies show that BACE1 is critical for Αβ generation, but the knockout mice show an otherwise normal phenotype, raising the possibility that therapeutic BACE1 inhibition could be accomplished without major mechanism-based toxicity. However, target-mediated toxicity of β-secretase inhibition cannot be ruled out based on the currently available data alone. While various peptidic β-secretase inhibitors have been published, the key challenge now is the generation of more drug-like compounds that could be developed for therapeutic purposes. Other current areas of investigation, including identification of BACE1 substrates and the potential role of BACE1 overexpression in AD, are discussed.
Identification of β-Secretase
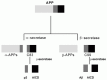
Since the cloning of amlyloid precursor protein (APP) revealed that Αβ must be excised from the middle of its large precursor protein,1 the two necessary proteolytic cleavage events, one at the N-terminus by an enzyme termed “β-secretase” and one at the C-terminus by an enzyme termed “γ-secretase”, have attracted a lot of attention. This is understandable because Αβ formation is the initial step in the hypothetical amyloid cascade2 and is thus supposed to be ultimately responsible for the pathology of Alzheimer's disease (AD). Moreover, the necessity of proteolytic cleavage for Αβ generation immediately suggested the existence of two potential therapeutic intervention targets which could be addressed using standard protease inhibition approaches. Consequently, APP processing and Αβ generation have been studied in a variety of systems by many investigators and their results are summarized in Figure 2.1. At least three distinct protease activities are involved in processing the membrane protein APP along two major pathways, the α-secretase and the amyloid-forming &β-secretase pathway. A relatively small minority of APP molecules enters the β-secretase pathway in which β-secretase cleaves APP and releases a soluble fragment, β-APPs. The C-terminal membrane-bound C99 peptide is then cleaved by γ-secretase within the transmembrane domain and two major isoforms of 40 and 42 amino acid length with different C-termini, Αβ40 and Αβ42, are generated. In the α-secretase pathway, α-secretase cleaves in the middle of the Αβ region (thus precluding Αβ formation) and releases a soluble fragment, γ-APPs. The remaining membrane-bound C-terminal fragment C83 is then cleaved by γ-secretase to give rise to p340 and p342, shortened versions of Αβ that do not appear to be major plaque components. Pharmacological and cell biological studies demonstrated early on that the three major activities, α-, β-, and γ-secretase were distinct. By the mid-1990s an approximate subcellular localization of these activities was established by defining where one can detect their respective cleavage products. Using this approach it was for example shown that β-secretase activity must reside both in endosomes3 and in the secretory pathway,4 whereas α-secretase activity could be clearly detected on the cell surface.5 However, it was far less clear whether all cleavages within each major class were carried out by one or several different, but related enzymes. Obviously, such questions could be addressed directly, and inhibitors could be developed in a rational way, if the relevant enzymes were identified and isolated. Therefore, isolating β- and γ-secretases has been a major goal for laboratories in academia and the pharmaceutical industry for a long time. It took more than 10 years to accomplish this, primarily because the biochemical purification of these enzymes using peptidic substrates proved exceedingly difficult. Upon homogenization of cells numerous active enzymes capable of cleaving peptidic substrates at the right position are released, and a variety of them have been suggested as candidates for β and/or γ-secretase over the years (for a detailed review of these efforts from the mid-90s see ref. 6). Clearly, there was a problem with irrelevant enzymes performing artifactual cleavages of short peptidic substrates that obscured the less robustly expressed secretases. We decided to circumvent the intrinsic problems of a biochemical purification approach by using an expression cloning strategy to identify genes that modulate Αβ production. We hypothesized that overexpression of α-secretase in cells overexpressing APP could lead to increased Αβ production. A cDNA library was prepared from 293 human embryonic kidney cells (which are known to express the complete APP processing machinery) and divided into pools of 100, and these pools were transfected into 293 human embryonic kidney cells overexpressing APP. Pools causing increased Αβ production were then subdivided into smaller pools and finally reduced to single clones. This expression cloning strategy ultimately led to the identification of beta-site APP-cleaving enzyme 1 (BACE1) as the major β-secretase.7 Subsequently, three other groups reported isolation of the same enzyme using different approaches. Hypothesizing that β-secretase belongs to the aspartic protease family and using a genomics approach and antisense studies, Yan et al isolated β-secretase.8 In contrast, Sinha et al used biochemical affinity purification to identify the enzyme.9 Finally, Hussain et al also reported identification of β-secretase, but they did not report why the particular candidate was selected initially.10
