

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Amyloid β peptide (Αβ), the pathogenic agent of Alzheimer's disease (AD), is a physiological metabolite in the brain. We focused our attention and effort on elucidation of the unresolved aspect of Αβ metabolism, proteolytic degradation. Among a number of Αβ-degrading enzyme candidates, we identified a member of the neutral endopeptidase family, neprilysin, as the major catabolic enzyme using a novel in vivo paradigm. Consistently, neprilysin deficiency resulted in defects in the metabolism of endogenous Αβ 40 and 42, notably, in a gene dose-dependent manner. Our observations suggest that even partial down-regulation of neprilysin activity, which could be caused by aging, can contribute to AD development by promoting Αβ accumulation. Moreover, we describe that an aging-dependent decline of neprilysin activity, leading to elevation of Αβ levels in the brain, is a natural process preceding AD pathology. We not only hypothesize in this chapter on the likely role of neprilysin down-regulation in sporadic AD (SAD) pathogenesis but also propose that the knowledge itself be used for developing novel preventive and therapeutic strategies. Finally, we also discuss the need for mathematical refinement of the current human genetic approaches to identify the risk and anti-risk factors for AD.
Introduction: Why Αβ Degradation?
As discussed in Chapter 1, the steady-state Αβ levels are determined by the dynamic and steady-state metabolic balances between the generation and clearance. The clearance mechanism consists of proteolytic degradation within brain parenchyma and transport to the cerebrospinal fluid (CSF) and to plasma followed by degradation outside the brain. In our laboratory, we chose to focus our attention and efforts on elucidation of the in-parenchyma degradation1,2 (see Fig. 6.1), assuming that the rate constants in Formula 1.1 (Chapter 1) were such that K2 > K3 and thus [Αβ42]3 K1/K2 × [APP] (almost). The basis of our reasoning is as follows:
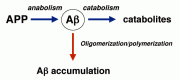
- Brains are rich sources of various peptidases which can proteolyze Αβ as long as they are accessible to the substrate.
- If reduced efficiency of transport out of brain to the circulatory system was a major cause of the pathological Αβ deposition, one would expect vascular amyloid deposition to arise earlier than parenchymal deposition, but the reality is generally the other way around particularly in humans.
- Human brains are approximately 1,000 times larger than mouse brains in size. Therefore, the advantage of in-parenchyma degradation over the transport-dependent clearance is likely to be even greater in humans than in mice particularly if the major transport pathway is via the interstitial fluid (ISF) and then to the CSF and plasma. Note that the primary function of proteolysis is recycling of amino acids, therefore in-parenchyma degradation is more economical from an energy metabolic point of view than out-of-brain transport especially in the case of humans.
These reasons do not necessarily rule out the importance of the transport mechanism. This is because, as discussed by Zlokovic and Holtzman in later chapters (Chapters 10 and 11), a dynamic balance seems to exist between the bran and plasma through the transport across the blood-brain barrier (BBB) and/or CSF under certain conditions at least in rodent models (particularly, when high-affinity anti-Αβ antibodies are injected into the plasma).6,7 If this also applies to humans to the extent that affects the risk of developing AD, physiological steady-state plasma and/or CSF Αβ levels could be an important parameter, and thus we may have to view the Αβdegradation issue in a more systemic manner rather than focusing only on the processes within the brain. Besides, it is quite possible that the mechanism closely associated with CAA formation may involve the transport mechanism rather than the in-parenchyma degradation. The latter point, however, is beyond the scope of this chapter.
Two Major Categories of Αβ-Degrading Mechanisms
The mechanism of Αβ degradation can be classified into at least two categories: “physiological degradation” that determines normal steady-state levels and “pathological degradation,” which may be mobilized to counterbalance the increased Αβ levels after the deposition starts. The distinction between these two categories is not necessarily clear, because peptidase(s) in the first category is likely to be involved in the second category degradation as well through alteration of the dynamic balances between monomers, oligomers and polymers of Αβ.
Although both categories are probably equally important in general terms, our impression is that the first category degradation is more likely to be associated with the etiology of SAD because the common in vivo phenotype of almost all the FAD-causing mutations is elevation of the steady-state Αβ42 levels in brain.5 We however had previously hypothesized that the plasmin system and matrix metalloproteinase (MMP) system might be candidates that could dispose of polymerized Αβ because they are the major proteinases capable of degrading fibrillized proteins.8 Consequently, we identified a novel brain-specific MMP, MT-5 MMP.9 Although our anti-MT5-MMP antibodies do in fact stain both senile plaques and neurofibrillary tangles in AD patients.9 We noted that NFT is more strongly stained (Iwata and Saido, unpublished data) and this was confirmed by Marion Maat-Schieman, Leiden University Medical Center, in hereditary cerebral hemorrhage with amyloidosis Dutch-type (HCHWA-D) brains and also by Takeshi Iwatsubo, University of Tokyo, in SAD brains (personal communications). Because MT-5 is a type 1 membrane-spanning protein, we do not yet know how important this proteinase is in AD pathogenesis. This project is presently suspended in our laboratory mainly because we did not succeed in observing any proteolytic action of MT-5 MMP on fibrillized Αβ even in an in vitro paradigm, because both the plasmin and MMP systems generally require activation of latent precursors, and because proteinases belonging to these systems could be destructive by degrading a number of matrix-consisting proteins and could worsen the pathological states through, for instance, inducing inflammation.
We also need to consider such cellular degradation mechanism as phagocytosis and pinocytosis conducted primarily by activated astrocytes and microglia.10,11 I left out this important cellular aspect of Αβ clearance just to avoid complexity. In Chapter 12, Cindy Lemere et al refer to this aspect of Αβ clearance in association with the vaccination topic. For further details, see reviews and articles published by such experts as P. McGeer, G. Cole, F. Maxwell, T. Weiss-Corary, etc.
