In photoreceptor cells cGMP is the second messenger that transduces light into an electrical response. Regulation of cGMP synthesis by Ca2+is one of the key mechanisms by which Ca2+ exerts negative feedback to the phototransduction cascade in the process of light adaptation. This Ca2+ feedback to retinal guanylyl cyclases (RetGCs) is conferred by the guanylate cyclaseactivating proteins (GCAPs). Mutations in GCAP1 that disrupt the Ca2+ regulation of RetGCs in vitro have been associated with severe human vision disorders. This chapter focuses on recent data obtained from biochemical and electrophysiological studies of GCAP1/GCAP2 knockout mice and other GCAP transgenic mice, addressing:
- the quantitative aspects of the Ca2+feedback to RetGCs in regulating the light sensitivity and adaptation in intact rods;
- functional differences between GCAP1 and GCAP2 in intact rod photoreceptors; and
- whether GCAP mutants with impaired Ca2+ binding lead to retinal disease in vivo by constitutive activation of RetGCs and elevation of intracellular cGMP, as predicted from in vitro studies.
Introduction
Visual excitation in retinal photoreceptor cells is mediated by a cascade that leads to the enzymatic hydrolysis of cGMP and the subsequent closure of cGMPgated channels in the plasma membrane. Recovery of the dark state requires the resynthesis of cGMP, which is catalyzed by particulate (membraneassociated) guanylate cyclases (RetGCs). RetGC activity has long been known to be stimulated with high cooperativity by the decrease in cytosolic [Ca2+] that follows light exposure.1 This cyclase stimulation has been proposed to be the major mechanism by which photoreceptors adjust their sensitivity according to background illumination.2,3 The soluble regulators that confer Ca2+sensitivity to RetGCs have been cloned and identified as 23kDa Ca2+binding proteins from the calmodulin superfamily,46 the guanylate cyclaseactivating proteins (GCAPs).
Two isoforms of RetGCs are expressed in mammalian retinas, RetGC1 and RetGC2.7,8 RetGC1 localizes to the outer segments and synaptic terminals of rods and cones,9,12 whereas the distribution of RetGC2 within photoreceptors is less well known. Although RetGC1 appears to be indispensable for phototransduction in human rods and cones,13 both RetGC1 and RetGC2 are expressed,14 and probably functional15 in murine rod outer segments. In photoreceptor cells, RetGCs are transmembrane proteins oriented in the disc membranes so that the “extracellular domain” is intradiscal, and are regulated intracellularly by the GCAPs.
Three isoforms of mammalian GCAPs, namely GCAP1, GCAP2 and GCAP3 have been isolated.5,6,16 GCAP1 and GCAP2 have both been localized to photoreceptors in numerous studies,46,17,18 although reports of the distribution of GCAPs between rods and cones in different species have been more ambiguous.17,19 In the human, monkey and bovine species, GCAP1 immunoreactivity appears stronger in the cone outer segments, whereas GCAP2 immunostaining is seen in the entire region of rods and cones.20 GCAP3 expression seems to be limited to humans16 and zebrafish,21 localizing to all cone types in human retinas.21 Whether individual GCAPs might play distinct roles in phototransduction is not known.