Characterization of β-Secretase
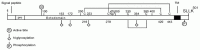
BACE1 is a 501 amino acid protein with an amino-terminal signal peptide of 21 amino acids followed by a proprotein domain spanning amino acids 22 to 45 (Fig. 2.2). The lumenal domain of the mature protein extends from residues 46 to 460 and is followed by a transmembrane domain of 17 residues and a short cytosolic tail of 24 amino acids. BACE1 contains two active site motifs at amino acids 93 to 96 and 289 to 292 in the lumenal domain, each containing the highly conserved signature sequence of aspartic proteases D T/S G T/S. Based on the amino acid sequence, BACE1 is predicted to be a type I transmembrane protein with the active site on the lumenal side of the membrane where β-secretase cleaves APP. At the amino acid level, BACE1 shows less than 30% sequence identity with human pepsin family members. The BACE1 gene is localized to chromosome 11q23.3, not associated with AD.
We have thoroughly demonstrated that BACE1 exhibits all the known properties of β-secretase7 and subsequent publications from other groups have confirmed this analysis. Tissue culture and animal studies indicated that β-secretase is expressed in all tissues, but higher in neurons of the brain. This is exactly what we found for BACE1 mRNA. However, we and others also found very high levels of BACE1 mRNA in the pancreas, leading to speculations about a role of BACE1 as a protease in this tissue, but recently this mRNA has been shown to encode a shortened BACE1 splice variant of unknown function which is deficient in protease activity.11 We also demonstrated the presence of BACE1 protein in brain.7 BACE1 has the right topological orientation to attack the β-secretase cleavage site of APP and is localized within acidic intracellular compartments, such as endosomes and trans-Golgi network (TGN) as expected for β-secretase.7 BACE1 is also detectable at the cell surface, and cycling of the protein between the cell membrane and endosomes was documented.12,13 Endosomal targeting of BACE1 depends on a cytoplasmic dileucine motif (residues 499 and 500).12 Endogenous BACE1 was shown to primarily localize to the TGN from which a small portion is delivered to the plasma membrane from which it then recycles to endocytic compartments. It appears that the BACE1 transmembrane domain contains a TGN targeting signal.14 Overexpression of BACE1 in cells increases the β-secretase products C99 (the major β-cleavage product starting with the Asp1 amino acid of Αβ), C89 (the β'-cleavage product starting with the Glu11 amino acid of Αβ) and APPsβ, while the α-secretase product APPsα is decreased. In cells expressing wild-type APP, this directly leads to increased Αβ generation.7 Under overexpression conditions the ratio of β'/β-cleavage correlates with BACE1 expression levels,15 both APP and C99 can undergo β'-cleavage16 and β-site proteolysis predominates in the endoplasmic reticulum, whereas β'-cleavage predominates in the trans-Golgi network.17 Antisense inhibition of BACE1 decreases β-secretase cleavage and Αβ generation.7,8 Purified forms of BACE1 cleave APP substrates in vitro at the correct site and with the same P1 specificity previously described for β-secretase in cell-based assays. Purified forms of BACE1 have an acidic pH optimum and are not inhibited by pepstatin, as expected for β-secretase. BACE1 undergoes a series of posttranslational modifications. All four potential N-glycsosylation sites in the ectodomain are occupied by carbohydrate; no O-glycosylation is detected.18 Protease activity is affected by the occupancy of glycosylation sites, but the effect is likely to be indirect, e.g., by enhancing correct folding or increasing solubility of the protein.19 The six cysteine residues in the ectodomain all form intramolecular disulfide bonds in a pattern which is not conserved in other aspartic proteases.18 In pulse chase experiments BACE1 protein (calculated MW 50 kDa) is initially detectable as a 60 kDa immature glycosylated form made in the ER which undergoes rapid maturation to a 70 kDa product which is stable.13,18 Ectodomain shedding of BACE1 upon overexpression has been reported.20 It is not known whether this ectodomain shedding occurs under physiological conditions, and its functional significance remains unclear. BACE1 is initially synthesized as a proprotein that is cleaved at residue E46 to form the mature enzyme. Interestingly, both the proprotein and the mature protein are proteolytically active and the Pro domain does not suppress activity as in a strict zymogen, but appears to facilitate proper folding of the active enzyme.21 The prodomain cleavage is not autocatalytic, as in some other aspartic proteases. Instead, furin, or a furin-like protease is likely responsible for the propeptide cleavage.22 After full maturation BACE1 can be phosphorylated within its cytoplasmic domain at Ser-498. The phosphorylation regulates retrieval of BACE1 from endocytosed vesicles, a mechanism reminiscent of furin trafficking.23