Identification of Neprilysin as a Major in vivo Αβ-Degrading Enzyme in Brain
A number of reports have described potential Αβ-degrading enzymes using either test-tube or in vitro paradigms. See Table 6.1 for the list of major candidates thus far described.12-33 The intrinsic problem of such paradigms is that, if you incubate Αβ with a given peptidase under the optimal conditions for long enough, almost any peptidase will degrade the substrate. Our quest to identify the physiologically relevant in vivo peptidase(s) was based on the philosophy expressed by the following metaphor.
Table 1
List of some candidates for Αβ-degrading enzymes.
“If you feed a hungry lion (almost any protease) in a cage (test tube or cell culture) a penguin (Αβ), the lion would probably eat the penguin, but such an observation obviously does not tell you that lions are natural predators of penguins. You have to capture the process in an environment that is as natural as possible (in the brain of live animals) without affecting the environment.” This metaphor is not meant to be sarcastic to scientists who have been mainly employing in vitro paradigms. It is just that the degradation problems cannot be dealt with properly in the same way as the production problems have been, for which the molecular and cellular approaches have indeed been powerful and relevant tools. Besides, it is not exactly our original metaphor but rather is partly a form of cultural transmission. The late Efraim Racker used to tell TCS something like “If you have sufficient amounts of a kinase, ATP, and necessary cofactors, you can even phosphorylate your grandmother,” when TCS was a visiting graduate student to Cornell University, working with a biophysicist, Watt W. Webb, in 1985-1986. Another factor that makes degradation research more difficult to perform is that the major degradation processes are likely to take place extracytoplasmically, i.e., in extracellular space or on the lumen side of intracellular vesicles represented by secretory vesicles axonally transported from soma to presynaptic terminals. This means that not only the cellular topology but also the complex structural organization of the brain tissue composed of various types of cells need to be taken into consideration.
Therefore, we started our series of degradation studies by establishing a novel in vivo experimental paradigm in which we injected synthetic internally multi-radio-labeled Αβ1–42 in to the hippocampus of anesthetized live rats and analyzed the degradation process by high-performance liquid chromatography (HPLC) directly connected a flow-type scintillation counter (Fig.6.2).1 Experiments using a panel of more than 20 peptidase inhibitors suggested that a neutral endopeptidase family member similar or identical to neprilysin appears to play a major role in the Αβ1–42 catabolism because thiorphan, a well-characterized neutral endopeptidase inhibitor, was the most potent inhibitor. In accordance, short-term and long-term infusions of rat hippocampus with thiorphan resulted in biochemical and pathological accumulation of endogenous Αβ, respectively.
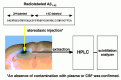
The next task was to identify the major responsible peptidase among members of the neutral endopeptidase family. We thus made efforts to determine its molecular identity by biochemical and molecular biological approaches and, consequently, predicted that neprilysin is likely to be the primary candidate.34,35 We thus examined the degradation of the radiolabeled Αβ in neprilysin-KO mouse brains using their wild-type litter mates as positive controls and confirmed our prediction as follows.2
Neprilysin is a type II membrane-associated peptidase with the active site facing the lumen or extracellular side of membranes36-39 (Fig. 6.3). This topology is favorable for the degradation of extracytoplasmic peptides such as Αβ. Furthermore, we devised to establish quantitative immunofluorescence visualization of neprilysin using neprilysin-KO40 mice as a negative control. (We actually had to test several tens of combinations of antibodies and protocols to optimize the experimental protocols). We have confirmed that neprilysin is essentially exclusively expressed in neurons, not in glia, and that the peptidase, after synthesis in the soma, is axonally transported to presynaptic terminals40 presumably in a manner similar to the way APP is transported. Therefore, presynaptic terminals and nearby intracellular (lumen-sided) locations are likely to be the sites of Αβ degradation by neprilysin (Fig. 6.4).

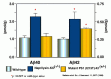
Based on the above observations, we examined the ability of neprilysin-KO mouse brains to degrade Αβ in vivo and quantified the endogenous Αβ levels in the brains.2 Due presumably to the redundancy in the neutral endopeptidase family,35,38,41 the KO mice are normal in reproduction, development, and adult anatomy to our knowledge.42 The ability to degrade the radio-labeled Αβ (see Fig. 6.2) was significantly reduced in the KO mouse brains. Consistently, both the endogenous Αβ40 and Αβ42 levels were elevated approximately 2-fold, in a manner comparable to or even greater than what has been described in FAD-causing mutant presenilin transgenic or knock-in mice43 46 (Fig. 6.5). More importantly, the elevation of Αβ levels was inversely correlated with the gene dose of neprilysin and thus with the enzyme activity (Fig. 6.6). These observations suggest that even partial loss of neprilysin expression/activity causes the elevation of Αβ42 levels and thus could induce Αβ amyloidosis on a long-term basis in a manner similar to that of FAD-causing gene mutations. These results also indicate that the rate constant for the in-parenchyma degradation conducted by neprilysin accounts for about 50% of all the clearance activity, or K2 + K3 in Formula 1.1 in Chapter 1. Therefore, K2 being negligibly smaller than K3 is unlikely and K2 is likely to be even greater in human brains for fourth of the reasons stated in the Introduction section.
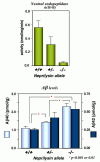
In a similar manner, some of the other Αβ-degrading enzyme candidates were examined by a similar reverse genetic approach, i.e., by measuring the brain Αβ levels in the brains of KO-mice (Table. 6.2). To our knowledge, neprilysin seems to be the only peptidase that actually regulates the steady-state level of the primarily pathogenic Αβ species, Αβ42, as the pathogenic FAD presenilin mutations do. This however does not necessarily rule out these other peptidases as relevant Αβ degrading enzymes in more general terms (see section Other Αβ-Degrading Enzyme Candidates in this chapter and Chapter 7).
Table 2
Effects of deficiency of various Αβ-degrading enzymes on brain Αβ levels.
Although we previously described the pathological deposition of the endogenous Αβ in mouse brains infused with thiorphan,1 we never observed such pathological structures in the brains of neprilysin-deficient mouse brains.2 Several reasons for this apparent discrepancy can be advanced as follows.