Extensive biochemical analyses have been done to characterize the molecular properties and biological activity of each GCAP. The extent and the mechanism by which GCAPs modulate RetGC activity in a bidirectional way in response to [Ca2+] is now better understood, thanks to a thorough mutational approach defining functional domains in both GCAPs and RetGC molecules.22,29 Both GCAP1 and GCAP2 possess three high affinity EF hand motifs that bind Ca2+. They appear to have similar Ca2+dependence and cooperativity for GC activation by Ca2+, with reported EC50s for the inhibitory effect of Ca2+ of ˜580 nM for GCAP1 and ˜250 nM for GCAP2.4,5,30,31 Consistent with the ability of GCAPs to activate and inhibit RetGCs, RetGCs and GCAPs appear to bind to each other independently of [Ca2+].25,32,33 Therefore, it has been proposed that GCAPs are constitutively bound to RetGCs, and that Ca2+ binding to the GCAP.RetGC complexes induces structural changes that stabilize the inactive state relative to the active state of the RetGC catalytic dimer31,32 (see Figure 1). Mutations in the RetGC1 or GCAP1 genes that have been shown to alter this finetuning of RetGC activity by Ca2+ in vitro have been linked to autosomal dominant cone or conerod dystrophies in human patients.30,31,34,38
The cloning of the GCAP proteins and the characterization of their gene structure17,39 have opened up the possibility of developing mouse models to study the normal functions of this Ca2+regulation of RetGCs by GCAPs in the context of intact, living rods. It has also made it possible to study the mechanism by which malfunctioning GCAPs lead to disease in vivo. This chapter describes new insights gained from mouse models developed to study the functions of the GCAP proteins in vivo. We have divided this chapter into three sections. In the first section we review the characterization of the GCAP1/GCAP2 knockout mice, in which the Ca2+feedback to RetGCs is specifically abolished. We discuss how disrupting this feedback loop affects the kinetics and the sensitivity of the light response in darkadapted rods and how it affects the ability of rod photoreceptors to adapt to light. In the second section we discuss the effect of restoring GCAP2 in the GCAP1/GCAP2 double knockout mice as a first step toward determining whether GCAP1 and GCAP2 have distinct functions in regulating the light response in rods. Individual restoration of GCAP1 and GCAP2 expression in GCAP1/GCAP2 knockout mice might show functional differences that would only be revealed when RetGC regulation is analyzed in intact cells under real time kinetics. The third section of this chapter summarizes the characterization of transgenic mice developed to study the mechanism by which mutant forms of GCAPs with impaired Ca2+binding (and in vitro altered Ca2+sensitivity) might lead to retinal disease in vivo. These transgenic mice express a mutant form of GCAP2 that is incapable of binding Ca2+. In vitro characterization of this mutant GCAP2 shows it to constitutively activate RetGCs.23 Because several mutations in the GCAP1 gene disrupt Ca2+ binding, these transgenic mice may reveal the molecular basis of the pathology caused by mutations in the GCAP1 gene that have been linked to autosomal dominant cone dystrophy.34,35,37
Functions of the GCAP Proteins in Intact Photoreceptors: Effects of the Ca2+Feedback to RetGCs on the Light Response
The vertebrate visual system can operate in a wide range of light intensities due, in part, to the capacity of rod and cone photoreceptors to light adapt. Rod photoreceptors, for instance, are specialized for the detection of dim light. In rods, activation of one rhodopsin molecule by a single photon gives rise to a measurable electrical response.40 This is due to a huge amplification of the signal in the transduction process, achieved in two successive steps: the activation of hundreds of transducin molecules (and cGMPPDE catalytic subunits) by a single photoactivated rhodopsin during its lifetime (120 s1 per R*), and the hydrolysis of thousands of cGMP molecules by each activated PDE holomer, whose catalytic efficiency is reflected in a Kcat/Km value that exceeds 108 M1 s1.41 This extraordinary signal amplification would lead to rod saturation at very dim illumination if the sensitivity of rods to light were fixed. But rods have the capacity to adjust their sensitivity in the presence of a background light, making it smaller as the background light is brighter. By shifting the responseintensity function toward brighter light stimuli, photoreceptors extend the range of light intensities in which they can operate.
It has been known since the late 1980s that light adaptation in vertebrate rods and cones can be abolished by slowing or preventing movements of Ca2+ across the plasma membrane.42,43 These studies established that Ca2+ is the major second messenger modulating transduction during light adaptation. Several steps in the transduction cascade are now known to be sensitive to Ca2+, responding to the lightdriven decrease in [Ca2+] by either reducing the gain or speeding the termination of the light response. The lightactivated PDE activity is reduced in response to a decrease in [Ca2+]44,47 through an effect that may be mediated by recoverin.48,50 In addition, a decrease in [Ca2+] is thought to cause calmodulin to dissociate from the b subunit of the cGMPgated channel, increasing the channel's sensitivity for cGMP.51,53 As mentioned, a decrease in [Ca2+] stimulates cGMP synthesis by RetGCs1,2,54,55 through the action of the GCAPs, speeding restoration of cGMP and recovery of the dark current. Lightdriven changes in [Ca2+] could also affect the response kinetics by affecting the phosphorylation state of RGS91;56 or by acting on as yet unidentified targets.