BACE2, the Closest Relative
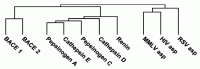
Immediately after the identification of BACE1, database mining led to the discovery of BACE2, an aspartic protease which has 64% amino acid sequence similarity to BACE1 and which also shows a C-terminal transmembrane domain. Together BACE1 and BACE2 define a novel family of aspartic proteases (Fig. 2.3). BACE2 localizes to the obligate Down's syndrome region of chromosome 21, making it attractive to speculate that it may have β-secretase activity and that its overexpression could contribute to the AD pathology observed in Down's syndrome. However, its brain expression is very low and it is primarily peripherally expressed.24 Several other studies also indicate that this enzyme does not play a major role in Αβ generation. First, in contrast to BACE1, antisense inhibition does not impact Αβ generation.8 Second, overexpression of BACE2 does not lead to increased Αβ production24-26 and very recent data suggest that BACE2 may even have an anti-amyloidogenic role.27 While BACE2 does recognize the β-site of APP, it primarily cleaves APP after the known α-secretase sites Phe-19 and Phe-20 which does not lead to increased Αβ generation.26,28 Third, BACE1 knockout alone is sufficient to abolish Αβ production (see below). Currently, the physiological role of BACE2 and its substrates are unknown, but because it is closely related to the important BACE1, it is interesting to analyze the properties of BACE2. Like BACE1, BACE2 undergoes posttranslational modifications and is transported through the secretory pathway to the plasma membrane, but in contrast to BACE1 it is hard to detect in endosomes.29 The transmembrane domain of BACE2 is only 22% identical to that of BACE1 and this may explain why BACE2 is not primarily localized to the TGN, but has a more diffuse localization pattern.28 In contrast to BACE1, BACE2 prodomain processing is autocatalytic.28,30 The crystal structure of BACE2 has not been disclosed and the phenotype of BACE2 knockout mice has not been reported yet.
BACE1 Transgenics and Knockouts
Mice transgenic for human BACE1 expressed under the mouse Thy-1 promoter have been generated. High BACE1 protein expression in the hippocampus and cortex was observed and stable lines were established, suggesting that neuronal BACE1 overexpression in vivo is not lethal. Detailed pathology of the transgenics has not been reported, and the focus of the study was on showing that in vivo BACE1 overexpression leads to increased amyloidogenic processing of APP, as demonstrated by reduced levels of full-length APP and increased levels of C99, C89 and APPsβ, ultimately resulting in increased Αβ production.31 This result does not come as a surprise, but was expected based on the tissue culture data. A recent report from another group suggests that BACE1 overexpression under the same promoter leads to a neurodegeneration phenotype with motor deficits.32 It is not yet known why this phenotype was not observed in the first study. For inhibitor development, knockout mouse studies are more important, and consequently knockout mouse results were available earlier than transgenic data. The finding that β-secretase knockout mice are deficient in Αβ production, independently reported by us and two other groups, was not unexpected, but it did provide ultimate in vivo validation of BACE1 as β-secretase and it demonstrated that in mice no compensatory mechanism for β-secretase cleavage exists.33-35 The more exciting and unexpected aspect of the knockout studies was the absence of major problems due to β-secretase ablation: We analyzed the phenotype of our BACE1 knockout mice and found them to be healthy and fertile. A detailed analysis demonstrated that the knockout mice are normal in terms of gross morphology and anatomy, tissue histology, hematology and clinical chemistry.33 In addition, behavioral analysis of the knockout mice generated by Roberds et al showed no obvious deficits in basal neurological and physiological functions.35 While no study has investigated aged knockout mice or knockout mice subjected to various challenges, the absence of distinct pathology is very encouraging for β-secretase drug development.