- Neprilysin is a member of a family of endopeptidases that includes neprilysin-like-endopeptidase, (NEPLP) α, β, and γ, PEX, XCE/DINE, and angiotensin-converting enzyme (ACE).25,35 This molecular redundancy could have compensated for the absence of neprilysin alone. Consistently, we observed the presence of thiorphan-sensitive non-neprilysin Αβ-degrading activities in the raft (see Chapters 8 and 9) fractions from mouse brains (Takaki Y et al, submitted for publication).
- The injection paradigm used in our initial study is accompanied by various surgical stresses because a cannula must be placed in the parenchyma for four weeks. These stresses combined with the neprilysin inhibitor infusion may have contributed to the pathological states in the brain. We actually observed astrocyte activation within two days after injection of the vehicle with or without the inhibitor (Hama and Saido, unpublished data).
- The experimental paradigm requiring long-term insertion of cannula into the brain parenchyma is likely to have caused at least slight leakage of plasma from the circulatory system directly into the brain parenchyma. Possible presence of plasma-derived amyloid-forming proteins such as amyloid P component in an unphysiological manner could have contributed to the pathological Αβ deposition.
In any case, our conclusion was that the initial approaches accompanying surgical procedures were, though more relevant than the solely in vitro paradigms, less appropriate than the reverse genetic approaches in understanding the mechanism of Αβ catabolism, particularly on a long-term basis. We have also been crossing the neprilysin-KO mice with the APP-overexpressing transgenic mice. The results seem to vary depending on which transgenic mouse strains are used. In one case, we observed that neprilysin deficiency caused more increases in soluble Αβ than insoluble Αβ (the differences were significant in both cases), in contrast to what we observed with the endogenous Αβ in nontransgenic mice; in another case, we observed a significant but lesser elevation of the Αβ levels (Iwata and Saido, unpublished data). Although not proven in any relevant way, the following serve as possible explanations for the differences in the effect of neprilysin deficiency in the transgenic mice (see also Fig. 6.4).
- Depending on the promoter used to drive the expression of transgenic APP, the degree of colocalization of the transgene-derived Αβ and neprilysin varies in a significant manner.
- An unphysiologically high overexpression of APP (i.e., 10-100-fold) particularly in soma may alter the way Αβ is metabolized, for instance, through the endosomal-lysosomal pathway instead of being transported via secretory vesicles to presynaptic terminals.
- Depending on the mouse strains and the cell types that express the transgene-derived APP, the transport of Αβ across the BBB or via the CSF to the circulatory system may vary to significant extent.
Region-Specific Decline of Neprilysin Expression and Activity in Brain Upon Aging
McGeer and colleagues reported that neprilysin mRNA levels are significantly reduced in the brain areas vulnerable to Αβ pathology in SAD patients at a relatively early stage (Braak stage II) as compared to the age-matched normal controls.47,48 These observations are consistent with our hypothesis described in Section 6.1. In this respect, Αβ resembles the garbage that needs to be constantly disposed of on a daily basis (see Fig. 6.7). Just imagine the situation if the public garbage collection service for large cities like New York or Tokyo was stopped for several months; the entire city would be burdened by extremely large amount of garbage and would lose the major city functions. In our view, AD brains resemble cities in such an analogy. This analogy actually also applies to the transport-dependent clearance mechanisms (Chapters 10-12).

Nevertherless, it still remained unclear till 2002 whether reduction of neprilysin expression or activity precedes Αβ pathology during the course of aging. We thus established two distinct methods to quantify the expression/activity of neprilysin in a relevant manner using neprilysin-KO mice as negative controls: (1) A biochemical assay to measure neutral endopeptidase activity in the brain (The advantage is that it is more quantitative than the latter, whereas the disadvantage is that the method fails to provide information regarding the local or spatial activity/expression of neprilysin); (2) An immunofluorescence detection using an antineprilysin antibody40 (see also the previous section).
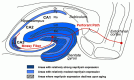
Representative immunofluorescence results are shown in Figure 6.8. The selective reduction of neprilysin expression upon aging is apparent in the polymorphic cellular, inner molecular layer, and outer molecular layer of the dentate gyrus and also in the stratum lucidum of the hippocampal CA3 sector.49 The differences were also proven to be statistically significant according to quantitative image analyses. The results of the enzymatic quantification were also consistent with these observations; we observed a statistically significant reduction of approximately 10% of the enzyme activity per year in the entire hippocampus for two years and a 10% reduction in the neocortex in two years. If the 1.5-fold increase of Αβ42, caused by the pathogenic mutations in the presenilin gene is truly sufficient for the development of early-onset FAD, then a 1% reduction of neprilysin activity per year, leading to 50% reduction of neprilysin activity at the age of 50 and thus about 1.5-fold elevation of the Αβ levels in the brain, would be sufficient to be causative of late-onset AD in humans. Obviously, this observation needs to be confirmed in human brains as well, but the problem is that almost all the proteins begin to be degraded within 30 min post-mortem and it is practically impossible to obtain samples prior to protein degradation in clinical situations. We therefore must indicate that proteomics using post-mortem human materials would make only a little sense if any. For reasons that we do not know, mRNAs are much more stable than proteins under such conditions. It probably is better to confirm the chronology using animals that show aging-associated Αβ amyloidosis, such as polar bears, monkeys, and dogs.50-52

The major implications of the immunofluorescence observations are schematized in Figure 6.9. The most important of these is that the areas where we see selective reduction of neprilysin expression correspond to the terminal zones of mossy fiber and of perforant path, which suggests that the local Αβ concentrations are particularly elevated at the presynaptic locations originally projecting from the entorhinal cortex. Note that entorhinal cortex is the region where the initial neurodegeneration takes in AD brains.
