

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Achromosomal double-strand break (DSB) can arise from multiple sources including ionizing radiation and DNA replication itself. An understanding of the intricate pro tein pathways that recognize DSBs and recruit the DNA repair and cell cycle checkpoint machinery is developing rapidly. The ATM kinase plays an early, pivotal role in the signaling process by detecting DSBs and relaying this information to numerous downstream transducer and effector proteins. Within minutes after DSBs occur, ATM undergoes inter-molecular autophosphorylation at Ser1981, which converts it to an active monomer. ATMSer1981-P immediately phosphorylates histone H2AX over a megabase region of DNA surrounding a DSB. Discrete nuclear foci of phosphorylated H2AX (γH2AX) are visible by immunofluorescence and appear to be true markers of DSBs. MDC1 and 53BP1, transducer proteins that contain two C-terminal BRCT domains, are also phosphorylated by ATM and colocalize faithfully with γH2AX. Subsequent transducers and effectors include the Mre11-Rad50-NBS1 complex (both transducer and effector), and the breast cancer susceptibility proteins BRCA1 (a transducer) and BRCA2 (an effector). BRCA2 interacts directly with DNA and the Rad51 strand-transferase to help initiate homologous recombination. When the DNA replication machinery is chemically inhibited or encounters a damaged template containing single-strand breaks or blocking lesions, replication forks may arrest, collapse into one-sided DSBs, and require recombinational repair to be reestablished. This recovery process is dependent on the ATR kinase acting in concert with the Rad17-Rfc clamp-loader complex and the Rad9-Rad1-Hus1 clamp complex. Modifiers of DNA topology, such as BLM and WRN helicases associated with Bloom and Werner syndromes, assist in preserving chromosomal continuity during replication. These proteins are thought to resolve anomalous replication intermediates that arise at stalled forks, thereby preventing aberrant recombination for unrepaired DSBs. Overall, the precise nature of a DSB likely determines whether ATM or ATR is utilized to initiate the damage-response pathways.
Replication-Independent Double-Strand Breaks (DSBs)
Origins of DSBs
Double-strand breaks (DSBs) are of fundamental importance in many fields of biology. The incorrect repair of DSBs often results in chromosomal rearrangements, which are considered to be a major initiating factor in carcinogenesis. Cancer cells generally exhibit numerous structural rearrangements (i.e., deletions, exchanges, duplications, and inversions) as well as increased numbers of chromosomes. Progression of malignancy often correlates with increased chromosomal instability and plasticity, which are driven by escalating defects in DNA repair processes1 and cell cycle checkpoint functions.24 The cellular lethality of ionizing radiation (IR) occurs largely through the production of DSBs. Many cancer treatments rely on the ability of IR and chemical agents (e.g., bleomycin) to produce DSBs that can be targeted to preferentially eradicate tumor cells versus damaging normal tissues. Thus, understanding the quantitative yields of DSBs and the molecular mechanisms that eliminate them is a central issue in cancer biology and radiation biology.
The yield of breaks produced by IR is estimated to be ˜35 DSBs per diploid G1 cell per Gy (measured at doses ≥ 20 Gy), compared with a value of ˜1000 single-strand breaks (SSBs) per Gy.58 Recent estimates of DSB yield measured by the frequency of IR-induced γH2AX foci9,10 at doses between 0.001 and 3 Gy give a value that is very similar to the ˜35 breaks per Gy determined at high doses by pulsed-field gel electrophoresis.11 However, estimates based on premature chromosome condensation (PCC), which allows visualization of chromosomes in G1 nuclei, are considerably lower at 5 to 6 DSB per Gy per cell.12,13 The reason(s) for this discrepancy is unclear. One possible explanation is that IR-generated DNA fragments arising from two or more DSBs in relatively close proximity would not be microscopically distinguishable from a single DSB. Second, in the PCC method some fraction of the rapidly repaired DSBs will be missed because of the 15–20 min post-IR incubation at 37°C required to produce cell fusion and chromosome condensation. This fraction could be as high as 65%.14 IR also produces oxidative base damage, but the amounts of damaged bases8 per Gy of radiation are estimated to be 10–100 fold lower than the steady state levels (˜1.5 ×105 oxidative base lesions per human cell15) produced by normal oxygen metabolism.16
Recently though, it has become apparent that IR produces clustered oxidative base damages and SSBs on opposite strands of the DNA molecule, which can develop into DSBs and represent potentially lethal lesions.1719 Clusters of closely opposed SSBs, oxidized purines, oxidized pyrimidines, or oxidized abasic sites within a few helical turns are estimated to comprise at least 70% of the complex lesions produced in cells.19 These clustered lesions are likely difficult to repair20 and may get converted to DSBs through processing by base-excision repair enzymes, or by interaction with DNA replication forks as they encounter clustered lesions.
DNA Repair Systems that Act on DSBs
Cells possess complex, highly efficient mechanisms for detecting DSBs and signaling their presence to the DNA repair and replication machinery. In this review, we address what is known about these recognition and information transfer systems outlined in (Fig. 1). Our understanding of how cells respond to DSBs has developed rapidly with respect to the enzymatic machinery that performs repair. Considerably less is known about the preceding events of detecting/sensing/recognizing breaks and the signaling processes that recruit DNA repair systems to the sites of damage. The two major pathways that repair DSBs are referred to as nonhomologous end joining (NHEJ)21,22 and homologous recombinational repair (HRR).2325 In this review, we address what is known about these recognition and information transfer systems, along with a brief, updated summary of HRR (Fig. 5).
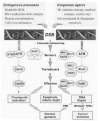

DSBs occur normally during meiosis to initiate strand exchange between homologous chromosomes and in hematopoietic cells during gene processing through V(D)J recombination, which mediates antibody diversity and gene rearrangements for T cell receptors. These highly regulated, programmed DSB-mediated processes are normally extremely accurate and utilize many of the same DNA break-processing enzymes that repair spontaneous or agent-induced DSBs. High levels of spontaneous DSBs and chromosomal rearrangements are observed in mouse cells carrying null mutations in the genes of the NHEJ complexes composed of Ku70-Ku86-DNA-PKcs or LIG4-XRCC4.2628 The levels of chromosomal breaks can be reduced by lowering the oxygen tension from 20% to 3%.29 Elevating the level of reactive oxidative species, rather surprisingly by overexpressing a transgene for the antioxidant enzyme superoxide dismutase 1 (SOD1), increases chromosome breakage. In SOD1-overexpressing cells, reducing oxygen to 3% also reduced chromosomal aberrations. The observation that oxidative damage results in spontaneous chromosome breaks may explain the neuronal degeneration and premature aging that typify mice having NHEJ mutations.3034
However, not all NHEJ-defective cell lines (i.e., Ku70 and Ku80 mutants in hamster CHO and chicken DT40 backgrounds) display markedly increased levels of spontaneous chromosomal aberrations.3537 The reason for the significant differences among cell types is not clear, but it is noteworthy that both the CHO and DT40 lines are defective for Tp53. Perhaps this defect allows for increased DNA-PKcs-independent end joining in the absence of the DNA-PK or LIG4-XRCC4 complexes.
Central Role of the ATM Kinase in DSB Signaling
The large ATM (ataxia telangiectasia mutated) and ATR (AT and Rad3-related) kinases have come into focus as early, central participants in the DNA damage recognition and signaling processes (Fig. 2 and Fig. 6).3,3841These functionally related proteins phosphorylate a multitude of substrates and appear to exist in vivo in high molecular weight complexes of >2 × 106 Da,42,43 which may contain many other damage-response proteins.44 Figure 2 expands the theme of (Fig. 1) by depicting numerous phosphorylation events as well as functionally important protein interactions. Exposure of cells to IR immediately activates the ATM kinase (3056 a.a.; Tel1Sp and Tel1Sc homologs in yeasts),45,46 and ATP can also induce activation of by a mechanism involving autophosphorylation.47 A major advance came with the discovery that IR-induced activation occurs through intermolecular autophosphorylation of Ser1981, which causes dissociation of ATM dimers and enhancement of kinase activity.48 After IR, phosphorylation of Ser1981 is maximal within 5 min and saturates at a dose of ˜40 cGy.48 Upon activation, ATM phosphorylates histone H2AX (modification referred to as γH2AX);49 DNA-PK and ATR also contribute to this modification.5053 Although ATM binds preferentially to DNA ends in vitro,54 the in vivo activation likely results from changes in chromatin structure instead of DNA binding.48 ATM phosphorylates numerous key proteins that often appear in nuclear foci (described below) and that mediate checkpoints and DNA repair: namely 53BP1, MDC1/NFBD1, Chk1, Chk2, NBS1, BRCA1, and FANCD2. Altogether, ATM has more than 20 substrates, as recently reviewed in more detail.25 Thus, throughout the cell cycle ATM acts as a master regulator and coordinator in the initial response to DSBs that are not associated with replication forks. ATM is also activated by agents such as methylating chemicals that do not directly cause DSBs, but lead to lesions that are subsequently converted to DSBs.55 The closely related ATR kinase discussed below may serve as a partial backup system for ATM and help to reinforce at later times the phosphorylating signaling initiated by ATM (see discussion in ref.41).

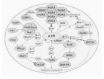
The fact that ATM is responsible for phosphorylating proteins that implement repair and checkpoint functions suggests that ATM itself might concentrate at sites of DSBs. Indeed, within 5 min after IR, the Ser1981-phosphorylated form of ATM begins to form foci that colocalize with γH2AX foci, and these become distinct foci by 60 min.48 Under conditions of detergent extraction to remove nucleoplasmic proteins, a portion of the total ATM pool becomes resistant to extraction and is detected in nuclear aggregates immediately after DSB formation.56 These aggregates are much more diffuse than the distinct foci formed by γH2AX.
Although the sensor proteins that first recognize DSBs are not well understood, ATM is a major candidate sensor protein. It could act alone or in combination with other proteins discussed below such as 53BP157 and MDC1/NFBD158 that localize within minutes to sites of DSBs. It is noteworthy that certain ATM mutations display a dominant negative phenotype in the heterozygous state, both in humans and mice.59,60 This situation could arise if mutant ATM binds and sequesters partner proteins into dysfunctional complexes that compete with normal complexes for DNA substrates. Although ATM is required to phosphorylate both NBS1 and Mre11,6165 genetic evidence suggests that the Mre11-Rad50-NBS1 (MRN) complex acts upstream of ATM, at least for some signaling events.66 Mre11-defective human cells show reductions in detergent-resistant retention of ATM protein, ATM kinase activity, and phosphorylation of downstream targets.
Origins of Nuclear Foci that Form in Response to DSBs
When cells are exposed to DNA damaging agents, the redistribution and subnuclear localization of specific proteins can be monitored to infer which proteins are important in damage recognition, signaling, checkpoint implementation, and repair. In mammalian cells, Rad51 protein, involved in homologous recombination, was one of the first proteins detected in discrete nuclear foci using immunofluorescence on mitotic and meiotic cells67,68 (Fig. 3A). The cytological visibility of these foci can be readily explained by the fact that Rad51 forms nucleoprotein filaments that can contain hundreds of Rad51 molecules. It is estimated that ˜100 fluorophore molecules localized within a very small volume are necessary for a visible focus.69 Many other proteins discussed below also form foci, but not all of these are expected to assemble en masse as multimeric functional complexes like Rad51. For example, the MRN complex is a key component in the processing of DSB termini,7072 and the MRN complex forms foci. Although the precise biochemical roles of this complex are not understood, only one or a few of these complexes, as a catalytic component, may be needed to produce single-stranded tails at the termini of DSBs prior to their repair by homologous recombination. Yet, focus formation may arise from the creation of multiple high-affinity binding sites for MRN in the vicinity of the DSB, thus causing numerous MRN complexes to concentrate at the modified site. This idea is illustrated by the observation that foci of Rad52 and Rad54 are highly dynamic structures.73 These foci exhibit rapid exchange of these proteins, which are recruited independently with differing mobility.
γH2AX Formation as a Marker of Radiation-Induced DSBs: Impact on Checkpoints and Repair
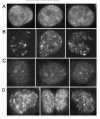
Recent developments suggest the possibility that units of higher order chromatin structure may facilitate the detection of DSBs within DNA. In response to the introduction of DSBs, the minor histone H2AX is rapidly phosphorylated in a dose-dependent manner on Ser139 (located four amino acids from the carboxyl terminus), yielding the form designated γH2AX.49 Each DSB produces ˜2000 γH2AX molecules and results in the modification of H2AX over a region corresponding to ˜ 2 Mbp,49 which is the equivalent of 0.03% of the chromatin. An antibody specific for the modified C-terminus of H2AX reveals that γH2AX appears as discrete nuclear foci within 1 min after exposure of cells to ionizing radiation.50 Cells in all phases of the cycle, including mitosis, show foci.50,53. Importantly, the number of foci agrees with the estimated number of induced DNA DSBs.10,11,50 Significant phosphorylation of H2AX was also observed after treatment with the DSB-inducing agents neocarzinostatin, bleomycin, and etoposide, whereas UV irradiation and the DNA methylating agent methyl methanesulfonate did not produce γH2AX.51 This pattern further supports the idea that γH2AX phosphorylation occurs specifically in response to DSBs. Recent studies show that the formation of γH2AX is severely reduced in DNA-damaged ataxia telangiectasia cells and that the residual level could be attributed to DNA-PK activity51,74 NBS cells (Nijmegen Breakage Syndrome; NBS1 is part of the MRN complex) have normal γH2AX focus formation,75 but depletion of the MDC1 signal transducer discussed below reduces focus formation.58 IR-induced H2AX phosphorylation is a highly conserved process that is present in vertebrates, Drosophila, and yeast.50 γH2AX is formed in response to DSBs arising by diverse means: directly from environmental insult by radiation or chemicals, collapse of DNA replication forks, and programmed processes that enzymatically introduce DSBs (e.g., meiosis). The trigger for γH2AX formation may involve topological changes in the DNA, such as the degree of super coiling.
The biological importance of γH2AX formation in maintaining chromosome stability is clearly revealed by the phenotypes of mice carrying knockout mutations in one or both copies the H2AX gene.76,77 H2AXΔ/Δ mice are growth retarded, radiation sensitive, immune deficient, and defective in spermatogenesis. The IR sensitivity of H2AXΔ/Δ ES (embryonic stem) cells is increased ˜3-fold,77 whereas the sensitivity of immortalized MEF (mouse embryonic fibroblast) cultures is increased only ˜1.6-fold, but nevertheless these latter cells were shown to have reduced DSB repair.76 Spontaneous chromosomal aberrations are also markedly elevated, e.g., from 5% in heterozygous H2AXFlox/Δ controls to 22% in H2AXΔ/Δ ES cells.77 Although checkpoint functions in all phases of the cell cycle were considered to be normal in H2AX/ cells following 10 Gy irradiation,76,77 at lower doses a clear G2 checkpoint defect was seen in both mouse B cells and MEFs.78 Thus, H2AX phosphorylation signals for both checkpoint activation and repair. In summary, the pleiotropic phenotype of H2AX/ mice is caused by defects in signaling that include impaired recruitment of MDC1, 53Bp1, NBS1, and Brca1 (but not Rad51) into IR-induced foci.58,76,77
Recruitment and CoLocalization of Signaling and Repair Proteins to Sites of γH2AX Foci
In this section we outline the characteristics of many proteins that are implicated in signaling, checkpoints, and repair through their redistribution within the nucleus in response to DNA breakage. Some of these proteins, such as MDC1/NFBD1 and 53BP1, appear to arise as quickly as, and coincident with, γH2AX foci while other foci (e.g., Rad51) arise much later. By further example, colocalizing γH2AX-BRCA1 foci were reported to appear sooner than γH2AX-Rad50 foci,75 implying that BRCA1 may act upstream of the Rad50 complex (MRN, discussed below). Figure 4 summarizes available information on the order of appearance of foci, including colocalizations. Certainly there are numerous pitfalls in deciphering the significance of focus formation. Foci studies performed at high doses (e.g., > 5 Gy) and many hours after exposure will be much more difficult to interpret than studies terminated minutes after irradiation at low doses (e.g., < 1 Gy). The remainder of this section summarizes the characteristics and significance of many proteins that have been shown to form nuclear foci.