β-Secretase Drug Development Is Under Way
On theoretical grounds inhibition of either β- or γ-secretase should be sufficient to block Αβ production (see Fig. 2.1), so the choice between these two targets could then be determined by technical feasibility and mechanism-based toxicity issues. In the absence of purified secretase enzymes, whole cell assays for inhibition of Αβ generation in the presence of compounds were run, and these assays delivered potent γ-secretase inhibitor leads, but no potent β-secretase inhibitors were disclosed in the scientific or patent literature. Consequently, during the 90s most drug development focused on the γ-secretase target. The recent insights into the potential liabilities of γ-secretase inhibition and the availability of pure β-secretase enzyme have led to a surge in interest in β-secretase inhibitor development. BACE1 is an attractive drug target: Its biology is relatively well understood, the knockout data show that the enzyme is necessary and sufficient for β-secretase cleavage and, most importantly, they suggest that mechanism-based toxicity of inhibitors may be nonexistent or manageable, and other aspartic proteases have been successfully targeted for drug development in the past.36 Moreover, BACE1 is predominantly expressed in the brain and because only very few aspartic proteases exist, the risk for cross-inhibition is limited. The crystal structure of the BACE1 ectodomain complexed with a peptidic inhibitor has been solved and this structural information is now available to guide inhibitor development. The overall structure of the enzyme is very similar to that of other known aspartic proteases. However, there are differences in the details of the active site which is generally more open and less hydrophobic than in other aspartic proteases.37 Potent peptidic inhibitors of BACE1 have already been described in several publications.9,38-40 The goal of ongoing studies is the design of smaller, more drug-like compounds. A recent kinetic study using synthetic peptide libraries and mass spectrometry for initial rate determination has led to a clearer understanding of the subsite specificity of BACE1. It was shown that a consensus peptide defined by the kinetic study as EIDLMVLD was cleaved with a kcat/KM value 14-fold better than the analogous APPsw-derived octamer. These data can be used to design better substrates and inhibitors. They also suggest that much better physiological substrates for β-secretase than APP may exist.41 The relatively large active site of β-secretase may pose challenges for the development of small molecule inhibitors. Based on the experience with other aspartic protease inhibitors, in particular renin and human immunodeficiency virus (HIV) protease drugs, one can predict that the generation of drug-like β-secretase inhibitors will be difficult and time consuming, but that it will be pursued by many companies, given the size of the AD problem and the limited number of validated alternative AD targets.