Utilization of Neprilysin Activity for Prevention and Therapy of AD
We have thus far described the possible role of neprilysin in the etiology of SAD. Our findings also indicate that regulation of neprilysin activity in a manner specific to brain regions vulnerable to Αβ deposition can be one of the strategies to reduce the Αβ burdens in the brain. Table 6.3 lists the advantages of utilizing neprilysin activity for the purpose of regulating brain Αβ levels. A relatively straight-forward approach in experimental terms, but not necessarily in clinical terms, is an application to gene therapy. Indeed, we demonstrated in an in vitro paradigm that overexpression of neprilysin, but not of an inactivated mutant form, in primary cultured neurons by Sindbis Virus leads to clearance of both the extracellular and cell-associated Αβ40 and Αβ42.53
Table 3
Advantages of utilizing neprilysin activity for clearance of Αβ from brain.
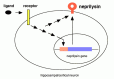
This observation by itself however is a matter of course as has been discussed using the “lion and penguin” metaphor. Besides, it will be many years from now, if ever, when gene therapy strategy becomes clinically realistic in the treatment of most neurological disorders. Instead, we have rather been focusing on the possible presence of specific peptide ligand-receptor system(s) that regulates the neprilysin activity/expression in neurons in a selective manner as shown in Figure 6.10. There are four major reasons upon which this assumption is based.
- Neprilysin expression is transcriptionally regulated in a tissue-specific manner.54
- The mechanisms schematized in Figure 6.10 have been shown to exist in neutrophils and fibroblasts.55 , 56 The ligands are morphine and substance P, respectively, indicating the possible presence of a specific peptide ligand(s)-regulated G-protein-coupled receptor(s) (GPCR) that may be involved in the regulation of neprilysin activity/expression in neurons.
- Several neuropeptides have been repeatedly reported to be down-regulated in AD brains.57-61 Some of them are also shown to be down-regulated upon aging.62 If any one of these happens to be the ligand, the aging-dependent decline of neprilysin activity resulting in elevation of Αβ levels in the brain can be accounted for by the scheme shown in Figure 6.10.
Identification of such a ligand-receptor system specifically present in hippocampus and neocortex would provide us with an idealistic pharmacological target. See our forthcoming publications for further discussions and details. Note that in the history of pharmaceutical sciences approximately 60% of cases of successful utilization of clinically effective relatively small molecules involve synthetic agonists or antagonists to such receptors. Enzyme inhibitors account for about 30%. Thus, all the other approaches, which may be interesting in experimental terms, account only for about 10%. On this basis, GPCRs would be pragmatic targets.
Other Αβ-Degrading Enzyme Candidates
As shown in Table 6.1, there have been a number of other Αβ-degrading enzyme candidates thus far described mostly by in vitro paradigms. Endothelin-converting enzyme (ECE) is similar to neprilysin in structural and biochemical properties (Fig. 6.11) and indeed seems to play some role in distinct cellular compartments (see Chapter 7 for details).
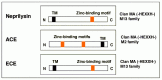
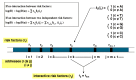
Figure 12
Multi-component analysis for identification of genetics risk and antirisk factors for AD. See the section on Human Genetics of Risk and Anti-Risk Factors for AD for details.
Among the enzyme candidates, the most extensively studied one is insulin-degrading enzyme (IDE). IDE has an interesting characteristic in that it has the tendency of degrading potentially amyloidogenic peptides. The enzyme is essentially cytoplasmic, but only a small part of the entire population has been shown to be present on the cell surface and in the extracellular space.64 The enzyme has also been shown to degrade the counterpart of the APP fragment (AICD) corresponding to the Notch intracellular domain (NICD).45 One of the difficulties in studying IDE is the lack of a specific inhibitor. Insulin is often used as a competitive inhibitor, but the problem is that insulin influences a number of cellular processes through activation of its receptor in cell culture and in vivo. The genome region containing the IDE gene has been claimed to be a candidate locus associated with the AD risk.66
Interestingly, not only neprilysin but a number of other Αβ-degrading enzyme candidates including IDE (see Table 6.1 and Fig. 6.11) are zinc-requiring enzymes. Besides, all the α-secretase candidates, a disintegrin and metalloproteinase (ADAMs) 9, 10, and 17 (see Chapters 3 and 5), which would contribute to reduction of Αβ synthesis, also require zinc for their proteolytic activities. Therefore, although too much zinc would be obviously harmful,67 a heightened zinc deficiency is likely to be a negative risk factor for AD.
Human Genetics of Risk and Anti-Risk Factors for AD: A Proposal for Multi-Component Analysis*
In the past, researchers including ourselves tended to be more concerned about human genetic evidence for the possible involvement of neprilysin activity in AD pathogenesis than they are now. This is because the aging-dependent decline of neprilysin activity in the brain, leading to local elevation of Αβ levels in areas relevant in pathogenic terms, has been shown to be a natural process prior to any apparent AD pathology at least in mice,49 and is, in our view, even programmed biologically (see forthcoming publications from our laboratory). Consistently, the association of neprilysin gene with the risk of developing AD does not seem to be very strong at least in the classical linkage analysis.
We have however been wondering why the apolipoprotein E ϵ4 allele is the only fully confirmable risk factor for SAD after hundreds or more of publications devoted to the human genetics of AD. There must be a reason. Although we are no specialists of human genetics, probably much worse than graduate students who have taken courses focusing mainly on the latest post-genome human genetics, there seems to be some limitation in the currently used techniques particularly in the search for risk and anti-risk factors for late-onset SAD. A simple question that led us to raise this issue is as follows: “Why has no one found risk or anti-risk single nucleotide polymorphisms (SNPs) or mutations in the promoter or promoter-associated regions of the APP gene in a consistent manner if the Αβ hypothesis is correct and if the pre-senile Αβ pathology in Down's syndrome brains is caused by only a 1.5-fold increase of the precursor of Αβ, APP, due to the trisomy of chromosome 21 carrying the APP gene?” (See also Formula 1.1 in Chapter 1).
