

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Myocardial function is compromised in several forms of heart disease due to the loss of cardiomyocytes and in part to the limited ability of surviving myocytes to re-enter the cell cycle. The ability to induce cardiomyocyte proliferation will improve myocardial function. There is scant information about the mechanisms regulating cardiomyocyte proliferation and cell death. In this Chapter, we have summarized various genetic manipulations impacting on DNA synthesis/proliferation of cardiomyocytes in the adult heart. In addition, we have described in detail our efforts to identify key cardiomyocyte cell cycle and survival regulators using cell lines derived from mice expressing SV40 large T antigen in the myocardium.
Introduction
It is clear that the adult myocardium lacks sufficient regenerative capacity to reverse most forms of heart disease. In light of this, it has been assumed that increasing the number of functional, coupled cardiomyocytes in a diseased heart will result in improved contractile function. Consequently, considerable effort has been invested to develop strategies aimed at restoring myocardial mass. Several approaches to accomplish this are currently under development. These include efforts to engraft donor myocytes prepared from a variety of sources,1 efforts to promote neocardiomyogenesis using cardiomyocyte lineage-determining genes,2 and efforts to promote proliferation of the surviving cardiomyocytes in diseased heart.3 In this Chapter, we present a brief overview of studies quantitating the rate of cardiomyocyte DNA synthesis in the adult heart, followed by a survey of transgenic models which have provided insight into the molecular regulation of the cardiomyocyte cell cycle. We then describe in detail studies from our group wherein cardiomyocyte cell lines derived from transgenic mice expressing the SV40 Large T Antigen (T-Ag) were utilized to identify key cardiomyocyte cell cycle and survival gene products.
Evidence for Cell Cycle Activity in the Adult Myocardium
Although it is clear that the innate regenerative capacity of the heart is limited, the absolute rate of cardiomyocyte DNA synthesis, karyokinesis and cytokinesis in the normal or injured adult heart remains somewhat controversial. A variety of markers have been employed to monitor cardiomyocyte DNA synthesis and/or cell cycle activity. For example, traditional cytologic markers such as the presence of mitotic figures (identified via histochemical analyses or via confocal microscopy) are indicative of karyokinesis and/or cytokinesis. Metabolic markers such as the incorporation of BrdU or tritiated thymidine have been employed to identify cells replicating their DNA (i.e., cells in S-phase). Finally, the presence of molecular markers, as for example proteins which are expressed only during discrete phases of the cell cycle, can be used to identify proliferating cells. Regardless of the approach used, the accuracy of the assay is dependent upon the fidelity with which cardiomyocyte (or cardiomyocyte nuclei) can be distinguished from non-myocytes (or non-myocyte nuclei). A priori cell type identification would seem to be a trivial task. However, cardiomyocytes constitute only 20% of the total number of cells present in the adult heart (despite the fact that they account for more than 90% of the mass of the heart). Given this, given the inherently low rate of cardiomyocyte DNA synthesis in the adult heart, and given the high propensity for non-myocyte proliferation (particularly in a diseased heart), accurate cell-type identification is critical.
Values reported in the literature for cardiomyocyte cell cycle activity in the normal adult heart range from as low as 0% to as high as ca. 3%, with even greater values reported for injured hearts. This range of values undoubtedly reflects in part the assay used to score cell cycle activity, as well as the fidelity of cell type identification. These issues have recently been reviewed in detail.4,5 Our laboratory has relied heavily upon a transgenic mouse model which expresses a nuclear localized β-galactosidase (nLAC) reporter gene under the regulation of the cardiomyocyte-restricted α-cardiac myosin heavy chain (MHC) promoter. Cardiomyocyte nuclei in histologic sections prepared from these animals (designated MHC-nLAC) can unequivocally be identified by simple staining with X-GAL (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside). When used in combination with tritiated thymidine incorporation, cardiomyocyte DNA synthesis is easily quantitated by simply scoring for the presence of silver grains over X-GAL-stained nuclei (Fig. 1).