Controversies and Open Questions
Compared to other areas of AD research, the β-secretase field has seen surprisingly little controversy. I would define three major landmarks of the field for which all publications agree or—if there is only one publication—everyone seems to agree with the published data and their interpretation. These landmarks are identification of BACE1 as the major β-secretase activity, the discovery that BACE1 knockout mice do not make Αβ, but seem otherwise normal and the description of the crystal structure of the enzyme. Several publications describing the detailed properties of BACE1 are also in agreement with each other with some minor differences on subcellular localization. While there is no more doubt about the identity of the long-sought after β-secretase, there are still numerous questions left to answer, and some of these questions may impact β-secretase inhibitor development: First, what are the other substrates of BACE1? Additional substrates are likely to exist, because it is counterintuitive to assume that β-secretase has evolved just to generate Αβ. The above-mentioned kinetic study suggesting that physiological β-secretase substrates could have several-hundred-fold better kcatK/m values than APP clearly supports this notion.41 This is interesting from a basic perspective, but also important to know in order to predict potential liabilities of β-secretase inhibitors. So far, two new potential β-secretase substrates, ST6Gal I, a sialyltransferase42 and most recently p-selectin glycoprotein ligand-1,43 have been proposed, mainly based on the finding that their secretion was increased upon BACE1 overexpression. It remains to be determined whether these proteins are actually cleaved by BACE1 at physiological expression levels. Second, what is the biological role of BACE2? What is the phenotype of a BACE2 knockout? What is the phenotype of a BACE1,2 double knockout? Again this is interesting from a basic perspective, but it would also be important to know, whether a β-secretase inhibitor drug would have to be BACE1 specific or whether cross-inhibition of BACE2 will be acceptable. Third, does overexpression of BACE1 play a role in some forms of AD? Three recent reports suggest that this may be the case,44-46 but more work is needed to find out whether BACE1 overexpression can be firmly linked to AD. Fourth, how is β-secretase expression regulated? This area still appears largely unexplored. Fifth, can β-secretase activity be modulated by mechanisms other than direct enzyme inhibition? A provocative recent paper claims that BACE1 partitions into lipid rafts and that this may underlie the cholesterol sensitivity of Αβ production, thus potentially linking BACE1 with the cholesterol field.47 And finally, the ultimate question addressing the amyloid hypothesis remains to be asked in the clinic—assuming a safe β-secretase inhibitor drug can be found, will it prevent AD, will it arrest progression, or will it even give symptomatic improvement?
References
- 1.
- Kang J, Lemaire HG, Unterbeck A. et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. [PubMed: 2881207]
- 2.
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. [PubMed: 1763432]
- 3.
- Koo EH, Squazzo S. Evidence that production and release of amyloid β-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed: 8021238]
- 4.
- Haass C, Lemere CA, Capell A. et al. The Swedish mutation causes early-onset Alzheimer's disease by β-secretase cleavage within the secretory pathway. Nat Med. 1995;1:1291–1296. [PubMed: 7489411]
- 5.
- Sisodia SS. β-Amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci USA. 1992;89:6075–6079. [PMC free article: PMC49440] [PubMed: 1631093]
- 6.
- Evin G, Beyreuther K, Masters CL. Alzheimer's disease amyloid precursor protein (ΑβPP): proteolytic processing, secretases and βΑ4 amyloid production. Amyloid: Int J Exp Clin Invest. 1994;1:263–280.
- 7.
- Vassar R, Bennett BD, Babu-Khan S. et al. β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. [PubMed: 10531052]
- 8.
- Yan R, Bienkowski MJ, Shuck ME. et al. Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase specificity. Nature. 1999;402:533–537. [PubMed: 10591213]
- 9.
- Sinha S, Anderson JP, Barbour R. et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature. 1999;402:537–540. [PubMed: 10591214]
- 10.
- Hussain I, Powell D, Howlett DR. et al. Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol Cell Neurosci. 1999;14:419–427. [PubMed: 10656250]
- 11.
- Bodendorf U, Fischer F, Bodian D. et al. A splice variant of β-secretase deficient in the amyloidogenic processing of the amyloid precursor protein. J Biol Chem. 2001;276:12019–12023. [PubMed: 11152683]
- 12.
- Huse JT, Pijak DS, Leslie GJ. et al. Maturation and endosomal targeting of β-site amyloid precursor protein-cleaving enzyme.The Alzheimer's disease β-secretase. J Biol Chem. 2000;275:33729–33737. [PubMed: 10924510]
- 13.
- Capell A, Steiner S, Willem M. et al. Maturation and pro-peptide cleavage of β-secretase. J Biol Chem. 2000;275:30849–30854. [PubMed: 10801872]
- 14.
- Yan R, Han P, Miao H. et al. The transmembrane domain of the Alzheimer's β-secretase (BACE1) determines its late Golgi localization and access to β-amyloid precursor protein (APP) substrate. J Biol Chem. 2001;276:36788–36796. [PubMed: 11466313]
- 15.