MDC1/NFBD1
Very recently a new nuclear human protein, MDC1 (mediator of DNA checkpoint; also called NFBD1 for nuclear factor containing two BRCT domains at the C-terminus; 2089 a.a.), which constitutively binds to chromatin, was identified as a very early participant in the recognition and signaling process.58,7982 Because of its C-terminal BRCT domains, MDC1 is a candidate functional homolog of Rad9Sc, one of the first checkpoint proteins to be identified in budding yeast.83,84 After IR damage, MDC1 becomes hyperphosphorylated in an ATM-, NBS1-, and Chk2-dependent manner,58,80,81 but MDC1 focus formation is still seen in the absence of ATM.81 Within 1 min after irradiation, MDC1 forms visible foci that peak in frequency at 30 min.58,82 MDC1 foci colocalize precisely with γH2AX foci, and γH2AX is needed for MDC1 focus formation.58,82 H2AX and MDC1 are mutually interdependent for phosphorylation and focus formation, and MDC1 forms complexes with γH2AX.58 However, there are conflicting data concerning whether MDC1 is required for 53BP1 focus formation.58,82 Suppression of MDC1 by siRNA results in decreased phosphorylation of SMC1S996 and Chk1S345.58 This defect in Chk1 phosphorylation results in defective intra-S phase and G2 checkpoints after IR exposure. Suppression of MDC1 expression by siRNA also causes reduced apoptosis in response to IR damage (because of the loss of the MDC1-Chk2P68 interaction80), but also decreases colony-forming ability.82
53BP1
Human 53BP1 (1972 a.a. and containing two C-terminal BRCT85 repeats like MDC1/NFBD1) was identified as a Tp53-interacting protein in a yeast two-hybrid screen86,87 and was found to have homology with Rad9Sc.88 53BP1-null (or 53BP1-truncated) mice have a phenotype resembling that of H2AX-deficient mice and that of AT, i.e., growth retardation, immune deficiency, radiation sensitivity, impaired Chk2 phosphorylation/activation (Fig. 2), and cancer proneness.89,90 Although embryo-derived cultures appear to have intact G1, S, and G2 checkpoints,89 other studies with different cell types report a requirement for 53BP1 in the S and G2 checkpoints.57,78
Cytologically, 53BP1 shows diffuse nuclear immunostaining in undamaged G1 cells but a punctate pattern in S phase,90 suggesting localization to sites of stalled or broken replication forks. 53BP1 localizes into nuclear foci in most cells as early as five min after IR exposure; doses as low as 0.5 Gy result in formation of these foci.88 After 1 Gy, the maximal numbers of foci per cell (≇ 20–35) and of foci-positive cells (˜92%) are seen between 15 and 120 min.78,88,91 This number of foci is consistent with the expected yield of DSBs, as discussed above. Significant colocalization of 53BP1 with γH2AX is seen between 10 and 240 min,78,88,91 and the two proteins show damage-dependent coimmunoprecipitation.91 As with γH2AX, 53BP1 focus formation is specific for agents that produce DSBs and occurs more rapidly than that of other proteins discussed below (BRCA1, MRN, and Rad51). Phosphorylation and focus formation of 53BP1 are controlled independently.92 One notable difference between 53BP1 and γH2AX is that only γH2AX forms foci in mitosis,50,93 where 53BP1 associates with kinetocores.94
Recent work shows that 53BP1 has a central role in IR-induced DSB signaling for the Sand G2-phase checkpoints [see editorial in ref. 95]. Upon inhibition of 53BP1 by siRNA, phosphorylation of Tp53 and BRCA1 by ATM is blocked, and the downstream formation of IR-induced BRCA1 foci is largely abolished.57 Studies with mouse knockout cells show that 53BP1 is required for a normal G2-M checkpoint at low IR doses.78 In AT cells, 53BP1 focus formation after 1 Gy was diminished at early times (10–20 min post irradiation) in one study,91 and the hyperphosphorylation that occurs after irradiation was absent or reduced.91,93 However, in other studies that used 3 or 8 Gy, 53BP1 focus formation appeared to be normal in AT cells,88,96 perhaps because of the higher doses used. ATM phosphorylates 53BP1 in vitro,78,91,92 further suggesting that ATM is responsible for directly phosphorylating 53BP1 within γH2AX foci. ATM and 53BP1 also show IR-dependent coimmunoprecipitation.96 53BP1 may either help recruit and activate ATM at sites of DSBs, or recruit ATM substrates to these sites.96 It is noteworthy that 53BP1, as well as Chk2, appears to be present in γH2AX foci before any of the proteins that participate in DSB repair. These results suggest that the initiation of checkpoints precedes the onset of DSB repair, which occurs over a period of several hours, depending on the dose.
HDAC4
Histone deacetylase 4 (HDAC4) is another early participant in IR-induced focus formation, and HDAC4 foci are seen in cells defective in ATM, DNA-PKcs, or NBS1.97 In contrast, HDAC2 or HDAC6 do not form foci. Interestingly, the stability of 53BP1, as well as its focus formation, shows a dependence on the presence of HDAC4, as shown by siRNA inhibition experiments. Depletion of HDAC4 abrogates the G2 delay and confers sensitization to killing by IR while also reducing cell viability.97 In this study the authors suggested that the degree of persistence of HDAC4 foci might be a measure of cellular radiosensitivity.
ChK2
Chk2/Cds1 is a key checkpoint kinase (for reviews see refs. 3,98,99), whose role appears to be primarily promoting apoptosis, not cell survival, after IR exposure.100,101 Chk2-deficient mice are radioresistant and defective in Tp53-mediated transcriptional changes.100,101 Thymocytes, splenocytes, skin, and neurons in the developing brain show protection from IR-induced apoptosis. The G1 checkpoint, but not the G2 or S-phase checkpoints, was substantially impaired in Chk2-/- embryonic fibroblasts and ES cells.100,101 IR-induced stabilization of Tp53 in Chk2-/- cells is ˜60% of that in wild-type cells.101 Caffeine further reduces Tp53 accumulation, suggesting the presence of another pathway for Tp53 stabilization that is ATM/ATR-dependent, but Chk2-independent. In spite of Tp53's partial stabilization and phosphorylation at Ser23 (Ser20 in human cells) in the absence of Chk2, Tp53-dependent transcriptional induction of target genes, such as CDKN1A/p21, was not observed in Chk2-/- cells.
IR treatment results in ATM-dependent activation and phosphorylation of Chk2 at Thr68.102 The complete activation of Chk2 requires NBS1.103,104 Immuno-staining using phospho-specific Chk2 antibodies suggests that Chk2-Thr68P localizes into discrete foci within 10 min after irradiation and colocalizes with both γH2AX and 53BP1 (Fig. 4).57,102 These Chk2-Thr68P foci did not appear in AT cells or in 53BP1-depleted cells, and nonphosphorylated Chk2 molecules remain distributed throughout the nucleus.57 However, a subsequent study using 53BP1 mouse knockout cells and a different Chk2-Thr68P antibody concluded that 53BP1 is not required for Chk2 activation.78 These different conclusions might be accounted for by inadequate specificity of the Chk2-Thr68P antibody102 used in the former studies (see commentary in ref. 95). In summary, phosphorylation and focus formation of Chk2 may be a major determinant in programming cells for elimination by apoptosis through Chk2's site-specific phosphorylation of Tp53.
Chk1
Few Chk1 foci studies have been reported, but Chk1 and BRCA1 foci colocalize in the absence of IR exposure.105 In comparison to Chk2 described above, after IR damage in chicken DT40 cells Chk1 promotes reproductive survival, apoptosis (in these Tp53-deficient cells), implementation of the G2 checkpoint, and phosphorylation of Cdc2.106 Notably, Chk1 null cells completely lose the G2 checkpoint in DT40 cells (see Fig. 2). Inhibition of human Chk1 with siRNA also results in a G2 checkpoint defect, and phosphorylation of Ser317 and Ser345 appears nonessential for IR-mediated activation of Chk1 and the G2 checkpoint.107 IR activation of Chk1 depends on BRCA1.105
MRN Complex
The analysis of nuclear focus formation by various damage-response proteins suggests that γH2AX formation plays a critical role in recruiting and assembling repair proteins, including the Mre11-Rad50-NBS1 (MRN) complex. This complex is essential for HRR in human cells since hypomorphic mutations confer radiosensitivity.108110 MRN localizes to sites of damage within 30 min after irradiation111 (Fig. 4), and recent genetic evidence suggests that it may be a primary DSB recognition factor.66 By using a 390-nm laser combined with BrdUrd incorporation and Hoechst dye 33258112 to produce striped regions of DSBs, Bonner and coworkers have shown in human breast tumor MCF7 cells that γH2AX stripes appear rapidly in all cells, and MRN colocalizes to these stripes within 30 min.75 Colocalization of NBS1 and γH2AX foci appears to involve a direct interaction between NBS1 and γH2AX (and not H2AX), which is mediated by the FHA/BRCT domain of NBS1.52
Initial observations of MRN foci indicated that the kinetics of appearance was too delayed to correspond with productive repair of DSBs.113 However, the patterns of focus formation of proteins such as Rad50 and Rad51 are strongly influenced by the method of preparing the cells. In earlier experiments using methanol fixation and acetone to permeabilize human diploid fibroblasts, the percentage of nuclei that were positive for Rad50 focus formation after 12 Gy γ-irradiation reached a maximum of ˜65% after 8 hr.113 In comparison, nuclei with Rad51 foci reached a broader maximum between 4–8 hr at ˜35%. Subsequent studies in detergent extracted cells, designed to reveal proteins tightly associated with chromatin, found that MRN foci were detectable within 10 min after irradiation and reached a maximum at 2 hr when 70–95% of cells were positive.114 Since MRN foci are present in AT cells, MRN focus formation does not depend on the phosphorylation of NBS1 by ATM.6164 These results suggest that the MRN complex may participate in a very early, ATM-independent step of DSB recognition.66 Larger aggregates of MRN foci that become apparent by 8 hr in normal cells were not seen in AT cells.114 These larger, more robust foci may represent sites of slow or abortive repair of complex DSBs. In unirradiated detergent-extracted cells, Mre11 shows a high degree of colocalization with immunostaining of PML bodies, a nuclear depot of many proteins that may help regulate cellular defense against insults such as viruses.115
BRCA1 and Rad51
Radiation-induced γH2AX focus formation occurs within several minutes in both MCF7 and IMR90 human cells after exposure to only 0.6 Gy.50 However, higher radiation doses are usually needed to visualize the foci formed by several key proteins that are recruited to the γH2AX foci. After 12 Gy of IR, the kinetics of BRCA1 focus formation is significantly more rapid than that of MRN focus formation.75 By 2 hr, 10–15% of IMR90 cells show BRCA1 foci, and these overlap extensively with γH2AX foci.50 While γH2AX-BRCA1 colocalization is maximal by 2 hr, γH2AX-MRN colocalization increases up to 8 hr. BRCA1 focus formation and hyperphosphorylation after IR is dependent on MDC1.116 In the absence of detergent extraction, Rad51 foci appear at about the same time as γH2AX-MRN foci, but in different cells.113 Since MRN acts upstream of Rad51 (see Fig. 5), this observation is paradoxical unless all DSB repair events in a given cell were to occur in a synchronous manner. Rad51 foci become prominent at 6 hr in 20–25% of the cells, when the majority of these foci colocalize with BRCA1 foci. Rad51 foci form relatively slowly, are much less numerous than the estimated numbers of DSBs, and require high doses for their detection. Therefore, their biological significance is still unclear.
In SV40-transformed fibroblasts, the kinetics of focus formation was faster (e.g., γH2AX-BRCA1 colocalization in 45 min) and more cells had Rad51 foci, but the order of foci appearance was the same as for IMR90 cells.75 The relatively early appearance of γH2AX-BRCA1 foci suggests that BRCA1 might interact directly with DNA breaks.117
Pretreatment of MCF7 breast carcinoma cells for 30 min with phosphatidylinositol-3-kinase inhibitor wortmannin, which inhibits ATM and DNA-PKcs, completely blocks γH2AX focus formation after irradiation and prevents the formation of repair-protein foci.75 Importantly, when added 5 min after irradiation wortmannin has no effect on γH2AX, BRCA1, and Rad51 focus formation. These results emphasize that γH2AX is an early, critical event that initiates DNA repair processes. Consistent with this idea is the finding in mouse H2AX/ knockout B cells or ES cells that BRCA1 and MRN foci cannot form in response to irradiation.76,77 However, Rad51 foci do form in both these mutant cell types although their intensity seems to be diminished in the mutant ES cells exposed to 20 Gy.76,77
DNA-PK
The kinase activity of DNA-PK is required for the efficient repair of DSBs by NHEJ, but the molecular mechanism underlying this activity is not understood.118,119 Very recently the catalytic subunit of DNA-PK (DNA-PKcs) was found to undergo autophosphorylation in vitro at the highly conserved Thr2609 and phosphorylation at this position occurred in vivo in response to IR damage.120 DNA-PKcs Thr2609 colocalizes within 30 min after IR with both γH2AX and 53BP1. After 10 Gy the phosphorylation of Thr2609 is maximal by 30 min and persists for up to 4 hr. The biological significance of this phosphorylation is indicated by the finding that a Thr2609Ala substitution mutation is associated with defective DSB rejoining and causes increased radiation sensitivity in CHO cells. DNA-PK autophosphorylation at Thr2609is reduced in the absence of ATM. Attempts to identify Ku70/86 foci in irradiated cells have generally been unsuccessful in seeing localization of this DNA end-binding complex.114,121 Immunostaining of Ku70/86, or total DNA-PKcs, in the absence of detergent extraction, does not reveal discrete foci. This specificity suggests that only DNA-PKcsThr2609 may be needed to recruit Ku70/86 to DSBs, and that a high local concentration of Ku70/86 at a break may be unnecessary because of its abundance in the nucleus.
RPA
The trimeric RPA complex binds single-stranded DNA and is an essential component of both HRR and DNA replication. RPA might provide an appropriate marker for foci in which recombination has been initiated through the processing of DSB ends into structures containing single-stranded DNA coated with RPA. In human cells, interaction between RPA and Rad51 is mediated by the 70-kDa subunit of RPA.122 RPA foci induced by irradiation are detectable at doses as low as 0.5 Gy and are present at 2 hr (and possibly earlier).123 In focus-positive cells, the number of RPA foci per nucleus reached a maximum at ˜3 hr. It would be of interest to examine the colocalization of RPA with Rad51 at low doses that allow relatively high cell survival (e.g., 0.5 Gy). This information might allow an estimate of the time required for Rad51 filament formation to occur.
TOPBP1
Human TopBP1 (1522 a.a.) has sequence similarity with other checkpoint proteins (Rad4/ Cut5Sc, Dpb11Sp, & Mus101Dm) and contains eight BRCT domains.124 TopBP1 binds to DNA ends through its BRCT domains,125 is required for DNA replication, and interacts with Polϵ.126 After radiation damage, ATM phosphorylates TopBP1, but TopBP1 focus formation occurs even in AT cells.127 TopBP1 colocalizes ˜50% with 53BP1 within one hr after irradiation and substantially colocalizes with NBS1 and BRCA1 at six hr after irradiation (Fig. 4). Inhibition of DNA synthesis by hydroxyurea results in relocalization of TopBP1 together with BRCA1 to replication forks, suggesting a role for TopBP1 in rescue of stalled forks.126
Kinetics of DSB Repair and Contributions of NHEJ Versus HRR
It is informative to compare the information on redistribution of repair proteins in (Fig. 4) with the published studies on the kinetics of DSB rejoining using pulsed-field gel electrophoresis. Rejoining experiments are generally conducted at even higher radiation doses than those used in foci studies. Therefore, the kinetics observed may underestimate the rates of DSB repair occurring at physiological levels of damage. Nevertheless, in a variety of vertebrate cell lines, DSB repair measured by electrophoresis or neutral filter-elution exhibits a rapid component with a half-life of ˜15 min and a slow component of ≥ 3–5 hr.14,128,129 SSBs are repaired, more rapidly than DSBs, and with a rapid component that has a half-life of ˜ 4 min.130,131 In a study using premature chromosome condensation to monitor the rate of repair of visible chromosomal breaks after 6 Gy of x-rays, normal fibroblasts eliminated breaks with a half-life of 1.7 hr,12 which is intermediate between the rapid and slow components determined by electrophoresis.
The analysis of mutant cell lines provides insight into the relative contributions of NHEJ and HRR pathways to the kinetics of DSB repair. In mouse scid cells, which have mutant DNA-PKcs, primarily the slow component of the biphasic curve was prolonged after a high IR dose. The final residual level of breaks was the same as in control cells.132 Similarly, an extensive study of NHEJ and HRR mutants of chicken DT40 cells found that the ku70 mutant, but not HRR mutants (rad51, rad52, rad54, and rad51b), had an increased half-time for DSB rejoining.14 However, since assays of DNA size typically do not distinguish between correctly and incorrectly rejoined ends, there could well be qualitative differences between the mutants in the two pathways. Correct rejoining events, measured by restriction-fragment analysis in normal human fibroblasts, occur primarily within the first two hours, and misjoining events occur more slowly.133 Recent work indicating the saturation of HRR at high IR doses (e.g., 20 Gy)134 suggests that DSB repair studies done at such doses have limited biological relevance. This saturation explains why HRR mutants have not shown defects in physical assays of DSB repair.