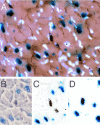
Experiments using this reporter gene assay revealed that only 0.0005% of the cardiomyocyte nuclei are actively synthesizing DNA in the normal adult mouse heart following a single injection of tritiated thymidine.6 Furthermore, only a slight increase in cardiomyocyte DNA synthesis was observed in animals with isoproterenol-induced hypertrophy or in peri-necrotic zone of animals with cautery injury of the left ventricular wall6,7. It is important to note that our transgenic reporter assay assumes that the MHC promoter remains transcriptionally active (or that the half-life of βGAL activity is sufficient to permit identification of the cell). Although silencing of the MHC promoter in cells synthesizing DNA would lead to an under-estimate of the number of cycling cells, the available data suggest that this is not likely to be a serious concern. Analysis of dispersed cell preparations from adult MHC-nLAC mice indicates that 100% of cardiomyocytes (identified based on morphologic attributes) express nLAC. Moreover, the nuclear βGAL activity in those cardiomyocytes which do synthesize DNA is extremely strong (Fig. 1),4 suggesting that α-MHC promoter activity is not altered in these cells. Finally, similar cardiomyocyte labeling indices were obtained in experiments with neonatal mice which directly compared cardiomyocyte labeling indices determined with the MHC-nLAC reporter system vs. those determined with dispersed cell preparations.6
Transgenic Models for Cardiomyocyte Cycle Deregulation
The development of efficient gene transfer techniques has permitted direct assessment of the effects of specific gene products in cardiomyocyte biology. In particular, the ability to generate gain- and loss-of-function transgenic animals enables one to directly examine the role of specific gene products in the regulation of cardiomyocyte proliferation and survival in vivo.8,9 Table 1 provides a summary of studies wherein transgenic manipulation has impacted upon cardiomyocyte cell cycle regulation. Our goal is to illustrate the breadth of transgenic approaches which have yielded information regarding cardiomyocyte cell cycle regulation; however this list is by no means exhaustive. Studies which enhanced cardiomyocyte proliferation relied on targeted expression of DNA tumor virus oncoproteins, targeted expression of cellular protooncogenes, altered expression of tumor suppressors or negative cell cycle regulatory molecules, or altered expression of signaling molecules. In addition, several examples of genetic manipulations which gave rise to hypoplastic hearts are presented. It is clear from this list that germ-line manipulation in transgenic animals provides a powerful approach with which to study cardiomyocyte proliferation in vivo.
Table 1
Genetic manipulations impacting cardiomyocyte proliferation/DNA synthesis.
Expression of the SV40 Large T Antigen in the Myocardium
Our laboratory has studied a number of transgenic mouse models in which expression of the T-Ag oncoprotein was targeted to the atrial or ventricular myocardium. For example, T-Ag expression under the regulation of the human atrial natriuretic factor (ANF) promoter gave rise to predominantly right atrial tumors (Fig. 2.) 10 The tumors were comprised of differentiated, proliferating cardiomyocytes. Typically, left atrial involvement in these animals was less pronounced, despite the fact that similar levels of T-Ag were expressed in the left vs. right atrium.10 The asymmetrical tumorigenic penetrance may reflect differences in the developmental timing of transgene expression, or alternatively differential rates of cell cycle withdrawal, in the left vs. right atria. These data suggest that T-Ag's ability to influence cardiomyocyte proliferation may be restricted to cells which are actively dividing, a notion which is supported by recent cell culture studies using atrial cardiomyocytes from transgenic mice expressing a temperature labile T-Ag.11 Other studies have revealed that mice expressing T-Ag under the regulation of the α-cardiac MHC promoter developed both atrial and ventricular tumors.12 Minimally, these observations indicated that cardiomyocytes from both chambers have a similar response to targeted T-Ag expression.

T-Ag Binding Proteins as a Paradigm for the Identification of Cardiomyocyte Cell Cycle Regulators
Studies from a number of groups in the late 1980's demonstrated that the DNA tumor virus oncoproteins (as exemplified by T-Ag and Adenoviral E1A) form a stable complex with the retinoblastoma gene product (RB), the prototypical tumor suppressor protein.13–15 These observations immediately suggested a mechanism by which the DNA tumor virus oncoproteins could subjugate cell cycle control. Specifically, it was proposed that the transforming activities of these oncoproteins resided largely in their ability to bind to and thereby alter the activity of endogenous cell cycle regulators.
We have used this paradigm to identify putative cardiomyocyte cell cycle regulators. The first step entailed the derivation of cardiomyocyte cell lines from the transgenic tumors expressing T-Ag.16,17 Once generated, the cells were cultured in the presence of 35S-methionine in order to radiolabel the proteins. The cells were then disrupted in the presence of non-ionic detergents such that protein-protein interactions were maintained, and the resulting solution of proteins was reacted with anti-T-Ag monoclonal antibodies to form an immune complex. The immune complex was absorbed onto protein A-sepharose beads, collected by centrifugation, denatured and displayed on polyacrylamide gels. In addition to T-Ag, proteins with molecular weights of 53, 120, 193 and 380 kd were present in the anti-T-Ag immune complex, but not in control immune complex (Fig. 3). These data indicate that the proteins bind to T-Ag directly, or alternatively, that they bind T-Ag indirectly as part of a multi-protein complex. Importantly, all four proteins were present in immune complex generated with multiple anti-T-Ag antibodies.17,18 Moreover, the same binding proteins were observed in atrial and ventricular cardiomyocytes, suggesting that T-Ag activates cardiomyocyte proliferation cycle via a similar mechanism in the atria and ventricle. The molecular activities of the 53, 120, 193 and 380 kd proteins are considered below.