- Creemers JW, Dominguez DI, Plets E. et al. Processing of β-secretase by furin and other members of the proprotein convertase family. J Biol Chem. 2001;276:4211–4217. [PubMed: 11071887]
- 16.
- Liu K, Doms RW, Lee VMY. Glu11 site cleavage and N-terminally truncated Αβ production upon BACE overexpression. Biochemistry. 2002;41:3128–3136. [PubMed: 11863452]
- 17.
- Huse JT, Liu K, Pijak DS. et al. β-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer's disease brain. J Biol Chem. 2002;277:16278–16284. [PubMed: 11847218]
- 18.
- Haniu M, Denis P, Young Y. et al. Characterization of Alzheimer's β-secretase protein BACE. A pepsin family member with unusual properties. J Biol Chem. 2000;275:21099–21106. [PubMed: 10887202]
- 19.
- Charlwood J, Dingwall C, Matico R. et al. Characterization of the glycosylation profiles of Alzheimer's β-secretase protein Asp-2 expressed in a variety of cell lines. J Biol Chem. 2001;276:16739–16748. [PubMed: 11278492]
- 20.
- Benjanne S, Elagoz A, Wickham L. et al. Posttranslational processing of β-secretase (β-amyloid-converting enzyme) and its ectodomain shedding. J Biol Chem. 2001;276:10879–10887. [PubMed: 11152688]
- 21.
- Shi XP, Chen E, Yin KC. et al. The pro domain of β-secretase does not confer strict zymogen-like properties but does assist proper folding of the protease domain. J Biol Chem. 2001;276:10366–10373. [PubMed: 11266439]
- 22.
- Bennett BD, Denis P, Haniu M. et al. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer's β-secretase. J Biol Chem. 2000;275:37712–37717. [PubMed: 10956649]
- 23.
- Walter J, Fluhrer R, Hartung B. et al. Phosphorylation regulates intracellular trafficking of β-secretase. J Biol Chem. 2001;276:14634–14641. [PubMed: 11278841]
- 24.
- Bennett BD, Babu-Khan S, Loeloff R. et al. Expression analysis of BACE2 in brain and peripheral tissues. J Biol Chem. 2000;275:20647–20651. [PubMed: 10749877]
- 25.
- Hussain I, Powell DJ, Howlett DR. et al. ASP1 (BACE2) cleaves the amyloid precursor protein at the β-secretase site. Mol Cell Neurosci. 2000;16:609–619. [PubMed: 11083922]
- 26.
- Farzan M, Schnitzler CE, Vasilieva N. et al. BACE2, a β-secretase homolog, cleaves at the β-site and within the amyloid-β region of the amyloid-β precursor protein. Proc Natl Acad Sci U S A. 2000;97:9712–9717. [PMC free article: PMC16930] [PubMed: 10931940]
- 27.
- Basi G, Frigon N, Tatsuno G, Xu M. et al. Cellular knockout of endogenous BACE2 reveals its role in APP processing homeostasis of Αβ production in cells coexpressing BACE1 and BACE2. Neurobiol Aging. 2002;23:S178–S179.
- 28.
- Yan R, Munzner JB, Shuck ME. et al. BACE2 functions as an alternative α-secretase in cells. J Biol Chem. 2001;276:34019–34027. [PubMed: 11423558]
- 29.
- Fluhrer R, Capell A, Westmeyer G. A non-amyloidogenic function of BACE2 in the secretory pathway. 2002. pp. 1011–1020. [PubMed: 12065613]
- 30.
- Hussain I, Christie G, Schneider K. et al. Prodomain processing of Asp1 (BACE2) is autocatalytic. J Biol Chem. 2001;276:23322–23328. [PubMed: 11316808]
- 31.
- Bodendorf U, Danner S, Fischer F. et al. Expression of human β-secretase in the mouse brain increases the steady-state level of β-amyloid. J Neurochem. 2002;80:799–806. [PubMed: 11948243]
- 32.
- Rockenstein E, Mante M, Adame A. et al. Overexpression of human β-secretase (BACE1) results in severe motor deficits and neurodegeneration in tg mice. Neurobiol Aging. 2002;23:S235–S236.
- 33.