HRR as an Error-Free Mechanism of DSB Repair in S and G2 Phases
HRR acts on DSBs that arise at broken replication forks or on DSBs occurring in segments of DNA that have already replicated. In the G1 phase of the cell cycle, homologous chromosomes do not participate in HRR at an appreciable frequency.135 Since the proteins that mediate HRR in mammalian cells were recently reviewed,24,25 they will not be discussed in detail. Figure 5 outlines the steps in HRR. BRCA1 and BRCA2 are essential for efficient HRR,136,137 but the precise biochemical role for BRCA1 remains unclear. The precise functions of the MRN complex are also poorly understood. It may have architectural functions besides an enzymatic role in end processing as shown in (Fig. 5)72,138140 The finding that null mutations in any of the MRN proteins, as well as in Rad51 and BRCA1/2, are incompatible with cell viability (see review in ref. 25) points to their having essential functions that likely coordinate HRR with DNA replication. (This feature of essentialness also imposes limitations in determining the quantitative contribution of HRR to DSB repair.) Hypomorphic mutations in BRCA2 are remarkably similar in phenotype to mutants of each of the five Rad51 paralogs (XRCC2, XRCC3, Rad51B, Rad51C, and Rad51D) (see review in ref. 25). Recent structural studies implicate BRCA2 directly in binding single-stranded DNA and in assembling the Rad51 nucleoprotein filament.141143
Replication Associated DSBs (One-Sided Breaks)
Overview
As cells proliferate, DNA-damaging biochemical reactions produce lesions that interact with the DNA replication machinery. Electron transfer during oxidative phosphorylation produces reactive oxidative species, which generate DNA single-strand breaks and oxidation products; enzymatic activation of procarcinogens generates species that form bulky adducts. Chromosomal discontinuities will arise during S-phase more frequently when replication encounters lesions (SSBs, adducts, oxidized bases, or abasic sites). Thus, a wide range of DNA damages likely give rise to DSBs during DNA replication. To deal with replication-interfering damages, cells possess an impressive array of safeguards that begin with the informational redundancy in the DNA duplex. Multiple, specialized polymerases with lesion-bypass activities help maintain the integrity of the DNA molecule,144146 but these error-prone polymerases have a finite coping capacity. Elegant interrelated checkpoint and repair systems are highly integrated with the replication and transcription machinery to prevent broken or rearranged chromosomes from being passed to daughter cells.
A variety of recent studies provide compelling evidence that DSBs normally arise during DNA replication. First, null mutations in ATR147 or the homologous recombination machinery (mre11, nbs1, rad50, and rad51 mutants) result in cell lethality that is associated with extensive chromosome breakage at metaphase.148153 In sperm nuclei replicating in Xenopus egg extracts, DSBs are detected as ends that label with terminal transferase and by the formation of γH2AX.154 These breaks are only detectable when DNA replicates in the absence of the MRN complex, indicating a vital role for MRN in repairing replication-associated breaks. Since low levels of γH2AX foci are normally present in S phase mammalian cells (less than one visible focus per cell in one study),134 and γH2AX foci directly correlate with DSBs,11 S-phase DSBs are likely rapidly repaired. Second, an extrapolation of findings in S. cerevisiae,155 based on relative genome size, indicates that ˜100 homologous recombination events, which would be initiated by DSBs, might occur in a diploid mammalian cell during each S phase. Third, this numerical estimate is similar to that (i.e., >90) derived from the frequency of sister-chromatid exchange and the very low frequency of crossing over during HRR of endonuclease-generated DSBs.25
DSBs arising at replication forks are thought to trigger checkpoint signaling by ATR156,157 and are dealt with by replication-fork restart and recombinational repair mechanisms (see reviews in refs. 158,159). ATR's burden of maintaining chromosome continuity during replication becomes heavier in cells lacking Tp53 (e.g., many kinds of tumor cells) because of a defective G1 checkpoint, which normally allows for the removal of damage before DNA replication.160 It should be emphasized that DSBs arising at replication forks differ topologically from those produced by IR in that they generally involve the creation of only one double-stranded end, i.e., a one-ended chromosome break.161,162 Such asymmetric DSBs may be preferentially recognized by the HRR machinery to accomplish error-free repair, with a lesser role played by the NHEJ machinery during S phase.
This section deals primarily with DSBs that are associated with replication and elicit the “replication checkpoint”, triggered by abnormal DNA structures arising as a consequence of a blocked or collapsed (broken) fork. However, another checkpoint pathway in S phase cells has been defined historically and is referred to as the “intra-S” or “S-phase” checkpoint, which occurs when cells are exposed to IR and the DSBs are not fork-associated. When the S-phase checkpoint is activated, the initiation of replication is preferentially inhibited compared with elongation of active replicons. The signal for this inhibition is presumably DSBs although the finding that deficiencies in Msh2 and Mlh1 compromise this checkpoint163 prompt the question as to whether other DNA lesions may initiate the process. The S phase checkpoint was originally identified by the finding that AT cells displayed “radioresistant DNA synthesis” caused by the lack of inhibition of replicon initiation.164 Besides ATM,165 the S-phase checkpoint requires MDC1,58,80,81 NBS1,61,70,108 Mre11,109 Chk1,107 SMC1,166 FANCD2,167 and Msh2/ Mlh1.163 Two complementary pathways for S-phase checkpoint activation, both of which require ATM, have been described. One subpathway operates through the Chk2 kinase and the other through phosphorylated NBS1 and SMC1 (see Fig. 2, lower left).166,168 The function of phosphorylated SMC1 is not yet known. Thus, the replication and S-phase checkpoints both act to slow the progression of cells through S phase, but act through different pathways (compare Fig. 2 and 6). The replication checkpoint depends primarily on ATR rather than ATM, as diagrammed in (Fig. 6) Despite these differences, the two pathways exhibit overlap in the activation of their downstream effectors. After IR damage, the G2/M checkpoint requires the cooperation of both ATM and ATR, as revealed from an elegant analysis of single and double mutants.169
Replication-Associated Dsbs Arising from a Damaged Template
In the simplest case, a DSB may occur in one daughter chromatid when a replication fork encounters a SSB, which can arise as an intermediate in base excision repair. Under conditions where excess SSBs are present, as during the repair of methylation damage (e.g., MMS exposure), the production of replication-associated DSBs will be exacerbated. When helicase and polymerase activities become uncoupled at the replication fork170172 and generate extended regions of single-stranded DNA (e.g., 1 kb), the likelihood of disrupting chromatid continuity may increase because of increased exposure of SSBs. Moreover, new SSBs might arise from the nicking activity of nucleases acting on single-stranded DNA.
When replication forks encounter polymerase-blocking lesions such as bulky adducts, DSBs can arise by several processes. A blocking lesion in the leading strand can result in the generation of extensive downstream ssDNA in that strand (Fig. 7A). These single-stranded gaps have been documented experimentally during SV40 replication that initiates upstream of a pyrimidine-dimer173,174 and are favored under conditions when damage bypass of specific lesions is inhibited.144,175 Under these conditions, ssDNA is generated as the replication fork proceeds while chain extension on the leading strand is restricted. The resultant DNA structures can become destabilized and cause the marked increase in DSBs seen after UV irradiation of both yeast176 and hamster cells177 containing photolytic lesions induced by UVA in BrdUrd-substituted DNA in the presence of a photosensitizing dye. This phenomenon is greatly exaggerated in bypass-deficient xeroderma pigmentosum variant (XP-V) cells, which are defective in the bypass polymerase Y/η (discussed further below).178,179 Regions of ssDNA are also candidates for the formation of secondary structures such as hairpins and cruciform structures that can be recognized and cleaved by the MRN complex or other enzymes.180183
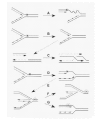
Under certain circumstances stalled replication forks can regress or reverse to generate a 4-stranded structure (often called a “chickenfoot”184) that can be considered a topologically masked DSB (Fig. 7E). Recent electron micrographs reveal the presence of such structures in yeast rad53 kinase checkpoint mutants.185 However, in mammalian cells early studies cast doubt on the existence of these structures. The appearance of doubly-dense DNA from cells pulse labeled with BrdUrd was found to decrease dramatically upon crosslinking with psoralen, suggesting that branch migration occurred as an artifact of DNA isolation.186,187 Regressed forks are structurally similar to, but topologically distinct from, Holliday junctions (HJs), a common intermediate produced during the repair of DSBs by HR (Fig. 7C). These 4-stranded structures generated by fork regression can be unwound by the structure specific helicases WRN and BLM188190 and cut by resolvases.191,192 Thus, branched intermediate structures that arise at sites of stalled replication can be converted to DSBs in a manner dependent upon their structural and topological context, as further addressed below.
Replication-Associated DSBs Arising from Inhibition of Replication
As in E. coli and yeast,193195 stalled replication forks caused by DNA synthesis inhibitors in mammalian cells are an efficient source of DSBs, cytotoxicity, and potentially deleterious recombination.196 Inhibition of DNA replication by hydroxyurea and aphidicolin produces chromatid discontinuities at replication forks.197,198 DSB-promoted intrachromosomal recombination of a partially duplicated HPRT gene in hamster cells was most strongly induced by inhibitors of DNA synthesis (hydroxyurea, methotrexate, aphidicolin, cytosine arabinoside), followed by the topoisomerase inhibitors (camptothecin, etoposide), then bifunctional alkylating agents (cisplatin, mitomycin C), and lastly spindle poisons (vincristine).199 Camptothecin-induced DSBs associated with replication forks in synchronized cells were detectable by PFGE and were repaired primarily by HRR, not NHEJ.199
Although both HRR and NHEJ mediate the resolution of stalled forks during replication arrest, analysis of mutant phenotypes suggests the two pathways play different roles, which likely depend on the nature of fork damage.197 There is some evidence that blocked forks that have not collapsed promote HRR,197 but this issue needs further study. Related studies found a differential involvement of NHEJ and HRR in the resolution of DSBs accumulated during extended (6–24 hr) arrest induced by replication inhibitors.200 The data suggest that NHEJ precedes HRR in the resolution of arrest-induced DSBs, and that HRR becomes more important with extended replication arrest induced by inhibitors.200,201
Recent studies in permeabilized cells treated with cisplatin provide insights into how chromosomal fork progression is controlled after DNA damage in vertebrates.202 Cisplatin DNA adducts actively slow fork progression by a process that requires XRCC3, a Rad51 paralog that participates in homologous recombination.203 The addition of purified human RAD51C-XRCC3 protein complex restores slowing of fork progression in permeabilized XRCC3-/- cells.202 Moreover, this requirement for XRCC3 is alleviated by adding human Rad51 protein, but not by Rad52 or the recombination complex Rad51B-Rad51C-Rad51D-XRCC2. These data demonstrate that XRCC3 and Rad51 cooperatively modulate the progression of replication forks on damaged vertebrate chromosomes.
Recent results suggest that the mode of DSB repair at arrested replication forks depends on the functional status of Tp53 (see also discussion on WRN and BLM below). The induction of HRR during the inhibition of replication is stimulated by mutant Tp53,204 and stalled replication forks produced by hydroxyurea and aphidicolin stimulate the accumulation of nuclear Tp53 that is impaired and altered in its transcriptional activity compared with Tp53 induced by IR.205 In XP-V cells following UV-induced replication arrest, inactivation of Tp53 by SV40 or HPV16 transformation stimulates DSB-dependent modification and relocalization of the MRN complex.178,179,206 High wortmannin concentrations, which inhibit PI3KKs (phosphatidylinositol-3-kinase related kinases), lead to a marked increase in the number of transformed XP-V cells exhibiting MRN foci following UV irradiation.207
Recognition and Signaling of Stalled and Collapsed Forks
ATR-Mediated Signaling
Creation of DSBs at sites of arrested forks can occur when enzymatic processing of the blocked structures leads to breakage of parental DNA strands, causing what is termed fork collapse (Fig. 7B). The details of how mammalian cells recognize and signal the presence of specific replication anomalies are beginning to be understood. Although the abnormal DNA structures that are initially recognized as critical replication fork “damage” are not yet well defined, the ATR kinase is a key early component in the signaling process.3,38,169 (The homologs of ATR in yeasts are Rad3Sp and Mec1Sc.) It is not entirely clear whether replication-associated γH2AX formation always requires collapse of a replication fork. However, the fact that a 3 hr aphidicolin treatment did not produce γH2AX foci strongly suggests that the signal arises from DSBs rather than stalled forks.53 (Much longer aphidicolin exposure causes γH2AX focus formation).134 In case of treatment with camptothecin, which binds Top1, DSBs associated with the cleavage complex at replication forks result in ATR-dependent γH2AX focus formation; DNA-PK and ATM also contributes to this phosphorylation.53 Examples of γH2AX foci seen in both CHO and human cells are given in (Fig. 3), panels B-D.
Even though ATR and ATM are functionally related, they differ very significantly. In contrast to ATM, which responds primarily to DSBs occurring outside the context of replication arrest, the ATR kinase prevents the accumulation of DSBs during replication arrest, as from aphidicolin treatment (Fig. 5).3,169,208 Unlike ATM, deletion of ATR function leads to early mouse embryonic and cellular lethality.147,209,210 ATR, but not ATM, plays a critical role in preventing chromosomal gaps and breaks at fragile sites (as well as at random sites), which are greatly enhanced by inhibiting replication with aphidicolin.198
The response of ATR to replication stress has provided many of the insights into its essential cellular functions as a regulator of replication arrest. Overexpression of kinase-inactive ATR mutants in human fibroblasts produces dominant negative phenotypes that show abnormalities in cell cycle progression, reduced phosphorylation of signaling and repair proteins, and elevated sensitivity to killing by DNA damaging agents (IR, UV, MMS, the topoisomerase I inhibitor topotecan, & the topoisomerase II inhibitor etoposide) and replication blocks (e.g., hydroxyurea).42,211,212 In Xenopus egg extracts and mammalian cells, ATR is required both to phosphorylate Chk1 in response to replication arrest and to properly activate the replication checkpoint.213215 In the Xenopus extracts, replication forks were shown to be an obligate intermediate for the activation of this checkpoint.216,217 In addition to phosphorylating Chk1, ATR plays a role in regulating the replication checkpoint through phosphorylation of Ser15 on Tp53, which is also a target of the Chk1 kinase (Fig. 5).3,214,215,218220 ATR and p53 can function independently, but loss of both can cause synergistic disruption of the replication checkpoint.219 In response to replication stress induced by UV-dimer photoproducts or replication inhibitors, ATR redistributes to form nuclear foci, presumably at stalled or broken replication forks.90,221,222 ATR interacts with 53BP1, phosphorylates it in vitro, and colocalizes with it after replication inhibition.90 ATR also phosphorylates BRCA1 with an overlapping spectrum of sites compared with ATM, and forms foci that partially colocalize with BRCA1 foci.221,222
ATR appears to act, not only in response to drug-imposed blocks to replication (e.g., aphidicolin, hydroxyurea), but also as an intrinsic replication fork checkpoint initiator that is normally active throughout S phase.222 Experiments using Xenopus extracts have shown that this checkpoint function depends on the replication-dependent chromatin-binding properties of ATR.223 After the initiation of replication, ATR binds to chromatin, where it can phosphorylate a range of downstream effectors and then dissociate upon the completion of replication.216,223 Nuclei in Xenopus extracts treated with replication inhibitors accumulate ssDNA, RPA, and γH2AX foci, consistent with the production of replication-associated DSBs.224 In the absence of inhibitors, replication-associated RPA coating ssDNA appears to facilitate the binding to chromatin of both ATR and the checkpoint protein Hus1 (hydroxyurea sensitive); the recruitment of Polα is also required for chromatin association of Hus1.172 Recruitment of the Hus1 protein complex to chromatin is independent of ATR binding, and checkpoint activation requires RNA synthesis by Polα.172,225 Since both ATR and Hus1 are required for the phosphorylation/activation of Chk1,172 their coincident binding to chromatin is likely critical in activating ATR either by interaction with Hus1 or by recruiting Chk1 to the chromatin.