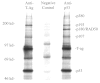
p53
The p53 tumor suppressor was originally identified by virtue of its ability to bind to T-Ag.19 It thus seemed likely that the 53 kd protein present in anti-T-Ag immune complex was p53. This was directly confirmed by additional immune precipitation analyses using radiolabelled proteins from the cadiomyocyte cell lines: immune complex generated with anti-p53 monoclonal antibodies contained the 53 kd protein, as well as T-Ag and the 120, 193 and 380 kd proteins (Fig. 3)It is now well established that p53 plays a critical role in triggering apoptosis in a wide variety of mammalian cells, largely via its ability to function as a transcriptional co-activator of pro-apoptosis as well as growth inhibitory genes. For example, p53 can activate transcription of Bax, a pro-apoptotic member of the Bcl-2 family. The resulting shift in the relative levels of pro-survival and pro-apoptosis Bcl-2 family members activates Apaf-1, which in turn activates caspase 8 and the apoptotic cascade. p53 can also induce expression of p21CIP, a negative regulator of the G1/S cyclin dependent kinases, and thereby directly influence cell cycle progression. These activities bestow a pivotal role for p53 in cell cycle regulation. Indeed, inheritance of a mutant p53 allele results in marked predisposition to a wide spectrum of cancers in Li-Fraumeni patients,20 as well as in genetically manipulated mice.21 Gene amplification and other anomalies are readily induced in fibroblasts lacking p53 (which fail to apoptose in response to genotypic stress), but not in cells with functional p53 (in which the pro-apoptotic pathway is active).22
In the mouse, Northern blot analyses of total RNA samples prepared from intact hearts revealed that p53 expression closely followed cardiomyocyte cell cycle activity, with relatively high levels of expression in fetal hearts and low levels in adult hearts.23,24 p53 also has been implicated in cardiomyocyte apoptosis. For example, increased levels of p53 and concomitant increases in cardiomyocyte apoptosis are observed in several models of myocardial injury in vitro and in vivo.25–30 However, it also appeared that other forms of cardiomyocyte apoptosis occur via p53-independent pathways.31–33 In addition to these descriptive studies, there is also functional data supporting a role for p53 in cardiomyocyte apoptosis. For example, adenoviral delivery of p53 induces apoptosis in cultured cardiomyocytes.34,35 Cardiomyocyte apoptosis is markedly decreased in allografts from p53 deficient animals as compared to wild type animals.36 Expression of a dominant negative p53 blocked stretch-mediated cardiomyocyte apoptosis, with concomitant inhibition of the renin-angiotensin system.37 These data collectively implicate induction of p53 activity with at least some forms of pathophysiologic cardiomyocyte apoptosis.
Finally, the 180-Kd protein present in immune complex generated with anti-p53 monoclonal antibody PAb-241(Fig. 3) but not in other anti-p53 nor anti-T-Ag immune complexes,17,18 deserves brief mention. Microsequence and molecular cloning studies revealed that this is the mouse homologue of RAD50, a protein required for the repair of double-stranded DNA breaks. 38RAD50 has limited epitopic homology to p53, and is directly immune precipitated by PAb-421.
p107
Immune precipitation/Western analyses revealed that the 120-Kd protein present in cardiomyocyte anti-T-Ag immune complex was p107, a member of the RB tumor suppressor family. p107 was initially identified by virtue of its ability to bind to E1A and T-Ag.39–41 Although sequence analysis of full-length human cDNAs indicated a deduced molecular weight of 119 Kd,42 the original designation of p107, which was based on the protein's apparent molecular weight in polyacrylamide gels, has persisted. Once cloned, sequence analyses revealed a striking degree of regional homology between p107 and the "pocket domain" of RB. Functional analyses indicated that over-expression of p107 inhibited proliferation in some cell lines, providing the first indication supporting a direct role of p107 in cell cycle regulation.42 This notion was further supported by the observation that p107 binds to many of the same key cell cycle regulators as RB, including E2F family members,43 as well as a number of cyclin:cyclin-dependent kinase complexes. Like p53, p107 mRNA and protein were expressed at relatively high levels in fetal mouse hearts where cardiomyocyte proliferation rates are high, and were essentially absent in the adult mouse heart.23,34,44
Although no overt phenotype was apparent in transgenic mice lacking p107, homozygous mutation of p130 (the third member of the RB tumor suppressor family) resulted in embryonic lethality with multiple developmental anomalies when crossed into a BALB/cJ genetic background.45 Of particular interest, the ventricular walls of these animals were abnormally thin, although it is not clear if this reflected a primary vs. secondary effect of p130 mutation. The prevalence of T-Ag/p107 immune complex in the cardiomyocyte cell lines, and in particular the absence of T-Ag/RB and T-Ag/p130 immune complex, deserves mention. This observation was particularly puzzling as our cardiomyocyte cell lines express hypophosphorylated RB (the isoform which preferentially binds to T-Ag). However, no RB was detected in anti-T-Ag immune complex generated from the cardiomyocyte cell lines, nor in immune complex generated from primary transgenic mouse myocardial tumors.17,23 This strongly suggested that abrogation of RB activity by direct T-Ag binding is not required for cardiomyocyte cell cycle activation in the transgenic mouse model. This notion is indirectly supported by a number of other observations. For example, the absence of primary myocardial tumors in retinoblastoma patients, as well as the absence of an overt cardiomyocyte phenotype in RB deficient transgenic mice, indicate that modulation of RB expression by itself is insufficient to markedly drive cardiomyocyte cell cycle activity. Similarly, lack of correlation between RB levels and phosphorylation status vs. cardiomyocyte cell cycle activity when whole heart extracts from mouse were examined further argues against a regulatory role for RB in the heart.23,24