If an individual happens to have both the risk and anti-risk factors with similar strengths but opposite effects on AD development within a given genome region, he or she would appear to be normal or average in human genetic terms in the presently used major approaches, i.e., (1) positional cloning through linkage analyses and (2) candidate gene-targeted approaches, which usually are case-control studies and occasionally longitudinal studies, focusing primarily on a single independent factor rather than a group of multiple genetic factors together. If such SNPs and mutations are randomly present in each family, the LOD (logarithm of odds) score in positional cloning would still be significantly high if a number of different families are employed but will probably appear smaller than they should be without taking the above-mentioned possibility into account. Besides, the situation will be even more complex if there is some interaction among such risk factors, which is possible. Our prediction is that AD human genetics needs to introduce the latest mathematically refined approaches such as have been successfully employed in search for risk factors in cancer research,68,69 led by the groups of E.S. Lander and others. (This may have actually begun in the AD research community without our knowledge).
We also predict that there probably will be a number of directly and indirectly influencing genetic factors not yet identified just because this could explain the presence of the substantial number of presenile cases that do not qualify as FAD by definition, not being autosomal dominantly inherited (See Table 1.1) and appearing like nongenetic SAD cases unless there exists a definitive environmental factor(s) specifically influencing the development of AD. Another fact that we have to keep in mind is that aging is the strongest risk factor; at the age of 85, one out of two people is affected either by AD or mild cognitive impairment (MCI) (see Chapter 1) whereas, at the age of 50, less than 0.1% of the population is affected. We would therefore look for risk factors in relatively young (< 60-65) SAD patients and for anti-risk factors in very old (> 80-100) non-AD subjects. Taking these observations together, we propose the following model as a simplest example (Fig. 6.8),
log(D) = log(D(a)) +Ý ric δc(ic) (Formula 6.1)
where D represents the degree of dementia, D(a) is a function of age, and indicates the effect of age on dementia, and the last term in the right-hand side is to indicate the effect of SNPs. For the sake of simplicity, the effect of gender is excluded here but could be easily incorporated if necessary. Also note that Formula 6.1 expresses the multiplicative effect between D(a) and Ý ric δc(ic), since Formula 6.1 is equivalent to D = D(a) exp{Ý ricδc(ic)}. Let us explain each term:
D: D can be any measure used to express the degree of dementia in principle but, preferably, should reflect the degree of dementia on a linear scale. One possibility is (30- MMSE (mini-mental state examination) score), in which the values 0 to 2 would correspond to normal people and those > 8–10 would correspond to demented patients and 30 would correspond to the terminal stage. (The MMSE values seem to be quite linear at least between 10 and 23 in clinical impressions [H. Arai, personal communication]). Another possibility is a quantity representing the pathological status, i.e., total Αβ levels, quantified images of the pathological structures, etc. This may become possible if, in the near future, we can observe the presence of Αβ amyloidosis or NFT by novel imaging techniques.
a: age
D(a): D(a) indicates the average “D” value at an age “a”. We can simply use a lookup table for D(a)
with respect to a, or use any function of “a”, where a simplest example is polynomial of “a” (with some degrees).
i: an address allocated to each SNP of a given locus (i =1, 2, 3,-------).
c = each nucleotide species: A, T, G, or C
ric: a risk constant for a given SNP nucleotide species “c” (A, T, G, or C) at the address “i.” . ric will be a positive value if the SNP is a risk factor and negative if an anti-risk factor.
δc(ic): 1 if the nucleotide species of SNP at the address “i” is “c” (A, T, G, or C) and 0 if it is not “c.” For simplicity, Formula 6.1 is given so as not to include any interaction between SNPs. If it is safer to consider the possible interactions between SNPs, we would expand Formula 6.1. as follows:
log(D) = log(D(a)) + Ý ricδc(ic) + Ý ricjc'δc,c' (ic,jc') (Formula 6.2)
where “j” corresponds to every SNP address other than “i,” and “ricjc'” to the risk generated by the interaction between “ic” and “jc”. Here, Formula 6.2 includes only the second-order interaction, but it is straightforward to include the third-order and higher order interactions, if necessary. We can also add into the above model some other features, such as specific etiological, pathological, and clinical features. In addition, we note that regarding “D” as a probability distribution of being demented at a given age with given SNPs would open further possibilities to refine the above model.
Given a set of samples, one should estimate the values of D(a) and ric in Formula 6.1. For example, if the number of all the SNP nucleotide species in the candidate locus is 50, the number of parameters ric, is 50 in Formula 6.1, while the number of parameters, ric and ricjc', is 50 + 50C2 = 1275 at the most in Formula 6.2. For example, there is no way to estimate, say, 50 parameters, given 10 samples, without any further assumptions. In other words, depending on the number of samples, one may need to modify the above model. Then, given a model, one must estimate the values of parameters. The SNPs that present distinctly larger or smaller “r” values than most of the others tested will be the major risk and anti-risk factors, respectively. For this purpose, one must examine the relevance of estimated values of parameters and also must validate a predictive power of the model using different data sets. Furthermore, it would be more thorough if one were to examine the predictive power of different models and select the best one.
In any case, these formula are consistent with our prediction that the risk factors should be easier to identify in young patients whereas the anti-risk factors should be easier to identify in very old, normal individuals because the contribution of D(a) becomes extremely large at ages >80-100, and thus even the strongest risk constants will be negligibly small. The effect of the apolipoprotein E ϵ4 allele on the incidence of AD is a good example; its effect becomes nonsignificant at ages > 80. If we were a human geneticist ourselves, we would first apply this approach to look for risk and anti-risk factors in the noncoding regions of the APP gene. In this post-genome era when more and more SNPs are being identfied, collaborations between human geneticists from all over the world may eventually make it possible to determine all the major genetic risk and anti-risk factors of AD, the knowledge of which would be very valuable not only for diagnosis but also for development of more preventive and therapeutic strategies. Remember that much of what seemed impossible even to professionals ten years ago are easily practiced now even by amateurs.