Improper execution of the replication checkpoint may account for the chromosomal fragmentation and early embryonic lethality observed in ATR null mouse embryos147,209 and in Chk1 null mice.226 Somewhat surprisingly, incomplete DNA replication in mouse cells after aphidicolin treatment can prevent M-phase entry independently of ATR and inhibitory phosphorylation of Cdc2.169 However, when the replication inhibitor is removed, ATR knockout cells proceed to mitosis with extensive chromosome breaks, indicating that ATR provides a key genome maintenance function in S phase.169
ATR may act directly as a DNA damage sensor during replication arrest. ATR preferentially binds to UV-damaged DNA in vitro.227 This binding depends on UV fluence and full-length ATR, and results in a stimulation of its kinase activity. The binding partner of ATR, ATRIP (hRad26), was recently identified as the human homolog of Ddc2Sc (also called Lcd1 or Pie1) and Rad26Sp.210 Observations that ATRIP associates with ATR, is a substrate of ATR, and is a phosphoprotein in vivo,210 are compatible with similar interactions found for the yeast homologs Mec1-Ddc2 in S. cerevisiae and Rad3-Rad26 in S. pombe.228231 DNA damage and replication inhibition cause ATRIP to colocalize with ATR.210 Deletion of ATRIP reduces the level of the ATR protein and generates checkpoint defects similar to those of an ATR deletion,210 which suggests that ATRIP and ATR are mutually dependent in signaling and checkpoint pathways. It remains unclear whether ATRIP recruits and/or stimulates the association of ATR to replication-associated DSBs.
The Rad17 and Rad9 Complexes
The ATR/ATRIP complex interacts with other early damage sensor elements during checkpoint activation. Specifically, the mammalian checkpoint proteins Rad17 (RFC1 homolog; RFC = replication factor C)232234 and the Rad9-Rad1-Hus1 (9-1-1) complex235237 are named after their counterparts in S. pombe.4 (The corresponding proteins in S. cerevisiae are Rad24, Rad17, Ddc1, and Mec3, respectively.) While direct evidence is lacking that the Rad17-RFC heteropentameric complex and the 9-1-1 complex actually recognize replication-associated DSBs or other abnormal DNA structures at arrested forks, their interactions with chromatin and ATR appear to be critical in the early recognition and signaling of replication arrest. The Rad17-RFC and 9-1-1 complexes show sequence and structural similarity to the clamp loader (RFC) and sliding clamp (PCNA) complexes required for replication.216,232,238243 The biological importance of Rad9 in IR sensitivity is illustrated by the strong phenotype of rad9 knockout mouse ES cells, which display increased spontaneous chromosomal aberrations, IR and UV sensitivity to killing (3-fold), and a partially defective G2 checkpoint.250 Homozygous mutant embryos die between E9.5 and E12.5.
It was recently inferred that the checkpoint role of the 9-1-1 complex is not restricted to S phase and replication blockage. DNA-damage-induced binding of Rad9 to chromatin occurred in noncycling cells after exposure to IR or a bulky-adduct mutagen.244 However, the dose of IR used (50 Gy) may produce interactions that are irrelevant to normal physiological responses. In cycling cell populations, the IR- and hydroxyurea-induced binding of Rad9 (and Rad1) to chromatin occurs independently of the ATM phosphorylation of Rad9 at Ser272 (Fig. 2) and PIKK activities. Phosphorylation is also not required for Rad9's interaction with Rad1, Hus1, and Rad17.244
The critical role of Rad17 in preventing accumulation of DNA DSBs during replication is revealed by the properties of a Rad17flox/- conditional mutant in human HCT116 cells.245 Loss of Rad17 causes rapid accumulation of chromosomal breaks and rearrangements as well as endoreduplication. However, the chromosomal breakage in rad17 null cells was less severe than that of atr null cells examined in parallel.245 Rad17 null cells have defective Chk1 phosphorylation after UV damage, normal phosphorylation of ATM targets including Chk2 after IR damage, and a partially defective G2 checkpoint after IR damage.
Rad17 is constitutively bound to chromatin in human cells (although this was not seen in Xenopus egg extracts without DNA damage217) and is phosphorylated by ATR on chromatin after treatment with UV radiation, γ-rays, and hydroxyurea.246 Rad17, but not its phosphorylation by ATR, is required for loading the 9-1-1 complex onto chromatin (Fig. 6) in a manner analogous to PCNA loading by RFC. However, phosphorylation of Rad17 is required for the downstream phosphorylation/activation of Chk1. Both ATM and ATR are required for Rad17 phosphorylation in response to IR at early times, but UV-induced Rad17 phosphorylation appears to be specifically produced by ATR.246 Hydroxyurea-induced Rad17 phosphorylation is partially dependent on ATR but independent of ATM in the presence of ATR.246 Similar to Xenopus mentioned above, the 9-1-1 and ATR-ATRIP complexes in human cells can be recruited to chromatin independently (in response to UV damage). Both complexes are present at sites of UV damage as indicated by the partial colocalization of ATR foci with Rad17-Ser635 foci. Thus, Rad17's interactions with the 9-1-1 complex may help determine the selection of substrates available to ATR.246 In S. pombe recent evidence indicates that the DinB damage-response polymerase physically interacts with the 9-1-1 complex and requires Rad17 to associate with chromatin, suggesting that the checkpoint response includes translesion synthesis. 247 One model of checkpoint activation is that the loading and interaction of the ATR-ATRIP and 9-1-1 complexes, mediated by Rad17, creates a higher-order chromatin structure to facilitate signaling and phosphorylation of Chk1.246 Such a chromatin change might be analogous to that which appears to be mediated by ATM already discussed.48
This idea is compatible with the finding that Hus1 acts upstream of Chk1 and is required for its optimal phosphorylation in mammalian cells.248 Hus1 is not required for Tp53 accumulation and activation or for Chk2 phosphorylation. Disrupted signaling during replication stress likely underlies the embryonic lethality in Hus1-deficient mice.249 Hus1-deficient embryonic fibroblasts have been rescued for in vitro viability by simultaneous disruption of CDKN1A/p21,249 and these cells exhibit chromosomal instability, heightened sensitivity to replication blocks, and altered cell cycle responses. They display high sensitivity to UV radiation and hydroxyurea but only slight IR sensitivity,248,249 which is consistent with the idea that the 9-1-1 complex functions in the response to replication-associated damage. The lesser IR sensitivity of hus1 mutant cells248 compared with rad9 cells250 suggests that these two complex members have overlapping but not identical functions.
Factors Promoting the Repair of Replication Associated DSBs
As already discussed, overt DSB production leads to the production of γH2AX, a large-scale chromatin modification that can be visualized as nuclear foci. Exposure of mammalian cells to UV radiation or hydroxyurea leads to ATR-dependent phosphorylation and γH2AX focus formation.251 Colocalization of these γH2AX foci with PCNA in S-phase synchronized cultures suggests that these foci are associated with sites of replication fork arrest.251 The idea that these γH2AX foci reflect DSBs is supported by the finding that both UV radiation and hydroxyurea efficiently induce sister-chromatid exchange,252 a manifestation of DSB repair by homologous recombination involving crossing-over.253 Replication-dependent formation of γH2AX foci is also seen in both human cells53 and nuclei incubated in Xenopus egg extracts224 after treatment with the topoisomerase I poison camptothecin. In the Xenopus system, the induction of γH2AX foci is inhibited by geminin, a replication licensing inhibitor.
Additional evidence supporting the importance of γH2AX after S-phase DSB formation comes from studies utilizing replication-defective xeroderma pigmentosum variant (XP-V) cells. Exposure of XP-V cells to UV radiation leads to a fluence-dependent increase in the fraction of cells showing γH2AX foci, which are illustrated in (Fig. 3) These foci are only observed in S-phase cells, and they coincide with PCNA and MRN foci, further supporting the concept that the chromatin modification associated with H2AX phosphorylation recruits HRR proteins that facilitate repair between sister chromatids.178,179 Moreover, cells derived from H2AX null mice show a decrease in the portion of proliferating cells and an increased level of S-phase-derived chromatid aberrations.76 These observations, coupled with data showing that H2AXΔ/Δ ES cells exhibit reduced HRR76 but normal NHEJ,77 suggest that chromatin modification involving H2AX is critical in protecting cells against replication-associated genomic instability associated with aberrant recombination.
The Mre11 complex plays an important role in repairing replication associated DSBs. MRN exhibits extensive colocalization with PCNA throughout S phase, and replication fork stalling imposed by hydroxyurea enhances the chromatin association of MRN.114,254 As in XP-V cells discussed above, these results further suggest that MRN loads onto chromatin at blocked or collapsed replication forks. This idea is supported by the observation that MRN preferentially localizes to single-stranded DNA arising in hydroxyurea-treated cells.254 In camptothecin treated cells, MRN focus formation requires γH2AX formation, and H2AX null mouse cells are hypersensitive to killing by camptothecin.53 The finding that aphidicolin blocks camptothecin-induced γH2AX focus formation shows that replication produces these DSBs.53
TopBP1 appears to be another key protein involved in preventing replication-associated chromosomal rearrangements. TopBP1contains eight BRCT domains, which mediate multiple interactions, and has sequence homologs in yeast255,256 and flies.257 These homologs play important roles in DNA replication, repair and checkpoints in lower organisms.257261 In addition to facilitating normal replication through its interaction with Pol-ϵ, TopBP1 responds to the inhibition of DNA synthesis by localizing with other repair proteins (BRCA1, PCNA) during S-phase, suggesting a possible role in rescuing stalled forks.126,127 TopBP1 localized at sites of replication arrest may act to relieve torsional stress developed during the generation of anomalous DNA structures. Replication stress also elicits focus formation for 53BP1,91 a protein discussed earlier in the context of IR damage.
Processing Abnormal Replication Intermediates and Associated DSBs
Helicases and topoisomerases are specialized enzymes that modify the three dimensional structure of DNA. Helicases increase accessibility of the replication and repair machinery to DNA by locally unwinding the duplex. Topoisomerases modulate the torsional strain of the DNA helix by catalyzing the interconversion of topological isomers. Known interactions between these two classes of proteins suggest a need to colocalize their activities to resolve abnormal DNA structures that can arise at stalled forks.
The RecQ family of helicases is critical to the maintenance of genomic integrity. These helicases (see reviews in refs. 190,262,263), named after the E. coli recQ gene product, include yeast Sgs1Sc264,265 and Rqh1Sp,266,267 and five members in humans: BLM,268 WRN,269 RecQ1/ RecQL,270,271 RecQ4,272 and RecQ5.272274 Deficiencies in the BLM, WRN, and RecQ4 helicases cause Bloom and Werner syndromes,268,269 and some cases of Rothmund-Thomson syndrome.275277,279 These rare genetic diseases manifest distinct yet overlapping clinical phenotypes of immunodeficiency, premature aging, chromosomal instability, and predisposition to cancer (see reviews in refs. 25,189,190,280282).
The mutations in BLM and WRN helicases are associated with replication defects, including impaired progression of replication forks, an accumulation of abnormal replication intermediates,283285 and aberrant homologous recombination.286,287 BLM- and WRN-defective cells display an abnormally high percentage of deletion mutations at specific loci.288,289 BLM and WRN helicases can suppress the increased homologous and illegitimate recombination in the S. cerevisiae sgs1 mutant.290 Elevated sensitivity to replication-blocking inhibitors is seen in Bloom syndrome (BS) cell lines291294 and in WRN-deficient cells.295299 Conflicting results are reported concerning altered sensitivity of BLM-deficient cells to inhibition by hydroxyurea.291,292,294 WRN mutant cell lines consistently show hypersensitivity to camptothecin and defective responses to hydroxyurea.295299
It has been proposed that DSBs formed during replication arrest can lead to the formation of HJs (see Fig. 7C), substrates recognized by the BLM and WRN helicases.188,300,301 The ability of these helicases to unwind HJs is dependent on Tp53, which binds to the enzymes and attenuates their branch migration activity, and possibly their anti-recombinase functions.302 Tp53 and BLM functionally interact during resolution of stalled DNA replication forks.278 The evidence supports a model in which the disruption of abnormal structures by the BLM and WRN helicases prevents aberrant recombination events that would result in chromosomal rearrangement (see Fig. 7G).188,287,300,301,303 The interaction between BLM and topoisomerase IIIα (TopIII) in human cells, which is highly conserved across eukaryotic species,264,304306 may also promote unlinking of the parental duplex by TopIII at sites of paused or convergent replication forks.
Once formed, HJs may elude the activity of helicases and be cleaved by endonucleolytic HJ resolvasesto generate DSBs. Much of our understanding of these reactions comes from the genetic studies in bacterial systems characterizing the branch migration complex RuvA/RuvB, and the RuvC and RusA resolvases (see ref. 192 for a brief review). Activities from mammalian cell extracts that resemble those expected for a true HJ resolvase have been identified,307,308 but it is unclear whether these activities correspond to the newly described mus81 proteins in yeast and humans.191,309311 Nonetheless, the Mus81 homologs appear to specifically cleave replication intermediates that possess branched structures, and Mus81 (i.e., the Mus81-Eme1 heterodimeric endonuclease) is particularly important in S. pombe in processing stalled or collapsed forks in a RecQ helicase-deficient background.311
Recent work shows an important link between the BLM helicase and the MRN complex during replication arrest. In response to hydroxyurea, the formation of MRN foci at sites of stalled forks was sharply reduced in Bloom syndrome cells.292 However, in nonBS cells, ATR-dependent phosphorylation of BLM was not required for subnuclear relocalization of MRN.292 Function(s) of the MRN complex are suggested by its unique architecture.72,139,140 The Rad50 zinc-hook motif, through the interaction of two Rad50 “tails”, provides a means of tethering sister chromatids for recombinational repair, thereby limiting the undesirable dissociation of broken DNA ends. After fork arrest, the MRN complex may assist in repairing DSBs both by promoting inter-sister connectivity lost during fork regression and by helping to restore chromatid continuity during fork breakdown or HJ resolution. Notably, MRN foci that appear in S phase detergent-extracted cells colocalize with PCNA foci during DNA replication and appear normal in H2AXΔ/Δ mutant cells.76
Outlook
We discussed what is known about the events of DSB formation, recognition, and signaling, which facilitate the recruitment of checkpoint and repair proteins to these lesions. Homologous recombination may well have arisen early in life forms as a mechanism for ensuring chromosome continuity in the face of DSBs that arise normally during replication. The large genomes of higher eukaryotes must achieve remarkable accuracy in rapidly detecting each DSB and announcing its presence through signal amplification and transduction in order to keep the genome intact in each daughter cell at mitosis. The discovery of γH2AX foci as a likely bona fide marker and sentinel for DSBs represents a major advance.11,49,50 It appears likely that γH2AX foci will provide a reliable method of quantifying DSBs and their repair.11 Just how this modification is triggered through ATM's chromatin sensing capacity remains to be determined. Additional candidates for the initial sensors, which help mediate γH2AX formation, are under intense investigation.
Recent research brings the promise of providing the cytological tools needed to accurately quantify the levels of DSBs in fixed cells, and perhaps eventually in living cells.73 Measuring the very early colocalization of 53BP1 and MDC1 with γH2AX may add robustness to experiments designed to accurately measure levels of DSBs at low IR doses. Better quantitative methods of image analysis are greatly needed to remove the subjectivity and labor inherent in the visual scoring of cytological foci. Since rapid progress has been made in recent years in this field, it seems very likely that we will soon have a detailed understanding of how cells cope so efficiently with DSBs.
Acknowledgments
We thank Karlene Cimprich, John Hinz, David Schild, Robert Tebbs, Alice Yamada, and Dawn Yean for providing valuable comments on the manuscript, and also Joel Bedford, Bill Dewey, and John Ward for helpful discussion. This work was prepared under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under contract No. W-7405-ENG-48 and funded by the Low Dose Radiation Research Program, Biological and Environmental Research (BER), U.S. Department of Energy, by NIH/NCI grant P01 CA92584-02, and by ACS grant # RPG-00-036-01 CNE (CLL).
References
- 1.
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. [PubMed: 11357144]
- 2.
- Zhou BB, Elledge SJ. The DNA damage response: Putting checkpoints in perspective. Nature. 2000;408:433–439. [PubMed: 11100718]
- 3.
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. [PubMed: 11544175]
- 4.
- Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol. 2002;14:237–245. [PubMed: 11891124]
- 5.
- Ruiz de Almodovar JM, Steel GG, Whitaker SJ. et al. A comparison of methods for calculating DNA double-strand break induction frequency in mammalian cells by pulsed-field gel electrophoresis. Int J Radiat Biol. 1994;65:641–649. [PubMed: 7912713]
- 6.
- Cedervall B, Wong R, Albright N. et al. Methods for the quantification of DNA double-strand breaks determined from the distribution of DNA fragment sizes measured by pulsed-field gel electrophoresis. Radiat Res. 1995;143:8–16. [PubMed: 7597148]
- 7.
- Ward JF. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog Nucl Acids Res Mol Biol. 1988;35:95–125. [PubMed: 3065826]
- 8.