p193
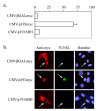
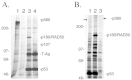
We have recently cloned and characterized the 193-Kd protein present in cardiomyocyte anti-T-Ag immune complex.18 Large-scale immune complex prepared from our transgenic cardiomyocyte cell lines was displayed on polyacrylamide gels, and the 193-Kd protein was excised and subjected to micro-sequence analysis. The resulting amino acid sequence was then used to clone cDNAs encoding the protein (designated p193). Analysis of the deduced amino acid sequence from a full-length p193 cDNA failed to reveal any obvious functional domains. However, a short amino acid motif (VRILKAHGDEGLHV) near the C-terminus of the protein suggested that p193 may be a new member of the Bcl-2 family of apoptosis regulators. Homology between Bcl-2 family members is restricted to the presence of short Bcl-2 homology domains (or BH domains46). The short amino acid motif in p193 matches the consensus sequence of a BH-3 domain (LXXXGDE). Of interest, members of the BH-3 only subgroup encode pro-apoptotic activity, and, as the name implies, their homology to Bcl-2 is limited to the presence of the BH-3 domain.47,48 Based on this observation, transfection experiments were initiated to determine if p193 encodes pro-apoptotic activity. As anticipated, expression of p193 in NIH-3T3 cells induced an apoptotic response (Fig. 4). Furthermore, deletion of the C-terminal motif encompassing the BH3 domain (VRILKAHGDEGLHV) abolished p193's apoptotic activity (Fig. 4.) Thus, p193 is a new member of the BH-3 only pro-apoptosis family.
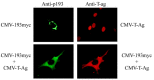
Additional analyses revealed that, like other BH-3 only family members, co-expression of Bcl-XL antagonizes p193's pro-apoptotic activity.18 Transfection studies with serum-synchronized cell cultures indicated that overexpression of p193 induced apoptosis at the G1/S boundary of the cell cycle. Surprisingly, transfection with epitope-tagged p193 expression constructs revealed that p193 is localized to the cytoplasm, a result which was not expected as p193 was isolated by virtue of its ability to bind to T-Ag, a nuclear oncoprotein. Immune cytologic analysis of co-transfected cells suggested an explanation of this paradox. Co-transfection of NIH-3T3 cells with expression constructs encoding p193 and T-Ag resulted in the cytoplasmic sequestration of both proteins (Fig. 5). This suggested that T-Ag/p193 binding might normally occur in the cytoplasm. In support of this, pulse-chase experiments revealed that, in our transgenic cardiomyocyte cell lines, T-Ag is sequestered in the cytoplasm from the onset of mitosis and remains in the cytoplasm well into G1 (the same point of the cell cycle where forced expression of p193 induces apoptosis). Finally, immune precipitation analyses of in vitro translated proteins mapped the p193 binding site to an N-terminal region of T-Ag overlapping with, but distinct from, the RB binding site. Previous studies have suggested that this region encodes transforming activity which is independent of RB family member binding.49
p380
Although we have not yet cloned the 380-Kd protein (which we have designated p380), some information regarding its molecular activity is available. As indicated above, p380 is present in both anti-T-Ag and anti-p53 immune complex generated from our tumor cardiomyocyte cell lines (Fig. 3). Given the difficulty in accurately assigning molecular weights for proteins larger than 200 Kd, our initial thought was that p380 might be p300 or a related molecule (p300 is a transcriptional co-activator with intrinsic histone acetylase activity). However, comparative analyses of immune complex generated with an anti-p300 antibody and with anti-T-Ag and anti-p53 antibodies clearly indicated that p380 is not p300 (Fig. 6A). Studies also indicated that our cardiomyocyte cell lines express high levels of p300. In a subsequent study, we compared anti-p53 immune complex generated from our cardiomyocyte cell lines to that generated from mouse embryonic stem (ES) cells (Fig. 6B). p380 was present in the immune complex from ES cells: since these cells do not express T-Ag, p380 must bind directly to p53. It is also of interest to note that the ES cells express high levels of RAD50 (see PAb-421 immune complex).
Future Applications, New Research, Anticipated Developments
We reasoned that since the activities encoded by T-Ag are sufficient to induce sustained cardiomyocyte proliferation in transgenic animals, identification of the T-Ag binding proteins would likely provide insight into the molecular regulation of the cardiomyocyte cell cycle. The T-Ag binding proteins identified in our studies are depicted schematically in (Figure 7A). To us, the observation that T-Ag binds to two pro-apoptotic proteins was quite surprising. This led to the suggestion that, in cardiomyocytes, it may be necessary to abrogate two pro-apoptotic pathways to induce proliferation.