Acknowledgements
We thank all the present and past members of the Laboratory for Proteolytic Neuroscience, RIKEN Brain Science Institute, for their participation in the projects described in the first six sections. In particular, Nobuhisa Iwata and Satoshi Tsubuki have made indispensable contributions to the series of works from the beginning of the laboratory. We express our gratitude to Shun-ichi Amari (RIKEN Brain Science Institute) for valuable discussions on Mathematics and Takeshi Iwatsubo (University of Tokyo) and Hiroyuki Arai (University of Tohoku) for providing us with clinical information. We also thank our collaborators that include Maho Morishima-Kawashima and Yasuo Ihara (University of Tokyo School of Medicine), Craig Gerard and his colleagues (Harvard Medical School), Kei Maruyama (Saitama Medical School), Masatoshi Takeda and his colleagues (University of Osaka School of Medicine), Steve Younkin (Mayo Clinics, Jacksonville), John Trojanowski and Virginia Lee (University of Pennsylvania), Greg Cole (UCLA), and Peter St. George-Hyslop (University of Toronto). TCS also thanks his secretary/technical assistant, Yukiko, not only for thoroughly helping him edit the entire book but also for kindly pushing him to finish everything necessary. TCS also thanks his ex-wife and still a friend, Shoko Ikuta, and his son, Ray Ikuta, for having motivated him to shift from basic biology to the more disease-oriented research through letting him know how precious one life is. Finally and most importantly, TCS thanks his wife, Tomoko, for supporting him in many ways particularly during the past difficult time he suffered with health problems after he had decided to edit this book. Our works have been supported by research grants from RIKEN BSI, Ministry of Education, Culture, Sports, Science and Technology, and Ministry of Health, Labor and Welfare of Japan, and also by a personal donation from Shigeru Sawada, whose mother died of Alzheimer's disease.
References
- 1.
- Iwata N, Tsubuki S, Takaki Y. et al. Identification of the major Αβ1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. [PubMed: 10655101]
- 2.
- Iwata N, Tsubuki S, Takaki Y. et al. Metabolic regulation of brain Αβ by neprilysin. Science. 2001;292:1550–1552. [PubMed: 11375493]
- 3.
- Vanderstichele H, Van Kerschaver E, Hesse C. et al. Standardization of measurement of β-amyloid(1-42) in cerebrospinal fluid and plasma. Amyloid. 2000;7(4):245–258. [PubMed: 11132093]
- 4.
- Scheuner D, Eckman C, Jensen M. et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. [PubMed: 8705854]
- 5.
- Hardy J, Hutton M, Farrer M. et al. The genetic causes of the tau and synucleinopathies. Neurosci News. 2000;3:21–27.
- 6.
- DeMattos RB, Bales KR, Cummins DJ. et al. Peripheral anti-Αβ antibody alters CNS and plasma Αβ clearance and decreases brain Αβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98: 8850–8855. [PMC free article: PMC37524] [PubMed: 11438712]
- 7.
- DeMattos RB, Bales KR, Cummins DJ. et al. Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295: 2264–2267. [PubMed: 11910111]
- 8.
- Saido TC. Degradation of amyloidbeta peptide: a key to Alzheimer pathogenesis, prevention and therapy. Neuroscience News. 2000;3(5):52–62.
- 9.
- Sekine-Aizawa Y, Hama E, Watanabe K. et al. Matrix metalloproteinase (MMP) system in brain: Identification and characterization of brain-specific MMP highly expressed in cerebellum. Eur J Neurosci. 2001;13(5):935–948. [PubMed: 11264666]
- 10.
- Akiyama H, Mori H, Saido TC. et al. Occurrence of the diffuse amyloid β-protein (Αβ) deposits with numerous Αβcontaining glial cells in the cerebral cortex of patients with Alzheimer's disease. Glia. 1999;25(4):324–331. [PubMed: 10028915]
- 11.
- Yamaguchi H, Sugihara S, Ogawa A. et al. Diffuse plaques associated with astroglial amyloid β protein, possibly showing a disappearing stage of senile plaques. Acta Neuropathol (Berl). 1998;95(3):217–222. [PubMed: 9542585]
- 12.
- McDermott JR, Gibson AM. Degradation of Alzheimer's β-amyloid protein by human cathepsin D. Neuroreport. 1996;7:2163–2166. [PubMed: 8930981]
- 13.
- Hamazaki H. Cathepsin D is involved in the clearance of Alzheimer's β-amyloid protein. FEBS Lett. 1996;396:139–142. [PubMed: 8914975]
- 14.
- Kurochkin IV, Goto S. Alzheimer's β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994;345:33–37. [PubMed: 8194595]
- 15.
- McDermott JR, Gibson AM. Degradation of Alzheimer's beta-amyloid protein by human and rat brain peptidases: involvement of insulin-degrading enzyme. Neurochem Res. 1997;22:49–56. [PubMed: 9021762]
- 16.
- Qiu WQ, Walsh DM, Ye Z. et al. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J Biol Chem. 1998;273:32730–32738. [PubMed: 9830016]
- 17.
- Vekrellis K, Ye Z, Qiu WQ. et al. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J Neurosci. 2000;20:1657–1665. [PMC free article: PMC6772918] [PubMed: 10684867]
- 18.
- Chesneau V, Vekrellis K, Rosner MR. et al. Purified recombinant insulin-degrading enzyme degrades amyloid β-protein but does not promote its oligomerization. Biochem J. 2000;351:509–516. [PMC free article: PMC1221388] [PubMed: 11023838]
- 19.
- Mukherjee A, Song E, Kihiko-Ehmann M. et al. Insulysin hydrolyzes amyloid β peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J Neurosci. 2000;20:8745–8749. [PMC free article: PMC6773064] [PubMed: 11102481]
- 20.
- Van Nostrand WE, Porter M. Plasmin cleavage of the amyloid β-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry. 1999;38:11570–11576. [PubMed: 10471309]
- 21.
- Tucker HM, Kihiko M, Caldwell JN. et al. J Neurosci. 2000;20:3937–3946. [PMC free article: PMC6772619] [PubMed: 10818128]
- 22.