- Ward JF. Nature of lesions formed by ionizing radiation, DNA damage and repair In: Nickoloff JA, Hoekstra M, eds.DNA Repair in Higher Eukaryotes Totowa, NJ: Humana Press,1998265–84.
- 9.
- Rogakou EP, Nieves-Neira W, Boon C. et al. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000;275:9390–9395. [PubMed: 10734083]
- 10.
- Sedelnikova OA, Rogakou EP, Panyutin IG. et al. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res. 2002;158:486–492. [PubMed: 12236816]
- 11.
- Rothkamm K, Lobrich M. Evidence for a lack of DNA double-stranded break repair in himan cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 2003;100:5057–5062. [PMC free article: PMC154297] [PubMed: 12679524]
- 12.
- Cornforth MN, Bedford JS. On the nature of a defect in cells from individuals with ataxia-telangiectasia. Science. 1985;227:1589–1591. [PubMed: 3975628]
- 13.
- Girard PM, Foray N, Stumm M. et al. Radiosensitivity in Nijmegen Breakage Syndrome cells is attributable to a repair defect and not cell cycle checkpoint defects. Cancer Res. 2000;60:4881–4888. [PubMed: 10987302]
- 14.
- Wang H, Zeng ZC, Bui TA. et al. Efficient rejoining of radiation-induced DNA double-strand breaks in vertebrate cells deficient in genes of the RAD52 epistasis group. Oncogene. 2001;20:2212–2224. [PubMed: 11402316]
- 15.
- Beckman KB, Saljoughi S, Mashiyama ST. et al. A simpler, more robust method for the analysis of 8-oxoguanine in DNA. Free radic. Biol Med. 2000;29:357–367. [PubMed: 11035265]
- 16.
- Beckman KB, Ames BN. Oxidative decay of DNA. J Biol Chem. 1997;272:19633–19636. [PubMed: 9289489]
- 17.
- Sutherland BM, Bennett PV, Sidorkina O. et al. Clustered damages and total lesions induced in DNA by ionizing radiation: Oxidized bases and strand breaks. Biochemistry. 2000;39:8026–8031. [PubMed: 10891084]
- 18.
- Sutherland BM, Bennett PV, Sidorkina O. et al. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc Natl Acad Sci USA. 2000;97:103–108. [PMC free article: PMC26623] [PubMed: 10618378]
- 19.
- Sutherland BM, Bennett PV, Sutherland JC. et al. Clustered DNA damages induced by x rays in human cells. Radiat Res. 2002;157:611–616. [PubMed: 12005538]
- 20.
- Dianov GL, O'Neill P, Goodhead DT. Securing genome stability by orchestrating DNA repair: Removal of radiation-induced clustered lesions in DNA. Bioessays. 2001;23:745–749. [PubMed: 11494323]
- 21.
- Barnes DE. Nonhomologous end joining as a mechanism of DNA repair. Curr Biol. 2001;11:R455–R457. [PubMed: 11448783]
- 22.
- Pierce AJ, Jasin M. NHEJ deficiency and disease. Mol Cell. 2001;8:1160–1161. [PubMed: 11885597]
- 23.
- Thompson LH, Schild D. The contribution of homologous recombination in preserving genome integrity in mammalian cells. Biochimie. 1999;81:87–105. [PubMed: 10214914]
- 24.
- Thompson LH, Schild D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat Res. 2001;477:131–153. [PubMed: 11376695]
- 25.
- Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res. 2002;509:49–78. [PubMed: 12427531]
- 26.
- Karanjawala ZE, Grawunder U, Hsieh CL. et al. The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr Biol. 1999;9:1501–1504. [PubMed: 10607596]
- 27.
- Difilippantonio MJ, Zhu J, Chen HT. et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–514. [PMC free article: PMC4721590] [PubMed: 10761921]
- 28.
- Ferguson DO, Sekiguchi JM, Chang S. et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci USA. 2000;97:6630–6633. [PMC free article: PMC18682] [PubMed: 10823907]
- 29.
- Karanjawala ZE, Murphy N, Hinton DR. et al. Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr Biol. 2002;12:397–402. [PubMed: 11882291]
- 30.
- Barnes DE, Stamp G, Rosewell I. et al. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998;8:1395–1398. [PubMed: 9889105]
- 31.
- Gao Y, Sun Y, Frank KM. et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. [PubMed: 9875844]
- 32.
- Li GC, Ouyang H, Li X. et al. Ku70: A candidate tumor suppressor gene for murine T cell lymphoma. Mol Cell. 1998;2:1–8. [PubMed: 9702186]
- 33.
- Vogel H, Lim DS, Karsenty G. et al. Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci USA. 1999;96:10770–10775. [PMC free article: PMC17958] [PubMed: 10485901]
- 34.
- Gu Y, Sekiguchi J, Gao Y. et al. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc Natl Acad Sci USA. 2000;97:2668–2673. [PMC free article: PMC15987] [PubMed: 10716994]
- 35.
- Kemp LM, Jeggo PA. Radiation-induced chromosome damage in X-ray-sensitive mutants (xrs) of the Chinese hamster ovary cell line. Mutat Res. 1986;166:255–263. [PubMed: 3785270]
- 36.
- Darroudi F, Natarajan AT. Cytological characterization of Chinese hamster ovary X-ray-sensitive mutant cells xrs 5 and xrs 6. I. Induction of chromosomal aberrations by X-irradiation and its modulation with 3-aminobenzamide and caffeine. Mutat Res. 1987;177:133–148. [PubMed: 3821761]
- 37.
- Takata M, Sasaki MS, Sonoda E. et al. Homologous recombination and nonhomologous end joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. [PMC free article: PMC1170875] [PubMed: 9736627]
- 38.
- Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: Variations on a theme? Curr Opin Cell Biol. 2001;13:225–231. [PubMed: 11248557]
- 39.
- Khanna KK, Lavin MF, Jackson SP. et al. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001;8:1052–1065. [PubMed: 11687884]
- 40.
- Shiloh Y, Kastan MB. ATM: Genome stability, neuronal development, and cancer cross paths. Adv Cancer Res. 2001;83:209–254. [PubMed: 11665719]
- 41.
- Shiloh Y. ATM and related protein kinases: Safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. [PubMed: 12612651]
- 42.
- Wright JA, Keegan KS, Herendeen DR. et al. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc Natl Acad Sci USA. 1998;95:7445–7450. [PMC free article: PMC22645] [PubMed: 9636169]
- 43.
- Shiloh Y. ATM and ATR: Networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11:71–77. [PubMed: 11163154]
- 44.
- Wang Y, Cortez D, Yazdi P. et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. [PMC free article: PMC316544] [PubMed: 10783165]
- 45.
- Banin S, Moyal L, Shieh S. et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. [PubMed: 9733514]
- 46.
- Canman CE, Lim DS, Cimprich KA. et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. [PubMed: 9733515]
- 47.
- Kozlov S, Gueven N, Keating K. et al. ATP activates ataxia-telangiectasia mutated (ATM) in vitro. Importance of autophosphorylation. J Biol Chem. 2003;278:9309–9317. [PubMed: 12645530]
- 48.
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. [PubMed: 12556884]
- 49.
- Rogakou EP, Pilch DR, Orr AH. et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. [PubMed: 9488723]
- 50.
- Rogakou EP, Boon C, Redon C. et al. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. [PMC free article: PMC2169482] [PubMed: 10477747]
- 51.
- Burma S, Chen BP, Murphy M. et al. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. [PubMed: 11571274]
- 52.
- Kobayashi J, Tauchi H, Sakamoto S. et al. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol. 2002;12:1846–1851. [PubMed: 12419185]
- 53.
- Furuta T, Takemura H, Liao ZY. et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks Induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem. 2003;278:20303–20312. [PubMed: 12660252]
- 54.
- Smith GC, Cary RB, Lakin ND. et al. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc Natl Acad Sci USA. 1999;96:11134–11139. [PMC free article: PMC17999] [PubMed: 10500142]
- 55.
- Adamson AW, Kim WJ, Shangary S. et al. ATM is activated in response to MNNG-induced DNA alkylation. J Biol Chem. 2002;277:38222–38229. [PubMed: 12151394]
- 56.
- Andegeko Y, Moyal L, Mitelman L. et al. Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem. 2001;276:38224–38230. [PubMed: 11454856]
- 57.
- Wang B, Matsuoka S, Carpenter PB. et al. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–1438. [PubMed: 12364621]
- 58.
- Stewart GS, Wang B, Bignell CR. et al. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. [PubMed: 12607005]
- 59.
- Scott SP, Bendix R, Chen P. et al. Missense mutations but not allelic variants alter the function of ATM by dominant interference in patients with breast cancer. Proc Natl Acad Sci USA. 2002;99:925–930. [PMC free article: PMC117407] [PubMed: 11805335]
- 60.
- Spring K, Ahangari F, Scott SP. et al. Mice heterozygous for mutation in Atm, the gene involved in ataxia-telangiectasia, have heightened susceptibility to cancer. Nat Genet. 2002;32:185–190. [PubMed: 12195425]
- 61.
- Lim DS, Kim ST, Xu B. et al. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. [PubMed: 10766245]
- 62.
- Wu X, Ranganathan V, Weisman DS. et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000;405:477–482. [PubMed: 10839545]
- 63.
- Gatei M, Young D, Cerosaletti KM. et al. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat Genet. 2000;25:115–119. [PubMed: 10802669]
- 64.
- Zhao S, Weng YC, Yuan SS. et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000;405:473–477. [PubMed: 10839544]
- 65.
- Dong Z, Zhong Q, Chen PL. The Nijmegen breakage syndrome protein is essential for Mre11 phosphorylation upon DNA damage. J Biol Chem. 1999;274:19513–19516. [PubMed: 10391882]
- 66.
- Shiloh Y. ATM: A sentry at the gate of genome instabilityThe Univ of Texas M D Anderson Cancer Center 55th Ann Symp Fund Cancer ResHouston, TX: Maintenance of genomic integrity Oct 15-18 200235Abstract.
- 67.
- Haaf T, Golub EI, Reddy G. et al. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci USA. 1995;92:2298–2302. [PMC free article: PMC42471] [PubMed: 7892263]
- 68.
- Ashley T, Plug AW, Xu J. et al. Dynamic changes in Rad51 distribution on chromatin during meiosis in male and female vertebrates. Chromosoma. 1995;104:19–28. [PubMed: 7587590]
- 69.
- Haaf T, Hayman DL, Schmid M. Quantitative determination of rDNA transcription units in vertebrate cells. Exp Cell Res. 1991;193:78–86. [PubMed: 1995304]
- 70.
- Petrini JH. The Mre11 complex and ATM: Collaborating to navigate S phase. Curr Opin Cell Biol. 2000;12:293–296. [PubMed: 10801460]
- 71.
- D'Amours D, Jackson SP. The Mre11 complex: At the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. [PubMed: 11988766]
- 72.
- Connelly JC, Leach DR. Tethering on the brink: The evolutionarily conserved Mre11-Rad50 complex. Trends Biochem Sci. 2002;27:410–418. [PubMed: 12151226]
- 73.
- Essers J, Houtsmuller AB, van Veelen L. et al. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 2002;21:2030–2037. [PMC free article: PMC125370] [PubMed: 11953322]
- 74.
- Li L, Sharipo A, Chaves-Olarte E. et al. The Haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and nonproliferating cells. Cell Microbiol. 2002;4:87–99. [PubMed: 11896765]
- 75.
- Paull TT, Rogakou EP, Yamazaki V. et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. [PubMed: 10959836]
- 76.
- Celeste A, Petersen S, Romanienko PJ. et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. [PMC free article: PMC4721576] [PubMed: 11934988]
- 77.
- Bassing CH, Chua KF, Sekiguchi J. et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA. 2002;99:8173–8178. [PMC free article: PMC123040] [PubMed: 12034884]
- 78.
- Fernandez-Capetillo O, Chen HT, Celeste A. et al. DNA damage-induced G(2)-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002;4:993–997. [PubMed: 12447390]
- 79.
- Shang YL, Bodero AJ, Chen PL. NFBD1, a novel nuclear protein with signature motifs of FHA and BRCT, and an internal 41 amino acid repeat sequence, is an early participant in DNA damage response. J Biol Chem. 2002;278:6323–6329. [PubMed: 12475977]
- 80.
- Lou Z, Minter-Dykhouse K, Wu X. et al. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–961. [PubMed: 12607004]
- 81.
- Goldberg M, Stucki M, Falck J. et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. [PubMed: 12607003]
- 82.
- Peng A, Chen PL. NFBD1, like 53BP1, is an early and redundant transducer mediating Chk2 phosphorylation in response to DNA damage. J Biol Chem. 2003;278:8873–8876. [PubMed: 12551934]
- 83.
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. [PubMed: 3291120]
- 84.
- Schiestl RH, Reynolds P, Prakash S. et al. Cloning and sequence analysis of the Saccharomyces cerevisiae RAD9 gene and further evidence that its product is required for cell cycle arrest induced by DNA damage. Mol Cell Biol. 1989;9:1882–1896. [PMC free article: PMC362979] [PubMed: 2664461]
- 85.
- Callebaut I, Mornon JP. From BRCA1 to RAP1: A widespread BRCT module closely associated with DNA repair. FEBS Lett. 1997;400:25–30. [PubMed: 9000507]
- 86.
- Iwabuchi K, Bartel PL, Li B. et al. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci USA. 1994;91:6098–6102. [PMC free article: PMC44145] [PubMed: 8016121]
- 87.
- Iwabuchi K, Li B, Massa HF. et al. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J Biol Chem. 1998;273:26061–26068. [PubMed: 9748285]
- 88.
- Schultz LB, Chehab NH, Malikzay A. et al. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151:1381–1390. [PMC free article: PMC2150674] [PubMed: 11134068]
- 89.
- Ward IM, Minn K, Van Deursen J. et al. p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol. 2003;23:2556–2563. [PMC free article: PMC150747] [PubMed: 12640136]
- 90.
- Morales JC, Xia Z, Lu T. et al. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J Biol Chem. 2003;278:14971–14977. [PubMed: 12578828]
- 91.
- Rappold I, Iwabuchi K, Date T. et al. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153:613–620. [PMC free article: PMC2190566] [PubMed: 11331310]
- 92.
- Ward IM, Minn K, Jorda KG. et al. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem. 2003 [PubMed: 12697768]
- 93.
- Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol. 2001;21:1719–1729. [PMC free article: PMC86718] [PubMed: 11238909]
- 94.
- Jullien D, Vagnarelli P, Earnshaw WC. et al. Kinetochore localisation of the DNA damage response component 53BP1 during mitosis. J Cell Sci. 2002;115:71–79. [PubMed: 11801725]
- 95.
- Abraham RT. Checkpoint signalling: Focusing on 53BP1. Nat Cell Biol. 2002;4:E277–E279. [PubMed: 12461529]
- 96.
- DiTullio RA, Mochan TA, Venere M. et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol. 2002;4:998–1002. [PubMed: 12447382]
- 97.
- Kao GD, McKenna WG, Guenther MG. et al. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol. 2003;160:1017–1027. [PMC free article: PMC2172769] [PubMed: 12668657]
- 98.
- Bartek J, Falck J, Lukas J. CHK2 kinase-a busy messenger. Nat Rev Mol Cell Biol. 2001;2:877–886. [PubMed: 11733767]
- 99.
- McGowan CH. Checking in on Cds1 (Chk2): A checkpoint kinase and tumor suppressor. Bioessays. 2002;24:502–511. [PubMed: 12111733]
- 100.
- Hirao A, Cheung A, Duncan G. et al. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol. 2002;22:6521–6532. [PMC free article: PMC135625] [PubMed: 12192050]
- 101.
- Takai H, Naka K, Okada Y. et al. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 2002;21:5195–5205. [PMC free article: PMC129029] [PubMed: 12356735]
- 102.
- Ward IM, Wu X, Chen J. Threonine 68 of Chk2 Is phosphorylated at sites of DNA strand breaks. J Biol Chem. 2001;276:47755–47758. [PubMed: 11668173]
- 103.
- Buscemi G, Savio C, Zannini L. et al. Chk2 activation dependence on NBS1 after DNA damage. Mol Cell Biol. 2001;21:5214–5222. [PMC free article: PMC87245] [PubMed: 11438675]
- 104.
- Girard PM, Riballo E, Begg AC. et al. Nbs1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene. 2002;21:4191–4199. [PubMed: 12082606]
- 105.
- Yarden RI, Pardo-Reoyo S, Sgagias M. et al. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002;30:285–289. [PubMed: 11836499]
- 106.
- Zachos G, Rainey MD, Gillespie DA. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 2003;22:713–723. [PMC free article: PMC140744] [PubMed: 12554671]
- 107.