The differential response of cardiomyocytes to T-Ag vs. E1A gene transfer in cardiomyocytes tends to support this notion. As demonstrated above, transgenic mice with targeted T-Ag expression develop tumors comprised of differentiated, proliferating cardiomyocytes. In contrast, although E1A expression activates DNA synthesis in cultured cardiomyocytes (as evidenced by BrdU incorporation), the cells undergo apoptosis prior to cytokinesis.50–54 The molecular basis for the differential response to T-Ag vs. E1A gene transfer can be deduced by comparison of their respective binding proteins (Fig. 7B). As we have seen, T-Ag binds to a regulator of restriction point transit (p107), and two pro-apoptotic proteins (p53 and p193). Experiments with primary cardiomyocyte cultures52 as a transcriptional co-activator (p300), but not to any pro-apoptotic proteins. If we assume that the activity of these proteins are altered or abrogated upon DNA tumor virus oncoprotein binding, it follows that T-Ag possesses anti-apoptotic activities (namely the ability to alter p53 and p193 activity) which are lacking in E1A.
The observation that E1A expression induces apoptosis in cardiomyocytes where the p53 and/or the p193 pro-apoptotic pathways are intact is consistent with our model.50–54 The failure of E1A and E1B co-expression to recapitulate the proliferative response seen with T-Ag expression in cardiomyocytes further underscores the importance of the p193 pathway.50 Based on our model, we would anticipate that expression of E1A in conjunction with blockade of both the p53 and the p193 pathways would induce cardiomyocyte proliferation. Indeed, this appears to be the case in embryonic stem cell-derived cardiomyocytes which co-express E1A and cDNAs encoding dominant negative p53 and p193 activities.55
Current studies are aimed at further characterizing the molecular activities of p193 in cardiomyocytes. For example, additional experiments are under way to determine if blockade of the p53 and p193 pathways renders ES-derived cardiomyocytes permissive to E2F-1 induced proliferation. This latter study was prompted by the observation that E2F-1 induced cell cycle activation in adult hearts was also followed by a prompt apoptotic response.56 Additional studies aimed at examining the roles of p53 and p193 in pathophysiologic apoptosis in the adult heart are also in progress. Of particular interest, preliminary results indicate that expression of a dominant negative p193 cDNA in transgenic mice renders the adult heart remarkably resistant to experimentally induced fibrosis (S.-C. Tsai and L.J. Field, unpublished observation). These studies should help to determine if manipulation of the p193 pathway can be exploited to effect cardioprotection and/or myocardial regeneration.
Acknowledgments
We thank the NHLBI (LJF) and American Heart Association, Indiana Affiliate (KBSP) for support, and Dr. Michael Rubart (Indiana University) for comments on the manuscript.
References
- 1.
- Reinlib L, Field LJ. Transplantation: Future therapy for cardiovascular disease? An NHLBI workshop. Circulation. 2000;101:e182–e187. [PubMed: 10801766]
- 2.
- Lin Q, Srivastava D, Olson EN. A transcriptional pathway for cardiac development. Cold Spring Harb Symp Quant Biol. 1997;62:405–411. [PubMed: 9598375]
- 3.
- Pasumarthi K B S, Field LJ. Strategies to identify cardiomyocyte cell cycle regulatory genes In: Hasenfuss G, Marban E, eds. Molecular Strategies to the Therapy of Heart Failure Darmstadt: Thieme Istein Kopff Publishers, 2000333–351.
- 4.
- Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. [PubMed: 9670914]
- 5.
- Anversa P, Kajstura J. Ventricular myocytes are not terminally differentiated in the adult mammalian heart. Circ Res. 1998;83:1–14. [PubMed: 9670913]
- 6.
- Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–H226. [PubMed: 9038941]
- 7.
- Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis during hypertrophy in adult mice. Am J Physiol. 1994;266:H1439–H1445. [PubMed: 8184922]
- 8.
- Field LJ. Transgenic mice in cardiovascular research. Annu Rev Physiol. 1993;55:97–114. [PubMed: 8466194]
- 9.
- Field LJ. Cardiovascular research in transgenic animals. Trends in Cardiovascular Medicine. 1991;1:141–146. [PubMed: 21239315]
- 10.
- Field LJ. Atrial natriuretic factor-SV40 T antigen transgenes produce tumors and cardiac arrhythmias in mice. Science. 1988;239:1029–1033. [PubMed: 2964082]
- 11.
- Pajak L, Field LJ. Expression of a temperature sensitive T Antigen transgene potentiates DNA synthesis in fetal but not neonatal cardiomyocytes Unpublished results. [PMC free article: PMC52490]
- 12.