- Luo Y, Bolon B, Kahn S. et al. Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci. 2001;4:231–232. [PubMed: 11224535]
- 34.
- Cai H, Wang Y, McCarthy D. et al. BACE1 is the major β-secretase for generation of Αβ peptides by neurons. Nat Neurosci. 2001;4:233–234. [PubMed: 11224536]
- 35.
- Roberds SL, Anderson J, Basi G. et al. BACE knockout mice are healthy despite lacking the primary β-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet. 2001;10:1317–1324. [PubMed: 11406613]
- 36.
- Leung D, Abbenante G, Fairlie DP. Protease inhibitors: current status and future prospects. J Med Chem. 2000;43:305–341. [PubMed: 10669559]
- 37.
- Hong L, Koelsch G, Lin X. et al. Structure of the protease domain of memapsin 2 (β-secretase) complexed with inhibitor. Science. 2000;290:150–153. [PubMed: 11021803]
- 38.
- Ghosh AK, Shin D, Downs D. et al. Design of potent inhibitors for human brain memapsin 2 (β-secretase). J Am Chem Soc. 2000;122:3522–3523. [PMC free article: PMC6233310] [PubMed: 30443047]
- 39.
- Ghosh AK, Bilcer G, Harwood C. et al. Structure-based design: potent inhibitors of human brain memapsin 2 (β-secretase). J Med Chem. 2001;44:2865–2868. [PubMed: 11520194]
- 40.
- Tung JS, Davis DL, Anderson JP. et al. Design of substrate-based inhibitors of human β-secretase. J Med Chem. 2002;45:259–262. [PubMed: 11784130]
- 41.
- Turner RT 3rd, Koelsch G, Hong L. et al. Subsite specificity of memapsin 2 (β-secretase): implications for inhibitor design. Biochemistry. 2001;40:10001–10006. [PubMed: 11513577]
- 42.
- Kitazume S, Tachida Y, Oka R. et al. Alzheimer's β-secretase, β-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci U S A. 2001;98:13554–13559. [PMC free article: PMC61079] [PubMed: 11698669]
- 43.
- Lichtenthaler S, Seed B. The aspartyl protease BACE regulates the shedding of pselectin glycoprotein ligand1. Neurobiol Aging. 2002;23:S175–S176.
- 44.
- Holsinger RMD, McLean CA, Masters CL. et al. BACE and β-secretase product CTFβ are increased in sporadic Alzheimer's disease brain. Neurobiol Aging. 2002;23:S177–.
- 45.
- Shen Y, Yang LB, Yang XL. et al. Alteration of beta secretase (BACE) expression in sporadic Alzheimer's brains. Neurobiol Aging. 2002;23:S190–.
- 46.
- Fukamoto H, Cheung B, Hyman BT. et al. β-site amyloid precursor protein cleaving enzyme (BACE) activity is increased in temporal neocortex of Alzheimer's disease. Neurobiol Aging. 2002;23:S181.
- 47.
- Riddell DR, Christie G, Hussain I. et al. Compartmentalization of β-secretase (Asp2) into low-buoyant density noncaveolar lipid rafts. Curr Biol. 2001;11:1288–1293. [PubMed: 11525745]
- β-Secretase: Progress and Open Questions - Madame Curie Bioscience Databaseβ-Secretase: Progress and Open Questions - Madame Curie Bioscience Database
- The Sarcoglycans - Madame Curie Bioscience DatabaseThe Sarcoglycans - Madame Curie Bioscience Database
- Nucleic Acids Are Not Boring Long Polymers of Only Four Types of Nucleotides: A ...Nucleic Acids Are Not Boring Long Polymers of Only Four Types of Nucleotides: A Guided Tour - Madame Curie Bioscience Database
- Surgical Stress Response and Cancer Metastasis: The Potential Benefit of Periope...Surgical Stress Response and Cancer Metastasis: The Potential Benefit of Perioperative Beta Blockade - Madame Curie Bioscience Database
- Branching Morphogenesis of the Prostate - Madame Curie Bioscience DatabaseBranching Morphogenesis of the Prostate - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...