- Gatei M, Sloper K, Sorensen C. et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem. 2003;278:14806–14811. [PubMed: 12588868]
- 107a.
- Yamane K, Chen J, Kinsella TJ. Both DNA topoisomerase II-binding protein 1 and BRCA1 regulate the G2-M cell cycle checkpoint. Cancer Res. 2003;63:3049–3053. [PubMed: 12810625]
- 108.
- Taalman RD, Jaspers NG, Scheres JM. et al. Hypersensitivity to ionizing radiation, in vitro, in a new chromosomal breakage disorder, the Nijmegen Breakage Syndrome. Mutat Res. 1983;112:23–32. [PubMed: 6828038]
- 109.
- Stewart GS, Maser RS, Stankovic T. et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. [PubMed: 10612394]
- 110.
- Kim YC, Koh JT, Shin BA. et al. An antisense construct of full-length human RAD50 cDNA confers sensitivity to ionizing radiation and alkylating agents on human cell lines. Radiat Res. 2002;157:19–25. [PubMed: 11754637]
- 111.
- Nelms BE, Maser RS, MacKay JF. et al. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–592. [PubMed: 9554850]
- 112.
- Limoli CL, Ward JF. A new method for introducing double-strand breaks into cellular DNA. Radiat Res. 1993;134:160–169. [PubMed: 7683818]
- 113.
- Maser RS, Monsen KJ, Nelms BE. et al. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol Cell Biol. 1997;17:6097–6104. [PMC free article: PMC232458] [PubMed: 9315668]
- 114.
- Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol. 2001;21:281–288. [PMC free article: PMC88801] [PubMed: 11113202]
- 115.
- Negorev D, Maul GG. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene. 2001;20:7234–7242. [PubMed: 11704851]
- 116.
- Lou Z, Chini CC, Minter-Dykhouse K. et al. Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J Biol Chem. 2003;278:13599–13602. [PubMed: 12611903]
- 117.
- Paull TT, Cortez D, Bowers B. et al. Direct DNA binding by Brca1. Proc Natl Acad Sci USA. 2001;98:6086–6091. [PMC free article: PMC33426] [PubMed: 11353843]
- 118.
- DeFazio LG, Stansel RM, Griffith JD. et al. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002;21:3192–3200. [PMC free article: PMC126055] [PubMed: 12065431]
- 119.
- Lee SH, Kim CH. DNA-dependent protein kinase complex: A multifunctional protein in DNA repair and damage checkpoint. Mol Cells. 2002;13:159–166. [PubMed: 12018836]
- 120.
- Chan DW, Chen BP, Prithivirajsingh S. et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16:2333–2338. [PMC free article: PMC187438] [PubMed: 12231622]
- 121.
- Balajee AS, Geard CR. Chromatin-bound PCNA complex formation triggered by DNA damage occurs independent of the ATM gene product in human cells. Nucleic Acids Res. 2001;29:1341–1351. [PMC free article: PMC29758] [PubMed: 11239001]
- 122.
- Golub EI, Gupta RC, Haaf T. et al. Interaction of human rad51 recombination protein with single-stranded DNA binding protein. RPA Nucleic Acids Res. 1998;26:5388–5393. [PMC free article: PMC148005] [PubMed: 9826763]
- 123.
- MacPhail SH, Olive PL. RPA foci are associated with cell death after irradiation. Radiat Res. 2001;155:672–679. [PubMed: 11302763]
- 124.
- Yamane K, Kawabata M, Tsuruo T. A DNA-topoisomerase-II-binding protein with eight repeating regions similar to DNA-repair enzymes and to a cell-cycle regulator. Eur J Biochem. 1997;250:794–799. [PubMed: 9461304]
- 125.
- Yamane K, Tsuruo T. Conserved BRCT regions of TopBP1 and of the tumor suppressor BRCA1 bind strand breaks and termini of DNA. Oncogene. 1999;18:5194–5203. [PubMed: 10498869]
- 126.
- Makiniemi M, Hillukkala T, Tuusa J. et al. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J Biol Chem. 2001;276:30399–30406. [PubMed: 11395493]
- 127.
- Yamane K, Wu X, Chen J. A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Mol Cell Biol. 2002;22:555–566. [PMC free article: PMC139754] [PubMed: 11756551]
- 128.
- DiBiase SJ, Zeng ZC, Chen R. et al. DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res. 2000;60:1245–1253. [PubMed: 10728683]
- 129.
- Stewart RD. Two-lesion kinetic model of double-strand break rejoining and cell killing. Radiat Res. 2001;156:365–378. [PubMed: 11554848]
- 130.
- Thompson LH, Brookman KW, Dillehay LE. et al. A CHO-cell strain having hypersensitivity to mutagens, a defect in strand-break repair, and an extraordinary baseline frequency of sister chromatid exchange. Mutat Res. 1982;95:427–440. [PubMed: 6889677]
- 131.
- vanAnkeren SC, Murray D, Meyn RE. Induction and rejoining of ?-ray-induced DNA single- and double-strand breaks in Chinese hamster AA8 cells and in two radiosensitive clones. Radiat Res. 1988;116:511–525. [PubMed: 3060896]
- 132.
- Nevaldine B, Longo JA, Hahn PJ. The scid defect results in much slower repair of DNA double-strand breaks but not high levels of residual breaks. Radiat Res. 1997;147:535–540. [PubMed: 9146698]
- 133.
- Löbrich M, Rydberg B, Cooper PK. Repair of x-ray-induced DNA double-strand breaks in specific Not I restriction fragments in human fibroblasts: Joining of correct and incorrect ends. Proc Natl Acad Sci USA. 1995;92:12050–12054. [PMC free article: PMC40294] [PubMed: 8618842]
- 134.
- Rothkamm K, Kruger I, Thompson LH. et al. Pathways of DNA double-strand break repair during the mammalian cell cycle Mol Cell Biol 2003. in press. [PMC free article: PMC166351] [PubMed: 12897142]
- 135.
- Moynahan ME, Jasin M. Loss of heterozygosity induced by a chromosomal double-strand break. Proc Natl Acad Sci USA. 1997;94:8988–8993. [PMC free article: PMC22995] [PubMed: 9256422]
- 136.
- Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–672. [PubMed: 11239455]
- 137.
- Moynahan ME, Cui TY, Jasin M. Homology-directed DNA repair, mitomycin-C resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–4850. [PubMed: 11406561]
- 138.
- de Jager M, van Noort J, van Gent DC. et al. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. [PubMed: 11741547]
- 139.
- Hopfner KP, Craig L, Moncalian G. et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–566. [PubMed: 12152085]
- 140.
- Wyman C, Kanaar R. Chromosome organization: Reaching out to embrace new models. Curr Biol. 2002;12:R446–R448. [PubMed: 12121633]
- 141.
- Yang H, Jeffrey PD, Miller J. et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. [PubMed: 12228710]
- 142.
- Kowalczykowski SC. Molecular mimicry connects BRCA2 to Rad51 and recombinational DNA repair. Nat Struct Biol. 2002;9:897–899. [PubMed: 12447354]
- 143.
- Pellegrini L, Yu DS, Lo T. et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. [PubMed: 12442171]
- 144.
- Woodgate R. A plethora of lesion-replicating DNA polymerases. Genes Dev. 1999;13:2191–2195. [PubMed: 10485842]
- 145.
- Patel PH, Loeb LA. Getting a grip on how DNA polymerases function. Nat Struct Biol. 2001;8:656–659. [PubMed: 11473246]
- 146.
- Friedberg EC. Why do cells have multiple error-prone DNA polymerases? Environ Mol Mutagen. 2001;38:105–110. [PubMed: 11746742]
- 147.
- Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article: PMC316378] [PubMed: 10691732]
- 148.
- Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997;25:2985–2991. [PMC free article: PMC146850] [PubMed: 9224597]
- 149.
- Sonoda E, Sasaki M, Buerstedde JM. et al. Rad51 deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. [PMC free article: PMC1170409] [PubMed: 9430650]
- 150.
- Luo G, Yao MS, Bender CF. et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci USA. 1999;96:7376–7381. [PMC free article: PMC22093] [PubMed: 10377422]
- 151.
- Yamaguchi-Iwai Y, Sonoda E, Sasaki MS. et al. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 1999;18:6619–6629. [PMC free article: PMC1171725] [PubMed: 10581236]
- 152.
- Zhu J, Petersen S, Tessarollo L. et al. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol. 2001;11:105–109. [PubMed: 11231126]
- 153.
- Tauchi H, Kobayashi J, Morishima K. et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature. 2002;420:93–98. [PubMed: 12422221]
- 154.
- Costanzo V, Robertson K, Bibikova M. et al. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell. 2001;8:137–147. [PubMed: 11511367]
- 155.
- Lisby M, Rothstein R, Mortensen UH. Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA. 2001;98:8276–8282. [PMC free article: PMC37432] [PubMed: 11459964]
- 156.
- Bentley NJ, Holtzman DA, Flaggs G. et al. The Schizosaccharomyces Pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article: PMC452488] [PubMed: 8978690]
- 157.
- Cimprich KA, Shin TB, Keith CT. et al. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci USA. 1996;93:2850–2855. [PMC free article: PMC39722] [PubMed: 8610130]
- 158.
- von Hippel PH. The recombination-replication interface. Trends Biochem Sci. 2000;25:155. [PubMed: 10754545]
- 159.
- Cox MM. Historical overview: Searching for replication help in all of the rec places. Proc Natl Acad Sci USA. 2001;98:8173–8180. [PMC free article: PMC37418] [PubMed: 11459950]
- 160.
- Nghiem P, Park PK, Kim Y. et al. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc Natl Acad Sci USA. 2001;98:9092–9097. [PMC free article: PMC55378] [PubMed: 11481475]
- 161.
- Pierce AJ, Hu P, Han M. et al. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. [PMC free article: PMC312854] [PubMed: 11751629]
- 162.
- Cromie GA, Connelly JC, Leach DR. Recombination at double-strand breaks and DNA ends: Conserved mechanisms from phage to humans. Mol Cell. 2001;8:1163–1174. [PubMed: 11779493]
- 163.
- Brown KD, Rathi A, Kamath R. et al. The mismatch repair system is required for S-phase checkpoint activation. Nat Genet. 2002;33:80–84. [PubMed: 12447371]
- 164.
- Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc Natl Acad Sci USA. 1980;77:7315–7317. [PMC free article: PMC350493] [PubMed: 6938978]
- 165.
- Costanzo V, Robertson K, Ying CY. et al. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol Cell. 2000;6:649–659. [PubMed: 11030344]
- 166.
- Yazdi PT, Wang Y, Zhao S. et al. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002;16:571–582. [PMC free article: PMC155356] [PubMed: 11877377]
- 167.
- Taniguchi T, Garcia-Higuera I, Xu B. et al. Convergence of the Fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109:459–472. [PubMed: 12086603]
- 168.
- Falck J, Petrini JH, Williams BR. et al. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet. 2002;30:290–294. [PubMed: 11850621]
- 169.
- Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–628. [PMC free article: PMC196009] [PubMed: 12629044]
- 170.
- Walter J, Newport J. Initiation of eukaryotic DNA replication: Origin unwinding and sequential chromatin association of Cdc45, RPA and DNA polymerase alpha. Mol Cell. 2000;5:617–627. [PubMed: 10882098]
- 171.
- Pages V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science. 2003;300:1300–1303. [PubMed: 12764199]
- 172.
- You Z, Kong L, Newport J. The role of single-stranded DNA and polymerase alpha in establishing the ATR, Hus1 DNA replication checkpoint. J Biol Chem. 2002;277:27088–27093. [PubMed: 12015327]
- 173.
- Cordeiro-Stone M, Zaritskaya LS, Price LK. et al. Replication fork bypass of a pyrimidine dimer blocking leading strand DNA synthesis. J Biol Chem. 1997;272:13945–13954. [PubMed: 9153257]
- 174.
- Cordeiro-Stone M, Makhov AM, Zaritskaya LS. et al. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. J Mol Biol. 1999;289:1207–1218. [PubMed: 10373362]
- 175.
- Wood RD. DNA repair. Variants on a theme. Nature. 1999;399:639–640. [PubMed: 10385109]
- 176.
- Kiefer J, Feige M. The significance of DNA double-strand breaks in the UV inactivation of yeast cells. Mutat Res. 1993;299:219–224. [PubMed: 7683089]
- 177.
- Limoli CL, Ward JF. Response of bromodeoxyuridine-substituted Chinese hamster cells to UVA light exposure in the presence of Hoechst dye #33258: Survival and DNA repair studies. Radiat Res. 1994;138:312–319. [PubMed: 7514307]
- 178.
- Limoli CL, Giedzinski E, Morgan WF. et al. Inaugural Article: Polymerase eta deficiency in the xeroderma pigmentosum variant uncovers an overlap between the S phase checkpoint and double-strand break repair. Proc Natl Acad Sci USA. 2000;97:7939–7946. [PMC free article: PMC16649] [PubMed: 10859352]
- 179.
- Limoli CL, Giedzinski E, Bonner WM. et al. UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA double-strand breaks, gamma-H2AX formation, and Mre11 relocalization. Proc Natl Acad Sci USA. 2002;99:233–238. [PMC free article: PMC117544] [PubMed: 11756691]
- 180.
- Paull TT, Gellert M. The 3' to 5' exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. [PubMed: 9651580]
- 181.
- Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. [PMC free article: PMC316715] [PubMed: 10346816]
- 182.
- Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. [PubMed: 11832209]
- 183.
- Ma Y, Pannicke U, Schwarz K. et al. Hairpin opening and overhang processing by an Artemis/ DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. [PubMed: 11955432]
- 184.
- Postow L, Ullsperger C, Keller RW. et al. Positive torsional strain causes the formation of a four-way junction at replication forks. J Biol Chem. 2001;276:2790–2796. [PubMed: 11056156]
- 185.
- Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. [PubMed: 12142537]
- 186.
- Tatsumi K, Strauss B. Production of DNA bifilarly substituted with bromodeoxyuridine in the first round of synthesis: Branch migration during isolation of cellular DNA. Nucleic Acids Res. 1978;5:331–347. [PMC free article: PMC341987] [PubMed: 147449]
- 187.
- Tatsumi K, Strauss BS. Accumulation of DNA growing points in caffeine-treated human lymphoblastoid cells. J Mol Biol. 1979;135:435–449. [PubMed: 537083]
- 188.
- Mohaghegh P, Karow JK, Brosh RM Jr. et al. The Bloom's and Werner's syndrome proteins are DNA structurespecific helicases. Nucleic Acids Res. 2001;29:2843–2849. [PMC free article: PMC55766] [PubMed: 11433031]
- 189.
- Mohaghegh P, Hickson ID. DNA helicase deficiencies associated with cancer predisposition and premature ageing disorders. Hum Mol Genet. 2001;10:741–746. [PubMed: 11257107]
- 190.
- Chakraverty RK, Hickson ID. Defending genome integrity during DNA replication: A proposed role for RecQ family helicases. Bioessays. 1999;21:286–294. [PubMed: 10377891]
- 191.
- Chen XB, Melchionna R, Denis CM. et al. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol Cell. 2001;8:1117–1127. [PubMed: 11741546]
- 192.
- Haber JE, Heyer WD. The fuss about Mus81. Cell. 2001;107:551–554. [PubMed: 11733053]
- 193.
- Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;16:430–408. [PMC free article: PMC1169647] [PubMed: 9029161]
- 194.
- Bidnenko V, Ehrlich SD, Michel B. Replication fork collapse at replication terminator sequences. EMBO J. 2002;21:3898–3907. [PMC free article: PMC126115] [PubMed: 12110601]
- 195.
- Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–606. [PubMed: 12142538]
- 196.
- Arnaudeau C, Tenorio Miranda E, Jenssen D. et al. Inhibition of DNA synthesis is a potent mechanism by which cytostatic drugs induce homologous recombination in mammalian cells. Mutat Res. 2000;461:221–228. [PubMed: 11056293]
- 197.
- Lundin C, Erixon K, Arnaudeau C. et al. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol. 2002;22:5869–5878. [PMC free article: PMC133974] [PubMed: 12138197]
- 198.
- Casper AM, Nghiem P, Arlt MF. et al. ATR regulates fragile site stability. Cell. 2002;111:779–789. [PubMed: 12526805]
- 199.
- Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–1245. [PubMed: 11292338]
- 200.
- Saintigny Y, Delacote F, Vares G. et al. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001;20:3861–3870. [PMC free article: PMC125539] [PubMed: 11447127]
- 201.
- Delacote F, Han M, Stamato TD. et al. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res. 2002;30:3454–3463. [PMC free article: PMC137076] [PubMed: 12140331]
- 202.