- Katz E, Steinhelper ME, Daud A. et al. Ventricular cardiomyocyte proliferation in transgenic mice expressing a-Cardiac Myosin Heavy Chain-SV40 T antigen fusion genes. Am J Physiol. 1992;262:H1867–1876. [PubMed: 1377879]
- 13.
- Whyte P, Buchkovich KJ, Horowitz JM. et al. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–129. [PubMed: 2968522]
- 14.
- DeCaprio JA, Ludlow JW, Figge J. et al. SV40 large T-antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. [PubMed: 2839300]
- 15.
- Moran E. A region of SV40 large T antigen can substitute for a tranforming domain of the adenovirus EIA products. Nature. 1988;334:168–170. [PubMed: 2968523]
- 16.
- Steinhelper ME, Lanson N Jr, Dresdner K. et al. Proliferation in vivo and in culture of differentiated adult atrial cardiomyocytes from transgenic mice. Am J Physio. 1990;259:H1826–H1834. [PubMed: 2175567]
- 17.
- Daud AI, Lanson N A Jr., Claycomb WC. et al. Identification of SV40 large T-antigen-associated proteins in cardiomyocytes from transgenic mice. Am J Physiol. 1993;264:H1693–700. [PubMed: 8498581]
- 18.
- Tsai S -C, Pasumarti K, Pajak L. et al. SV40 Large T Antigen binds a novel BH3 containing pro-apoptosis protein in the cytoplasm. J Biol Chem. 2000;275:3239–3246. [PubMed: 10652310]
- 19.
- Lane DP, Crawford LV. T antigen is bound to a host protein in SV40 transformed cells. Nature. 1979;278:261–263. [PubMed: 218111]
- 20.
- Srivastava S, Zou Z, Pirollo K. et al. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348:747–749. [PubMed: 2259385]
- 21.
- Donehower LA, Harvey M, Slagle BL. et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. [PubMed: 1552940]
- 22.
- Yin Y, Tainsky MA, Bischoff FZ. et al. Wild-type p53 reatores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–948. [PubMed: 1525830]
- 23.
- Kim KK, Soonpaa MH, Daud AI. et al. Tumor suppressor gene expression during normal and pathologic myocardial growth. J Biol Chem. 1994;269:22607–22613. [PubMed: 8077211]
- 24.
- Soonpaa MH, Kim KK, Pajak L. et al. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–H2189. [PubMed: 8945939]
- 25.
- Xie Z, Koyama T, Abe K. et al. Upregulation of P53 protein in rat heart subjected to a transient occlusion of the coronary artery followed by reperfusion. Jpn J Physiol. 2000;50:159–162. [PubMed: 10866709]
- 26.
- Oskarsson HJ, Coppey L, Weiss RM. et al. Antioxidants attenuate myocyte apoptosis in the remote non-infarcted myocardium following large myocardial infarction. Cardiovasc Res. 2000;45:679–687. [PubMed: 10728389]
- 27.
- Song H, Conte J V Jr, Foster AH. et al. Increased p53 protein expression in human failing myocardium. J Heart Lung Transplant. 1999;18:744–749. [PubMed: 10512520]
- 28.
- Ikeda S, Hamada M, Hiwada K. Cardiomyocyte apoptosis with enhanced expression of P53 and Bax in right ventricle after pulmonary arterial banding. Life Sci. 1999;65:925–933. [PubMed: 10465352]
- 29.
- Long X, Crow MT, Sollott SJ. et al. Enhanced expression of p53 and apoptosis induced by blockade of the vacuolar proton ATPase in cardiomyocytes. J Clin Invest. 1998;101:1453–1461. [PMC free article: PMC508701] [PubMed: 9502788]
- 30.
- Leri A, Liu Y, Malhotra A. et al. Pacing-induced heart failure in dogs enhances the expression of p53 and p53-dependent genes in ventricular myocytes. Circulation. 1998;97:194–203. [PubMed: 9445173]
- 31.
- Nakamura T, Ueda Y, Juan Y. et al. Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: In vivo study. Circulation. 2000;102:572–578. [PubMed: 10920071]
- 32.
- Fortuno MA, Zalba G, Ravassa S. et al. p53-mediated upregulation of BAX gene transcription is not involved in Bax-alpha protein overexpression in the left ventricle of spontaneously hypertensive rats. Hypertension. 1999;33:1348–1352. [PubMed: 10373214]
- 33.
- Bialik S, Geenen DL, Sasson IE. et al. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest. 1997;100:1363–1372. [PMC free article: PMC508314] [PubMed: 9294101]
- 34.
- Kirshenbaum LA, de Moissac D. The bcl-2 gene product prevents programmed cell death of ventricular myocytes. Circulation. 1997;96:1580–1585. [PubMed: 9315550]
- 35.
- Long X, Boluyt MO, Hipolito ML. et al. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. 1997;99:2635–2643. [PMC free article: PMC508109] [PubMed: 9169493]
- 36.