- Exley C, Korchazhkina OV. Neuroreport. 2001;12:2967–2970. [PubMed: 11588612]
- 23.
- Howell S, Nalbantoglu J, Crine P. Neutral endopeptidase can hydrolyze β-amyloid(140) but shows no effect on β-amyloid precursor protein metabolism. Peptides. 1995;16:647–652. [PubMed: 7479298]
- 24.
- Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer's amyloid β peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–24548. [PubMed: 11337485]
- 25.
- Hu J, Igarashi A, Kamata M. et al. J Biol Chem. 2001;276:47863–47868. [PubMed: 11604391]
- 26.
- Miyazaki K, Hasegawa M, Funahashi K. et al. A metalloproteinase inhibitor domain in Alzheimer amyloid protein precursor. Nature. 1993;362:839–841. [PubMed: 8479521]
- 27.
- Roher AE, Kasunic TC, Woods AS. et al. Proteolysis of Αβ peptide from Alzheimer disease brain by gelatinase A. Biochem Biophys Res Commun. 1994;205:1755–1761. [PubMed: 7811262]
- 28.
- Yamada T, Miyazaki K, Koshikawa N. et al. Selective localization of gelatinase A, an enzyme degrading β-amyloid protein, in white matter microglia and in Schwann cells. Acta Neuropathol (Berl). 1995;89:199–203. [PubMed: 7538720]
- 29.
- Backstrom JR, Lim GP, Cullen MJ. et al. Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-β peptide (1-40). J Neurosci. 1996;16:7910–7919. [PMC free article: PMC6579235] [PubMed: 8987819]
- 30.
- Saporito-Irwin SM, Van Nostrand W. Coagulation factor XIa cleaves the RHDS sequence and abolishes the cell adhesive properties of the amyloid β-protein. J Biol Chem. 1995;270:26265–26269. [PubMed: 7592834]
- 31.
- Hamazaki H. Carboxy-terminal truncation of long-tailed amyloid β-peptide is inhibited by serine protease inhibitor and peptide aldehyde. FEBS Lett. 1998;424:136–138. [PubMed: 9539136]
- 32.
- Carvalho KM, Franca MS, Camarao GC. et al. A new brain metalloendopeptidase which degrades the Alzheimer β-amyloid 1-40 peptide producing soluble fragments without neurotoxic effects. Braz J Med Biol Res. 1997;30:1153–1156. [PubMed: 9496430]
- 33.
- Yamin R, Malgeri EG, Sloane JA. et al. Metalloendopeptidase EC 3.4.24.15 is necessary for Alzheimer's amyloid-β peptide degradation. J Biol Chem. 1999;274:18777–18784. [PubMed: 10373494]
- 34.
- Takaki Y, Iwata N, Tsubuki S. et al. Biochemical identification of the neutral endopeptidase family member responsible for the catabolism of amyloid βpeptide in the brain. J Biochem (Tokyo). 2000;128:897–902. [PubMed: 11098130]
- 35.
- Shirotani K, Tsubuki S, Iwata N. et al. Neprilysin degrades both amyloid β peptides 1-40 and 1-42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem. 2001;276:21895–21901. [PubMed: 11278416]
- 36.
- Roques BP, Noble F, Dauge V. et al. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. 1993;45:87–146. [PubMed: 8475170]
- 37.
- Turner AJ. NeprilysinIn: Barrett AJ, Rawlings ND, Woessner JF, eds. Handbook of Proteolytic Enzymes.London: Academic Press,19981080-1085.
- 38.
- Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays. 2001;23(3):261–269. [PubMed: 11223883]
- 39.
- Carson JA, Turner AJ. β-Amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J Neurochem. 2002;81(1):1–8. [PubMed: 12067222]
- 40.
- Fukami S, Watanabe K, Iwata N. et al. Αβ-degrading endopeptidase, neprilysin, in mouse brain: Synaptic and axonal localization inversely correlating with Αβ pathology. Neurosci Res. 2002;43:39–56. [PubMed: 12074840]
- 41.
- Kiryu-Seo S, Sasaki M, Yokohama H. et al. Damage-induced neuronal endopeptidase (DINE) is a unique metallopeptidase expressed in response to neuronal damage and activates superoxide scavengers. Proc Natl Acad Sci USA. 2000;97:4345–4350. [PMC free article: PMC18244] [PubMed: 10759559]
- 42.
- Lu B, Gerard NP, Kolakowski Jr LF. et al. Neutral endopeptidase modulation of septic shock. J Exp Med. 1995;181:2271–2275. [PMC free article: PMC2192063] [PubMed: 7760013]
- 43.
- Duff K, Eckman C, Zehr C. et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. [PubMed: 8878479]
- 44.
- Borchelt DR, Thinakaran G, Eckman CB. et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Αβ1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. [PubMed: 8938131]
- 45.
- Oyama F, Sawamura N, Kobayashi K. et al. Mutant presenilin 2 transgenic mouse: effect on an age-dependent increase of amyloid β-protein 42 in the brain. J Neurochem. 1998;71(1):313–322. [PubMed: 9648880]
- 46.
- Nakano Y, Kondoh G, Kudo T. et al. Accumulation of murine amyloid-β42 in a gene-dosage-dependent manner in PS1 ‘knock-in’ mice. Eur J Neurosci. 1999;11:2577–2581. [PubMed: 10383647]
- 47.
- Yasojima K, Akiyama H, McGeer EG. et al. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett. 2001;297:97–100. [PubMed: 11121879]
- 48.
- Yasojima K, McGeer EG, McGeer PL. Relationship between β amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 2001;919:115–121. [PubMed: 11689168]
- 49.
- Iwata N, Takaki Y, Fukami S. et al. Region-specific reduction of Αβdegrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J Neurosci Res. 2002;70:493–500. [PubMed: 12391610]
- 50.
- Tekirian TL, Saido TC, Markesbery WR. et al. N-Terminal heterogeneity of parenchymal and cerebrovascular Αβ deposits. J Neuropathol Exp Neurol. 1998;57(1):76–94. [PubMed: 9600199]
- 51.