- Henry-Mowatt J, Jackson D, Masson JY. et al. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell. 2003;11:1109–1117. [PubMed: 12718895]
- 203.
- Liu N, Lamerdin JE, Tebbs RS. et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA crosslinks and other damages. Mol Cell. 1998;1:783–793. [PubMed: 9660962]
- 204.
- Saintigny Y, Lopez BS. Homologous recombination induced by replication inhibition, is stimulated by expression of mutant p53. Oncogene. 2002;21:488–492. [PubMed: 11821962]
- 205.
- Gottifredi V, Shieh S, Taya Y. et al. p53 accumulates but is functionally impaired when DNA synthesis is blocked. Proc Natl Acad Sci USA. 2001;98:1036–1041. [PMC free article: PMC14704] [PubMed: 11158590]
- 206.
- Cleaver J, Bartholomew J, Char D. et al. Polymerase eta and p53 jointly regulate cell survival, apoptosis and Mre11 recombination during S phase checkpoint arrest after UV irradiation. DNA Repair. 2001;1:41–57. [PubMed: 12509296]
- 207.
- Limoli CL, Laposa R, Cleaver JE. DNA replication arrest in XP variant cells after UV exposure is diverted into an Mre11-dependent recombination pathway by the kinase inhibitor wortmannin. Mutat Res. 2002;510:121–129. [PubMed: 12459448]
- 208.
- Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–551. [PubMed: 12142523]
- 209.
- de Klein A, Muijtjens M, van Os R. et al. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–482. [PubMed: 10801416]
- 210.
- Cortez D, Guntuku S, Qin J. et al. ATR and ATRIP: Partners in checkpoint signaling. Science. 2001;294:1713–1716. [PubMed: 11721054]
- 211.
- Cliby WA, Roberts CJ, Cimprich KA. et al. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. [PMC free article: PMC1170367] [PubMed: 9427750]
- 212.
- Cliby WA, Lewis KA, Lilly KK. et al. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J Biol Chem. 2002;277:1599–1606. [PubMed: 11700302]
- 213.
- Guo Z, Kumagai A, Wang SX. et al. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. [PMC free article: PMC317027] [PubMed: 11069891]
- 214.
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cel Biol. 2001;21:4129–4139. [PMC free article: PMC87074] [PubMed: 11390642]
- 215.
- Feijoo C, Hall-Jackson C, Wu R. et al. Activation of mammalian Chk1 during DNA replication arrest: A role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–923. [PMC free article: PMC1255922] [PubMed: 11535615]
- 216.
- Lupardus PJ, Byun T, Yee MC. et al. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 2002;16:2327–2332. [PMC free article: PMC187437] [PubMed: 12231621]
- 217.
- Stokes MP, Van Hatten R, Lindsay HD. et al. DNA replication is required for the checkpoint response to damaged DNA in Xenopus egg extracts. J Cell Biol. 2002;158:863–872. [PMC free article: PMC2173144] [PubMed: 12213834]
- 218.
- Tibbetts RS, Brumbaugh KM, Williams JM. et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. [PMC free article: PMC316393] [PubMed: 9925639]
- 219.
- Nghiem P, Park PK, Kim YS. et al. ATR is not required for p53 activation but synergizes with p53 in the replication checkpoint. J Biol Chem. 2002;277:4428–4434. [PubMed: 11711532]
- 220.
- Hammond EM, Denko NC, Dorie MJ. et al. Hypoxia links ATR and p53 through replication arrest. Mol Cell Biol. 2002;22:1834–1843. [PMC free article: PMC135616] [PubMed: 11865061]
- 221.
- Tibbetts RS, Cortez D, Brumbaugh KM. et al. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14:2989–3002. [PMC free article: PMC317107] [PubMed: 11114888]
- 222.
- Gatei M, Zhou BB, Hobson K. et al. Ataxia telangiectasia mutated (ATM) kinase and ATM and Rad3 related kinase mediate phosphorylation of Brca1 at distinct and overlapping sites. In vivo assessment using phospho-specific antibodies. J Biol Chem. 2001;276:17276–17280. [PubMed: 11278964]
- 223.
- Hekmat-Nejad M, You Z, Yee M. et al. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol. 2000;10:1565–1573. [PubMed: 11137007]
- 224.
- Kobayashi T, Tada S, Tsuyama T. et al. Focus-formation of replication protein A, activation of checkpoint system and DNA repair synthesis induced by DNA double-strand breaks in Xenopus egg extract. J Cell Sci. 2002;115:3159–3169. [PubMed: 12118071]
- 225.
- Michael WM, Ott R, Fanning E. et al. Activation of the DNA replication checkpoint through RNA synthesis by primase. Science. 2000;289:2133–2137. [PubMed: 11000117]
- 226.
- Takai H, Tominaga K, Motoyama N. et al. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000;14:1439–1447. [PMC free article: PMC316691] [PubMed: 10859163]
- 227.
- Unsal-Kacmaz K, Makhov AM, Griffith JD. et al. Preferential binding of ATR protein to UV-damaged DNA. Proc Natl Acad Sci USA. 2002;99:6673–6678. [PMC free article: PMC124461] [PubMed: 12011431]
- 228.
- Rouse J, Jackson SP. LCD1: An essential gene involved in checkpoint control and regulation of the MEC1 signalling pathway in Saccharomyces cerevisiae. EMBO J. 2000;19:5801–5812. [PMC free article: PMC305794] [PubMed: 11060031]
- 229.
- Edwards RJ, Bentley NJ, Carr AM. A Rad3-Rad26 complex responds to DNA damage independently of other checkpoint proteins. Nat Cell Biol. 1999;1:393–398. [PubMed: 10559981]
- 230.
- Clerici M, Paciotti V, Baldo V. et al. Hyperactivation of the yeast DNA damage checkpoint by TEL1 and DDC2 overexpression. EMBO J. 2001;20:6485–6498. [PMC free article: PMC125310] [PubMed: 11707419]
- 231.
- Rouse J, Jackson SP. Lcd1p recruits Mec1p to DNA lesions in vitro and in vivo. Mol Cell. 2002;9:857–869. [PubMed: 11983176]
- 232.
- Green CM, Erdjument-Bromage H, Tempst P. et al. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr Biol. 2000;10:39–42. [PubMed: 10660302]
- 233.
- Rauen M, Burtelow MA, Dufault VM. et al. The human checkpoint protein hRad17 interacts with the PCNA-like proteins hRad1, hHus1, and hRad9. J Biol Chem. 2000;275:29767–27971. [PubMed: 10884395]
- 234.
- Lindsey-Boltz LA, Bermudez VP, Hurwitz J. et al. Purification and characterization of human DNA damage checkpoint Rad complexes. Proc Natl Acad Sci USA. 2001;98:11236–11241. [PMC free article: PMC58713] [PubMed: 11572977]
- 235.
- Volkmer E, Karnitz LM. Human homologs of Schizosaccharomyces pombe Rad1, Hus1, and Rad9 form a DNA damage-responsive protein complex. J Biol Chem. 1999;274:567–570. [PubMed: 9872989]
- 236.
- Hang H, Lieberman HB. Physical interactions among human checkpoint control proteins HUS1p, RAD1p, and RAD9p, and implications for the regulation of cell cycle progression. Genomics. 2000;65:24–33. [PubMed: 10777662]
- 237.
- Burtelow MA, Roos-Mattjus PM, Rauen M. et al. Reconstitution and molecular analysis of the hRad9-hHus1-hRad1 (9-1-1) DNA damage responsive checkpoint complex. J Biol Chem. 2001;276:25903–25909. [PubMed: 11340080]
- 238.
- Shimomura T, Ando S, Matsumoto K. et al. Functional and physical interaction between Rad24 and Rfc5 in the yeast checkpoint pathways. Mol Cell Biol. 1998;18:5485–5491. [PMC free article: PMC109133] [PubMed: 9710632]
- 239.
- Shimada M, Okuzaki D, Tanaka S. et al. Replication factor C3 of Schizosaccharomyces pombe, a small subunit of replication factor C complex, plays a role in both replication and damage checkpoints. Mol Biol Cell. 1999;10:3991–4003. [PMC free article: PMC25738] [PubMed: 10588638]
- 240.
- Thelen MP, Venclovas C, Fidelis K. A sliding clamp model for the Rad1 family of cell cycle checkpoint proteins [letter] Cell. 1999;96:769–770. [PubMed: 10102265]
- 241.
- Venclovas C, Thelen MP. Structurebased predictions of rad1, rad9, hus1 and rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res. 2000;28:2481–2493. [PMC free article: PMC102700] [PubMed: 10871397]
- 242.
- Caspari T, Dahlen M, Kanter-Smoler G. et al. Characterization of Schizosaccharomyces pombe Hus1: A PCNA-related protein that associates with Rad1 and Rad9. Mol Cell Biol. 2000;20:1254–1262. [PMC free article: PMC85258] [PubMed: 10648611]
- 243.
- Griffith JD, Lindsey-Boltz LA, Sancar A. Structures of the human rad17-replication factor C and checkpoint rad 9-1-1 complexes visualized by glycerol spray/low voltage microscopy. J Biol Chem. 2002;277:15233–15236. [PubMed: 11907025]
- 244.
- Roos-Mattjus P, Vroman BT, Burtelow MA. et al. Genotoxin-induced Rad9-Hus1-Rad1 (9-1-1) chromatin association is an early checkpoint signaling event. J Biol Chem. 2002;277:43809–43812. [PubMed: 12228248]
- 245.
- Wang X, Zou L, Zheng H. et al. Genomic instability and endoreduplication triggered by RAD17 deletion. Genes Dev. 2003
- 246.
- Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. [PMC free article: PMC155323] [PubMed: 11799063]
- 247.
- Kai M, Wang TS. Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev. 2003;17:64–76. [PMC free article: PMC195967] [PubMed: 12514100]
- 248.
- Weiss RS, Matsuoka S, Elledge SJ. et al. Hus1 acts upstream of Chk1 in a mammalian DNA damage response pathway. Curr Biol. 2002;12:73–77. [PubMed: 11790307]
- 249.
- Weiss RS, Enoch T, Leder P. Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev. 2000;14:1886–1898. [PMC free article: PMC316817] [PubMed: 10921903]
- 250.
- Hopkins KM, Auerbach W, Wang XY. et al. Deletion of mouse Rad9 causes abnormal cellular responses to DNA damage, genomic instability and embryonic lethality 2003. submitted. [PMC free article: PMC479733] [PubMed: 15282322]
- 251.
- Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. [PubMed: 11673449]
- 252.
- Ishii Y, Bender MA. Effects of inhibitors of DNA synthesis on spontaneous and ultraviolet light-induced sister-chromatid exchanges in Chinese hamster cells. Mutat Res. 1980;79:19–32. [PubMed: 6448958]
- 253.
- Sonoda E, Sasaki MS, Morrison C. et al. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol Cell Biol. 1999;19:5166–5169. [PMC free article: PMC84359] [PubMed: 10373565]
- 254.
- Mirzoeva OK, Petrini JH. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res. 2003;1:207–218. [PubMed: 12556560]
- 255.
- Fenech M, Carr AM, Murray J. et al. Cloning and characterization of the rad4 gene of Schizosaccharomyces pombe; a gene showing short regions of sequence similarity to the human XRCC1 gene. Nucleic Acids Res. 1991;19:6737–7641. [PMC free article: PMC329303] [PubMed: 1762905]
- 256.
- Araki H, Leem SH, Phongdara A. et al. Dpb11, which interacts with DNA polymerase II(epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc Natl Acad Sci USA. 1995;92:11791–11795. [PMC free article: PMC40488] [PubMed: 8524850]
- 257.
- Yamamoto RR, Axton JM, Yamamoto Y. et al. The Drosophila mus101 gene, which links DNA repair, replication and condensation of heterochromatin in mitosis, encodes a protein with seven BRCA1 C-terminus domains. Genetics. 2000;156:711–721. [PMC free article: PMC1461266] [PubMed: 11014818]
- 258.
- Wang H, Elledge SJ. DRC1, DNA replication and checkpoint protein 1, functions with DPB11 to control DNA replication and the S-phase checkpoint in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1999;96:3824–3829. [PMC free article: PMC22379] [PubMed: 10097122]
- 259.
- McFarlane RJ, Carr AM, Price C. Characterisation of the Schizosaccharomyces pombe rad4/cut5 mutant phenotypes: Dissection of DNA replication and G2 checkpoint control function. Mol Gen Genet. 1997;255:332–340. [PubMed: 9268024]
- 260.
- Saka Y, Esashi F, Matsusaka T. et al. Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev. 1997;11:3387–3400. [PMC free article: PMC316798] [PubMed: 9407031]
- 261.
- Saka Y, Fantes P, Sutani T. et al. Fission yeast cut5 links nuclear chromatin and M phase regulator in the replication checkpoint control. EMBO J. 1994;13:5319–5329. [PMC free article: PMC395488] [PubMed: 7957098]
- 262.
- van Brabant AJ, Stan R, Ellis NA. DNA helicases, genomic instability, and human genetic disease. Annu Rev Genomics Hum Genet. 2000;1:409–459. [PubMed: 11701636]
- 263.
- Nakayama H. RecQ family helicases: Roles as tumor suppressor proteins. Oncogene. 2002;21:9008–9021. [PubMed: 12483516]
- 264.
- Gangloff S, McDonald JP, Bendixen C. et al. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: A potential eukaryotic reverse gyrase. Mol Cell Biol. 1994;14:8391–8398. [PMC free article: PMC359378] [PubMed: 7969174]
- 265.
- Watt PM, Louis EJ, Borts RH. et al. Sgs1: A eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell. 1995;81:253–260. [PubMed: 7736577]
- 266.
- Stewart E, Chapman CR, Al-Khodairy F. et al. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 1997;16:2682–2692. [PMC free article: PMC1169879] [PubMed: 9184215]
- 267.
- Murray JM, Lindsay HD, Munday CA. et al. Role of Schizosaccharomyces pombe RecQ homolog, recombination, and checkpoint genes in UV damage tolerance. Mol Cell Biol. 1997;17:6868–6875. [PMC free article: PMC232543] [PubMed: 9372918]
- 268.
- Ellis NA, Groden J, Ye TZ. et al. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. [PubMed: 7585968]
- 269.
- Yu CE, Oshima J, Fu YH. et al. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. [PubMed: 8602509]
- 270.
- Seki M, Miyazawa H, Tada S. et al. Molecular cloning of cDNA encoding human DNA helicase Q1 which has homology to Escherichia coli Rec Q helicase and localization of the gene at chromosome 12p12. Nucleic Acids Res. 1994;22:4566–4573. [PMC free article: PMC308502] [PubMed: 7527136]
- 271.
- Puranam KL, Blackshear PJ. Cloning and characterization of RECQL, a potential human homologue of the Escherichia coli DNA helicase RecQ. J Biol Chem. 1994;269:29838–29845. [PubMed: 7961977]
- 272.
- Kitao S, Ohsugi I, Ichikawa K. et al. Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics. 1998;54:443–452. [PubMed: 9878247]
- 273.
- Sekelsky JJ, Brodsky MH, Rubin GM. et al. Drosophila and human RecQ5 exist in different isoforms generated by alternative splicing. Nucleic Acids Res. 1999;27:3762–3769. [PMC free article: PMC148633] [PubMed: 10471747]
- 274.
- Shimamoto A, Nishikawa K, Kitao S. et al. Human RecQ5beta, a large isomer of RecQ5 DNA helicase, localizes in the nucleoplasm and interacts with topoisomerases 3alpha and 3beta. Nucleic Acids Res. 2000;28:1647–1655. [PMC free article: PMC102787] [PubMed: 10710432]
- 275.
- Kitao S, Lindor NM, Shiratori M. et al. Rothmund-Thomson syndrome responsible gene, RECQL4: Genomic structure and products. Genomics. 1999;61:268–276. [PubMed: 10552928]
- 276.
- Kitao S, Shimamoto A, Goto M. et al. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999;22:82–84. [PubMed: 10319867]
- 277.
- Balraj P, Concannon P, Jamal R. et al. An unusual mutation in RECQ4 gene leading to Rothmund-Thomson syndrome. Mutat Res. 2002;508:99–105. [PubMed: 12379465]
- 278.
- Sengupta S, Linke SP, Pedeux R. et al. BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J. 2003;22:1210–1222. [PMC free article: PMC150347] [PubMed: 12606585]
- 279.
- Lindor NM, Furuichi Y, Kitao S. et al. Rothmund-Thompson syndrome due to RECQ4 helicase mutations: Report and clinical and molecular comparisons with Bloom syndrome and Werner syndrome. Am J Med Genet. 2000;90:223–228. [PubMed: 10678659]
- 280.