- Hu Y, Zou Y, Hala M. et al. Prolonged survival of heart allografts from p53-deficient mice. Transplantation. 2000;69:2634–2640. [PubMed: 10910287]
- 37.
- Leri A, Fiordaliso F, Setoguchi M. et al. Inhibition of p53 function prevents renin-angiotensin system activation and stretch-mediated myocyte apoptosis. Am J Pathol. 2000;157:843–857. [PMC free article: PMC1885708] [PubMed: 10980124]
- 38.
- Kim KK, Daud AI, Wong SC. et al. Mouse RAD50 has limited epitopic homology to p53 and is expressed in the adult myocardium. J Biol Chem. 1996;271:29255–29264. [PubMed: 8910585]
- 39.
- Ewen ME, Ludlow JW, Marsilio E. et al. An N-terminal transformation governing sequence of SV40 large T antigen contributes to the binding of both p110Rb and a second cellular protein. Cell. 1989;58:257–267. [PubMed: 2526683]
- 40.
- Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell. 1989;56:67–75. [PubMed: 2521301]
- 41.
- Dyson N, Buchkovich K, Whyte P. et al. The cellular 107K protein that binds to adenovirus E1A also associates with the large T antigens of SV40 and JC virus. Cell. 1989;58:249–255. [PubMed: 2546678]
- 42.
- Zhu L, van den Heuvel S, Helin K. et al. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Devel. 1993;7:1111–1125. [PubMed: 8319904]
- 43.
- Shirodkar S, Ewen M, DeCaprio JA. et al. The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell. 1992;68:157–166. [PubMed: 1531040]
- 44.
- Kim KK, Soonpaa MH, Wang H. et al. Developmental expression of p107 mRNA and evidence for alternative splicing of the p107 (RBL1) gene product. Genomics. 1995;28:520–529. [PubMed: 7490090]
- 45.
- LeCouter JE, Kablar B, Whyte PF. et al. Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related p130 gene. Development. 1998;125:4669–4679. [PubMed: 9806916]
- 46.
- Adams JM, Cory S. The Bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. [PubMed: 9735050]
- 47.
- Kelekar A, Thompson CB. Bcl-2-family proteins: The role of the BH3 domain in apoptosis. Trends Cell Biol. 1998;8:324–330. [PubMed: 9704409]
- 48.
- Lutz RJ. Role of the BH3 (Bcl-2 homology 3) domain in the regulation of apoptosis and Bcl-2-related proteins. BioChem Soc Trans. 2000;28:51–56. [PubMed: 10816098]
- 49.
- Kohrman DC, Imperiale MJ. Simian Virus 40 large T antigen stably complexes with a 185 kd host protein. J Virol. 1992;66:1752–1760. [PMC free article: PMC240927] [PubMed: 1310776]
- 50.
- Kirshenbaum LA, Schneider MD. Adenovirus E1A represses cardiac gene transcription and reactivates DNA synthesis in ventricular myocytes, via alternative pocket protein- and p300-binding domains. J Biol Chem. 1995;270:7791–7794. [PubMed: 7713869]
- 51.
- Liu Y, Kitsis RN. Induction of DNA synthesis and apoptosis in cardiac myocytes by E1A oncoprotein. J Cell Biol. 1996;133:325–334. [PMC free article: PMC2120791] [PubMed: 8609165]
- 52.
- Bishopric NH, Zeng G -Q, Sato B. et al. Adenovirus E1A inhibits cardiac myocyte-specific gene expression through its amino terminus. J Biol Chem. 1997;272:20584–20594. [PubMed: 9252373]
- 53.
- Kirshenbaum LA, Abdellatif M, Chakraborty S. et al. Human E2F-1 reactivates cell cycle progression in ventricular myocytes and represses cardiac gene transcription. Dev Biol. 1996;179:402–411. [PubMed: 8903355]
- 54.
- Akli S, Zhan S, Abdellatif M. et al. E1A can provoke G1 exit that is refractory to p21 and independent of activating Cdk2. Circ Res. 1999;85:319–328. [PubMed: 10455060]
- 55.
- Pasumarthi K B S, Tsai S -C, Field LJ. Co-expression of mutant p53 and p193 renders embryonic stem cell-derived cardiomyocytes responsive to the growth-promoting activities of adenoviral E1A. Circ Res. 2001;88:1004–1011. [PubMed: 11375269]
- 56.
- Agah R, Kirshenbaum LA, Abdellatif M. et al. Adenoviral delivery of E2F-1 directs cell cycle reentry and p53 independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest. 1997;100:2722–2728. [PMC free article: PMC508475] [PubMed: 9389735]
- 57.
- Behringer RR, Peschon JJ, Messing A. et al. Heart and bone tumors in transgenic mice. Proc Natl Acad Sci USA. 1988;85:2648–2652. [PMC free article: PMC280055] [PubMed: 2833748]
- 58.