- Satou T, Cummings BJ, Head E. et al. The progression of β-amyloid deposition in the frontal cortex of the aged canine. Brain Res. 1997;774:35–43. [PubMed: 9452189]
- 52.
- Kanemaru K, Iwatsubo T, Ihara Y. Comparable amyloid β-protein (Αβ) 42(43) and Αβ 40 deposition in the aged monkey brain. Neurosci Lett. 1996;214:196–198. [PubMed: 8878117]
- 53.
- Hama E, Shirotani K, Masumoto H. et al. Clearance of extracellular and cell-associated amyloid β peptide through viral expression of neprilysin in primary neurons. J Biochem (Tokyo). 2001;130:721–726. [PubMed: 11726269]
- 54.
- Li C, Booze RM, Hersh LB. Tissue-specific expression of rat neutral endopeptidase (neprilysin) mRNAs. J Biol Chem. 1995;270:5723–5728. [PubMed: 7890699]
- 55.
- Wang TL, Chang H, Hung CR. et al. Morphine preconditioning attenuates neutrophil activation in rat models of myocardial infarction. Cardiovasc Res. 1998;40(3):557–563. [PubMed: 10070497]
- 56.
- Bae SJ, Matsunaga Y, Takenaka M. et al. Substance P induced preprotachykininA mRNA, neutral endopeptidase mRNA and substance P in cultured normal fibroblasts. Int Arch Allergy Immunol. 2002;127(4):316–321. [PubMed: 12021551]
- 57.
- Davies P, Katzman R, Terry RD. Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature. 1980;288(5788):279–280. [PubMed: 6107862]
- 58.
- Fujiyoshi K, Suga H, Okamoto K. et al. Reduction of arginine-vasopressin in the cerebral cortex in Alzheimer type senile dementia. J Neurol Neurosurg Psychiatry. 1987;50(7):929–932. [PMC free article: PMC1032137] [PubMed: 3625218]
- 59.
- Beal MF, Mazurek MF, Chattha GK. et al. Neuropeptide Y immunoreactivity is reduced in cerebral cortex in Alzheimer's disease. Ann Neurol. 1986;20(3):282–288. [PubMed: 3767313]
- 60.
- Crystal HA, Davies P. Cortical substance P-like immunoreactivity in cases of Alzheimer's disease and senile dementia of the Alzheimer type. J Neurochem. 1982;38(6):1781–1784. [PubMed: 6176686]
- 61.
- Bissette G, Reynolds GP, Kilts CD. et al. Corticotropin-releasing factor-like immunoreactivity in senile dementia of the Alzheimer type. Reduced cortical and striatal concentrations. JAMA. 1985;254(21):3067–3069. [PubMed: 3877182]
- 62.
- Fliers E, Swaab DF. Neuropeptide changes in aging and Alzheimer's disease. Prog Brain Res. 1986;70:141–152. [PubMed: 2883702]
- 63.
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6(4):347–470. [PubMed: 8915675]
- 64.
- Roth RA. InsulysinIn: Barrett AJ, Rawlings ND, Woessner JF, eds. Handbook of Proteolytic Enzymes. London: Academic Press,19981362–1367.
- 65.
- Edbauer D, Willem M, Lammich S. et al. Insulin-degrading enzyme rapidly removes the β-amyloid precursor protein intracellular domain (AICD). J Biol Chem. 2002;277(16):13389–13393. [PubMed: 11809755]
- 66.
- Bertram L, Blacker D, Mullin K. et al. Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science. 2000;290: 2302–2303. [PubMed: 11125142]
- 67.
- Cherny RA, Atwood CS, Xilinas ME. et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. [PubMed: 11430801]
- 68.
- Shipp MA, Ross KN, Tamayo P. et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8:68–74. [PubMed: 11786909]
- 69.
- Tibshirani R, Hastie T, Narasimhan B. et al. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci USA. 2002;99:6567–6572. [PMC free article: PMC124443] [PubMed: 12011421]
- 70.
- Lafrance MH, Vezina C, Wang Q. et al. Role of glycosylation in transport and enzymic activity of neutral endopeptidase-24.11. Biochem J. 1994;302(2):451–454. [PMC free article: PMC1137249] [PubMed: 8092997]
- 71.
- Suzuki N, Cheung TT, Cai XD. et al. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717)mutants. Science. 1994;264(5163):1336–1340. [PubMed: 8191290]
- Introduction: Why Αβ Degradation?
- Two Major Categories of Αβ-Degrading Mechanisms
- Identification of Neprilysin as a Major in vivo Αβ-Degrading Enzyme in Brain
- Region-Specific Decline of Neprilysin Expression and Activity in Brain Upon Aging
- Utilization of Neprilysin Activity for Prevention and Therapy of AD
- Other Αβ-Degrading Enzyme Candidates
- Human Genetics of Risk and Anti-Risk Factors for AD: A Proposal for Multi-Component Analysis*
- Acknowledgements
- References
- Proteolytic Degradation of Αβ by Neprilysin and Other Peptidases - Madame Curie ...Proteolytic Degradation of Αβ by Neprilysin and Other Peptidases - Madame Curie Bioscience Database
- The Role of IL-10 in Autoimmune Pathology - Madame Curie Bioscience DatabaseThe Role of IL-10 in Autoimmune Pathology - Madame Curie Bioscience Database
- Mating Cell-Cell Channels in Conjugating Bacteria - Madame Curie Bioscience Data...Mating Cell-Cell Channels in Conjugating Bacteria - Madame Curie Bioscience Database
- FROM STRUCTURE TO FUNCTION OF RNA BINDING DOMAINS - Madame Curie Bioscience Data...FROM STRUCTURE TO FUNCTION OF RNA BINDING DOMAINS - Madame Curie Bioscience Database
- Regulation and Coordination of Intracellular Trafficking: An Overview - Madame C...Regulation and Coordination of Intracellular Trafficking: An Overview - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...