- Shen J, Loeb LA. Unwinding the molecular basis of the Werner syndrome. Mech Ageing Dev. 2001;122:921–944. [PubMed: 11348659]
- 281.
- Brosh RM Jr, Bohr VA. Roles of the Werner syndrome protein in pathways required for maintenance of genome stability. Exp Gerontol. 2002;37:491–506. [PubMed: 11830352]
- 282.
- Mohaghegh P, Hickson I. Premature aging in RecQ helicase-deficient human syndromes. Int J Biochem Cell Biol. 2002;34:1496–1501. [PubMed: 12200042]
- 283.
- Gianneli F, Benson PF, Pawsey SA. et al. Ultraviolet light sensitivity and delayed DNA-chain maturation in Bloom's syndrome fibroblasts. Nature. 1977;265:466–469. [PubMed: 834301]
- 284.
- Hanaoka F, Yamada M, Takeuchi F. et al. Autoradiographic studies of DNA replication in the Werner syndrome In: Salk D, Fujiwara Y, Martin GM, eds.Werner's syndrome and human aging New York: Plenum Press,1985439–457. [PubMed: 4083159]
- 285.
- Lonn U, Lonn S, Nylen U. et al. An abnormal profile of DNA replication intermediates in Bloom's syndrome. Cancer Res. 1990;50:3141–3145. [PubMed: 2110504]
- 286.
- Chaganti RSK, Schonberg S, German J. A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci USA. 1974;71:4508–4512. [PMC free article: PMC433916] [PubMed: 4140506]
- 287.
- Saintigny Y, Makienko K, Swanson C. et al. Homologous recombination resolution defect in Werner syndrome. Mol Cell Biol. 2002;22:6971–6978. [PMC free article: PMC139822] [PubMed: 12242278]
- 288.
- Fukuchi K, Martin GM, Monnat RJ Jr. Mutator phenotype of Werner syndrome is characterized by extensive deletions Proc Natl Acad Sci USA 1989865893–5897.[published erratum appears in Proc Natl Acad Sci USA 1989 867994] [PMC free article: PMC297737] [PubMed: 2762303]
- 289.
- Lebel M. Increased frequency of DNA deletions in pink-eyed unstable mice carrying a mutation in the Werner syndrome gene homologue. Carcinogenesis. 2002;23:213–216. [PubMed: 11756244]
- 290.
- Yamagata K, Kato J, Shimamoto A. et al. Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: Implication for genomic instability in human disease. Proc Natl Acad Sci USA. 1998;95:8733–8738. [PMC free article: PMC21145] [PubMed: 9671747]
- 291.
- Imamura O, Fujita K, Shimamoto A. et al. Bloom helicase is involved in DNA surveillance in early S phase in vertebrate cells. Oncogene. 2001;20:1143–1151. [PubMed: 11313858]
- 292.
- Franchitto A, Pichierri P. Bloom's syndrome protein is required for correct relocalization of RAD50/ MRE11/NBS1 complex after replication fork arrest. J Cell Biol. 2002;157:19–30. [PMC free article: PMC2173275] [PubMed: 11916980]
- 293.
- Franchitto A, Pichierri P. Protecting genomic integrity during DNA replication: Correlation between Werner's and Bloom's syndrome gene products and the MRE11 complex. Hum Mol Genet. 2002;11:2447–2453. [PubMed: 12351580]
- 294.
- Ababou M, Dumaire V, Lecluse Y. et al. Bloom's syndrome protein response to ultraviolet-C radiation and hydroxyurea-mediated DNA synthesis inhibition. Oncogene. 2002;21:2079–2088. [PubMed: 11960380]
- 295.
- Okada M, Goto M, Furuichi Y. et al. Differential effects of cytotoxic drugs on mortal and immortalized B-lymphoblastoid cell lines from normal and Werner's syndrome patients. Biol Pharm Bull. 1998;21:235–239. [PubMed: 9556152]
- 296.
- Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci USA. 1998;95:13097–13102. [PMC free article: PMC23722] [PubMed: 9789047]
- 297.
- Poot M, Gollahon KA, Rabinovitch PS. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum Genet. 1999;104:10–14. [PubMed: 10071186]
- 298.
- Pichierri P, Franchitto A, Mosesso P. et al. Werner's syndrome cell lines are hypersensitive to camptothecin-induced chromosomal damage. Mutat Res. 2000;456:45–57. [PubMed: 11087895]
- 299.
- Pichierri P, Franchitto A, Mosesso P. et al. Werner's syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol Biol Cell. 2001;12:2412–2421. [PMC free article: PMC58603] [PubMed: 11514625]
- 300.
- Karow JK, Constantinou A, Li JL. et al. The Bloom's syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci USA. 2000;97:6504–6508. [PMC free article: PMC18638] [PubMed: 10823897]
- 301.
- Constantinou A, Tarsounas M, Karow JK. et al. Werner's syndrome protein (WRN) migrates holliday junctions and colocalizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. [PMC free article: PMC1083680] [PubMed: 11256630]
- 302.
- Yang Q, Zhang R, Wang XW. et al. The processing of Holliday junctions by BLM and WRN helicases is regulated by p53. J Biol Chem. 2002;277:31980–31987. [PubMed: 12080066]
- 303.
- Wang W, Seki M, Narita Y. et al. Possible association of BLM in decreasing DNA double strand breaks during DNA replication. EMBO J. 2000;19:3428–3435. [PMC free article: PMC313960] [PubMed: 10880455]
- 304.
- Ng SW, Liu Y, Hasselblatt KT. et al. A new human topoisomerase III that interacts with SGS1 protein. Nucleic Acids Res. 1999;27:993–1000. [PMC free article: PMC148278] [PubMed: 9927731]
- 305.
- Wu L, Davies SL, North PS. et al. The Bloom's syndrome gene product interacts with topoisomerase III. J Biol Chem. 2000;275:9636–9644. [PubMed: 10734115]
- 306.
- Johnson FB, Lombard DB, Neff NF. et al. Association of the Bloom syndrome protein with topoisomerase IIIalpha in somatic and meiotic cells. Cancer Res. 2000;60:1162–1167. [PubMed: 10728666]
- 307.
- Hyde H, Davies AA, Benson FE. et al. Resolution of recombination intermediates by a mammalian activity functionally analogous to Escherichia coli RuvC resolvase. J Biol Chem. 1994;269:5202–5209. [PubMed: 8106502]
- 308.
- Constantinou A, Davies AA, West SC. Branch migration and Holliday junction resolution catalyzed by activities from mammalian cells. Cell. 2001;104:259–268. [PubMed: 11207366]
- 309.
- Kaliraman V, Mullen JR, Fricke WM. et al. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 2001;15:2730–2740. [PMC free article: PMC312806] [PubMed: 11641278]
- 310.
- Boddy MN, Lopez-Girona A, Shanahan P. et al. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol. 2000;20:8758–8566. [PMC free article: PMC86503] [PubMed: 11073977]
- 311.
- Doe CL, Ahn JS, Dixon J. et al. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J Biol Chem. 2002;277:32753–32759. [PubMed: 12084712]
- 312.
- Beamish H, Kedar P, Kaneko H. et al. Functional link between BLM defective in Bloom's syndrome and the ataxia-telangiectasia mutated protein, ATM. J Biol Chem. 2002;277:30515–30523. [PubMed: 12034743]
- 313.
- Cortez D, Wang Y, Qin J. et al. Requirement of ATM-dependent phosphorylation of Brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. [PubMed: 10550055]
- 314.
- Gatei M, Scott SP, Filippovitch I. et al. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000;60:3299–3304. [PubMed: 10866324]
- 315.
- Xu B, Kim ST, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001;21:3445–3450. [PMC free article: PMC100266] [PubMed: 11313470]
- 316.
- Xu B, O'Donnell AH, Kim ST. et al. Phosphorylation of serine 1387 in Brca1 is specifically required for the Atm-mediated S-phase checkpoint after ionizing irradiation. Cancer Res. 2002;62:4588–4591. [PubMed: 12183412]
- 317.
- Okada S, Ouchi T. Cell cycle differences in DNA-damage-induced BRCA1 phosphorylation affect its subcellular localization. J Biol Chem. 2003
- 318.
- Khanna KK, Keating KE, Kozlov S. et al. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat Genet. 1998;20:398–400. [PubMed: 9843217]
- 319.
- Chehab NH, Malikzay A, Appel M. et al. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article: PMC316357] [PubMed: 10673500]
- 320.
- Matsuoka S, Rotman G, Ogawa A. et al. Ataxia telangiectasia-mutated phosphorylates chk2 in vivo and in vitro. Proc Natl Acad Sci USA. 2000;97:10389–01394. [PMC free article: PMC27034] [PubMed: 10973490]
- 321.
- Melchionna R, Chen XB, Blasina A. et al. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat Cell Biol. 2000;2:762–765. [PubMed: 11025670]
- 322.
- Ahn JY, Schwarz JK, Piwnica-Worms H. et al. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–5936. [PubMed: 11085506]
- 323.
- Xu X, Tsvetkov LM, Stern DF. Chk2 activation and phosphorylation-dependent oligomerization. Mol Cell Biol. 2002;22:4419–4432. [PMC free article: PMC133858] [PubMed: 12024051]
- 324.
- Ahn J, Prives C. Checkpoint Kinase 2 (Chk2) Monomers or Dimers Phosphorylate Cdc25C after DNA Damage Regardless of Threonine 68 Phosphorylation. J Biol Chem. 2002;277:48418–48426. [PubMed: 12386164]
- 325.
- Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. [PMC free article: PMC155347] [PubMed: 11877376]
- 326.
- Chen MJ, Lin YT, Lieberman HB. et al. Atm-dependent phosphorylation of human rad9 is required for ionizing radiation-induced checkpoint activation. J Biol Chem. 2001;276:16580–16586. [PubMed: 11278446]
- 327.
- Gately DP, Hittle JC, Chan GK. et al. Characterization of ATM expression, localization, and associated DNA-dependent protein kinase activity. Mol Biol Cell. 1998;9:2361–2374. [PMC free article: PMC25502] [PubMed: 9725899]
- 328.
- Chan DW, Son SC, Block W. et al. Purification and characterization of ATM from human placenta. A manganese-dependent, wortmannin-sensitive serine/threonine protein kinase. J Biol Chem. 2000;275:7803–7810. [PubMed: 10713094]
- 329.
- Wang H, Guan J, Wang H. et al. Replication protein A2 phosphorylation after DNA damage by the coordinated action of ataxia telangiectasia-mutated and DNA-dependent protein kinase. Cancer Res. 2001;61:8554–8563. [PubMed: 11731442]
- 330.
- Sapkota GP, Deak M, Kieloch A. et al. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem J. 2002;368:507–516. [PMC free article: PMC1223019] [PubMed: 12234250]
- 331.
- Stewart GS, Last JI, Stankovic T. et al. Residual ataxia telangiectasia mutated protein function in cells from ataxia telangiectasia patients, with 5762ins137 and 7271T—>G mutations, showing a less severe phenotype. J Biol Chem. 2001;276:30133–30141. [PubMed: 11382771]
- 332.
- Lee JS, Collins KM, Brown AL. et al. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature. 2000;404:201–204. [PubMed: 10724175]
- 333.
- Wu L, Davies SL, Levitt NC. et al. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem. 2001;276:19375–19381. [PubMed: 11278509]
- 334.
- Davies AA, Masson JY, McIlwraith MJ. et al. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7:273–282. [PubMed: 11239456]
- 335.
- Wu LC, Wang ZW, Tsan JT. et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–440. [PubMed: 8944023]
- 336.
- Chen J, Silver DP, Walpita D. et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2:317–328. [PubMed: 9774970]
- 337.
- Chai YL, Cui J, Shao N. et al. The second BRCT domain of BRCA1 proteins interacts with p53 and stimulates transcription from the p21WAF1/CIP1 promoter. Oncogene. 1999;18:263–268. [PubMed: 9926942]
- 338.
- Zhong Q, Chen CF, Li S. et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999;285:747–750. [PubMed: 10426999]
- 339.
- Garcia-Higuera I, Taniguchi T, Ganesan S. et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. [PubMed: 11239454]
- 340.
- Fujimori A, Tachiiri S, Sonoda E. et al. Rad52 partially substitutes for the Rad51 paralog XRCC3 in maintaining chromosomal integrity in vertebrate cells. EMBO J. 2001;20:5513–5520. [PMC free article: PMC125654] [PubMed: 11574483]
- 341.
- Lakin ND, Hann BC, Jackson SP. The ataxia-telangiectasia related protein ATR mediates DNA-dependent phosphorylation of p53. Oncogene. 1999;18:3989–3995. [PubMed: 10435622]
- 342.
- Liu Q, Guntuku S, Cui XS. et al. Chk1 is an essential kinase that is regulated by ATR and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article: PMC316686] [PubMed: 10859164]
- 343.
- Bao S, Tibbetts RS, Brumbaugh KM. et al. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–974. [PubMed: 11418864]
- 344.
- Wang X, Wang L, Callister MD. et al. Human Rad17 is phosphorylated upon DNA damage and also overexpressed in primary nonsmall cell lung cancer tissues. Cancer Res. 2001;61:7417–7421. [PubMed: 11606373]
- 345.
- Post S, Weng YC, Cimprich K. et al. Phosphorylation of serines 635 and 645 of human Rad17 is cell cycle regulated and is required for G1/S checkpoint activation in response to DNA damage. Proc Natl Acad Sci USA. 2001;98:13102–13107. [PMC free article: PMC60831] [PubMed: 11687627]
- 346.
- Karmakar P, Piotrowski J, Brosh RM Jr. et al. Werner protein is a target of DNA-PK in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J Biol Chem. 2002;277:18291–18302. [PubMed: 11889123]
- 347.
- Wang XW, Tseng A, Ellis NA. et al. Functional interaction of p53 and BLM DNA helicase in apoptosis. J Biol Chem. 2001;276:32948–32955. [PubMed: 11399766]
- 348.
- Garkavtsev IV, Kley N, Grigorian IA. et al. The Bloom syndrome protein interacts and cooperates with p53 in regulation of transcription and cell growth control. Oncogene. 2001;20:8276–8280. [PubMed: 11781842]
- 349.
- Von Kobbe C, Karmakar P, Dawut L. et al. Colocalization, physical, and functional interaction between Werner and Bloom syndrome proteins. J Biol Chem. 2002;277:22035–22044. [PubMed: 11919194]
- 350.
- Brosh J, Karmakar P, Sommers JA. et al. p53 modulates the exonuclease activity of Werner syndrome protein. J Biol Chem. 2001;276:35093–35102. [PubMed: 11427532]
- 351.
- Baynton K, Otterlei M, Bjoras M. et al. WRN interacts physically and functionally with the recombination mediator protein Rad52 J Biol Chem 2003. in press. [PubMed: 12750383]
- 352.
- Dahm K, Hubscher U. Colocalization of human Rad17 and PCNA in late S phase of the cell cycle upon replication block. Oncogene. 2002;21:7710–7719. [PubMed: 12400013]
- 353.
- Sagata N. MOLECULAR BIOLOGY: Untangling checkpoints. Science. 2002;298:1905–1907. [PubMed: 12471241]
- 354.
- Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA. 2002;99:14795–14800. [PMC free article: PMC137498] [PubMed: 12399544]
- 355.
- Donzelli M, Squatrito M, Ganoth D. et al. Dual mode of degradation of Cdc25A phosphatase. EMBO J. 2002;21:4875–4884. [PMC free article: PMC126287] [PubMed: 12234927]
- 356.
- Foray N, Marot D, Gabriel A. et al. A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J. 2003;11:2860–2871. [PMC free article: PMC156770] [PubMed: 12773400]
- Origin, Recognition, Signaling and Repair of DNA Double-Strand Breaks in Mammali...Origin, Recognition, Signaling and Repair of DNA Double-Strand Breaks in Mammalian Cells - Madame Curie Bioscience Database
- Protein Misassembly: Macromolecular Crowding and Molecular Chaperones - Madame C...Protein Misassembly: Macromolecular Crowding and Molecular Chaperones - Madame Curie Bioscience Database
- p53's Dilemma in Transcription: Analysis by Microarrays - Madame Curie Bioscienc...p53's Dilemma in Transcription: Analysis by Microarrays - Madame Curie Bioscience Database
- Genetics, Mutations, and Polymorphisms - Madame Curie Bioscience DatabaseGenetics, Mutations, and Polymorphisms - Madame Curie Bioscience Database
- Tumor-Initiating Cells, Cancer Metastasis and Therapeutic Implications - Madame ...Tumor-Initiating Cells, Cancer Metastasis and Therapeutic Implications - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...