- Chalifour LE, Gomes ML, Wang NS. et al. Polyomavirus large T-antigen expression in heart of transgenic mice causes cardiomyopathy. Oncogene. 1990;5:1719–1726. [PubMed: 2176284]
- 59.
- De Leon Jr, Federoff HJ, Dickson DW. et al. Cardiac and skeletal myopathy in beta myosin heavy-chain simian virus 40 tsA58 transgenic mice. Proc Natl Acad Sci USA. 1994;91:519–523. [PMC free article: PMC42980] [PubMed: 8290557]
- 60.
- Jackson T, Allard MF, Sreenan CM. et al. The c-myc proto-oncogene regulates cardiac development in transgenic mice. Mol Cell Biol. 1990;10:3709–3716. [PMC free article: PMC360819] [PubMed: 1694017]
- 61.
- Machida N, Brissie N, Sreenan CM. et al. Inhibition of cardiac myocyte division in c-myc transgenic mice. J Mol Cell Cardiol. 1997;29:1895–1902. [PubMed: 9236143]
- 62.
- Soonpaa MH, Koh GY, Pajak L. et al. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest. 1997;99:2644–2654. [PMC free article: PMC508110] [PubMed: 9169494]
- 63.
- Pajak L, Jin F, Xiao GH. et al. Sustained cardiomyocyte DNA synthesis in whole embro cultures lacking the TSC2 gene product. Am J Physiol. 1997;273:H1619–1627. [PubMed: 9321857]
- 64.
- Kobayashi T, Minowa O, Kuno J. et al. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line TSC2 mutation in mice. Cancer Res. 1999;59:1206–1211. [PubMed: 10096549]
- 65.
- Pasumarthi K B S, Nakajima J, Nakajima HO. et al. Enhanced cardiomyocyte DNA synthesis during myocardial hypertrophy in mice expressing a modified TSC2 transgene. Circ Res. 2000;86:1069–1077. [PubMed: 10827137]
- 66.
- Poolman RA, Li JM, Durand B. et al. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res. 1999;85:117–127. [PubMed: 10417393]
- 67.
- Gruver CL, DeMayo F, Goldstein MA. et al. Targeted developmental expression of calmodulin induces proliferative and hypertrophic growth of cardiomyocytes in transgenic mice. Endocrinology. 1993;133:376–388. [PubMed: 8319584]
- 68.
- Reiss K, Cheng W, Ferber A. et al. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci USA. 1996;93:8630–8635. [PMC free article: PMC38724] [PubMed: 8710922]
- 69.
- Hein L, Stevens ME, Barsh GS. et al. Overexpression of angiotensin AT1 receptor transgene in the mouse myocardium produces a lethal phenotype associated with myocyte hyperplasia and heart block. Proc Natl Acad Sci USA. 1997;94:6391–6396. [PMC free article: PMC21060] [PubMed: 9177228]
- 70.
- Shou W, Aghdasi B, Armstrong DL. et al. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. [PubMed: 9461216]
- 71.
- Sucov HM, Dyson E, Gumeringer CL. et al. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994;8(9):1007–1018. [PubMed: 7926783]
- 72.
- Charng MJ, Frenkel PA, Lin Q. et al. A constitutive mutation of ALK5 disrupts cardiac looping and morphogenesis in mice. Dev Biol. 1998;199:72–79. [PubMed: 9676193]
- 73.
- Nebigil CG, Choi DS, Dierich A. et al. Serotonin 2B receptor is required for heart development. Proc Natl Acad Sci USA. 2000;97:9508–9513. [PMC free article: PMC16895] [PubMed: 10944220]
- 74.
- Svensson EC, Huggins GS, Lin H. et al. A syndrome of tricuspid atresia in mice with a targeted mutation of the gene encoding Fog-2. Nat Genet. 2000;25(3):353–356. [PubMed: 10888889]
- Introduction
- Transgenic Models for Cardiomyocyte Cycle Deregulation
- Expression of the SV40 Large T Antigen in the Myocardium
- T-Ag Binding Proteins as a Paradigm for the Identification of Cardiomyocyte Cell Cycle Regulators
- p53
- p107
- p193
- p380
- Future Applications, New Research, Anticipated Developments
- Acknowledgments
- References
- Regulation of Cardiomyocyte Proliferation and Apoptosis - Madame Curie Bioscienc...Regulation of Cardiomyocyte Proliferation and Apoptosis - Madame Curie Bioscience Database
- S-Modulin - Madame Curie Bioscience DatabaseS-Modulin - Madame Curie Bioscience Database
- Development of Biological Potential - Madame Curie Bioscience DatabaseDevelopment of Biological Potential - Madame Curie Bioscience Database
- Neurons, Neurotrophins and Ceramide Signaling: Do Domains and Pores Contribute t...Neurons, Neurotrophins and Ceramide Signaling: Do Domains and Pores Contribute to the Dichotomy? - Madame Curie Bioscience Database
- Dopamine and Parkinson's Disease - Madame Curie Bioscience DatabaseDopamine and Parkinson's Disease - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...