The dependence of several of the transduction steps on Ca2+ has made it difficult in the past to assess the individual contributions of these mechanisms to the overall process of adaptation in vivo, where conditions designed to minimize Ca2+ dynamics would affect them all. More recently, a quantitative characterization of each of these pathways was undertaken in truncated salamander rods, by manipulation of the internal ionic conditions and introduction of inhibitors by dialysis to dissect the pathway under study.2,3,46,53 These studies concluded that over most of the range of background light intensities, the most important mechanism regulating the sensitivity adjustment to light was the Ca2+dependent modulation of the guanylyl cyclase.3
In this section we discuss the effect of disrupting the GCAPs mediated Ca2+feedback loop to RetGCs in intact vertebrate photoreceptors. Based on a mouse genetics approach, this analysis presents the advantage that ionic conditions and concentration of soluble components are under true physiological conditions. The analysis of the effect of disrupting the Ca2+feedback loop to RetGCs on the light response of intact, living rods, represents one of the most direct assessments of the functions of the GCAP proteins in vivo.
Development of a Mouse Model to Assess the Contribution of Ca2+Regulation of RetGCs in the Light Response
Given that both GCAP1 and GCAP2 appear to be present in murine rod outer segments17 and that they behave similarly in vitro in their ability to stimulate RetGCs, a genetic approach intended to abolish Ca2+ regulation of RetGC activity in rods required targeting the expression of both genes simultaneously. The fact that GCAP1 and GCAP2 genes are arranged in a tailtotail array made it possible to disrupt both genes in embryonic stem cells using a single homologous recombination step.57 The recombination strategy outlined in Figure 2A led to the removal of exons 2 to 4 of GCAP1 and exons 3 and 4 of GCAP2 in the disrupted allele. Heterozygous mice for the disrupted allele showed the corresponding reduction of expression of GCAP1 and GCAP2 in the retina, whereas homozygous knockout mice showed no expression at all (Fig. 2C). Importantly, the expression level of proteins directly involved in cGMP turnover, such as RetGC1, RetGC2, and PDE catalytic and inhibitory subunits, were unchanged (Fig. 2C). Immunoblot analysis of several other phototransduction proteins and expression profiling of ˜ 11000 murine genes and expressed sequence tags similarly showed little, if any changes resulting from the absence of GCAPs. Consistent with the lack of expression changes, the morphology of GCAPs/ retinas was largely normal (Fig. 2D). Removing GCAP1 and GCAP2 effectively abolished Ca2+ regulation of rod RetGC activity, as shown by the absence of Ca2+dependent GC activity throughout the entire physiological range of [Ca2+] in rod outer segments from GCAPs/ mice (Fig. 2E). These results show that disruption of GCAPs expression by gene targeting was highly specific and did not lead to compensatory changes in the expression of other photoreceptor genes or disruption of retinal morphology. These mice therefore constitute an ideal model to study the functions of Ca2+feedback to RetGCs in intact photoreceptors.

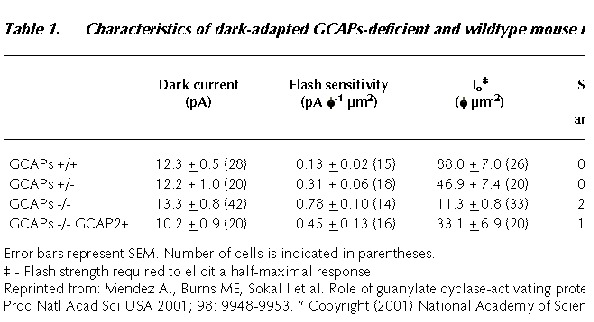
Basal RetGC Activity in Rods Lacking GCAP1 and GCAP2
One of the first questions addressed in the GCAP1/GCAP2 knockout mice was whether GCAP1 and GCAP2 regulate the basal activity of RetGCs in darkadapted rods. This can be done by comparing the free [cGMP] in wildtype and GCAPs/ rods, assuming that the dark PDE activity is not affected by the absence of GCAPs. The free [cGMP] in the outer segments is proportional to the outer segment lightsensitive membrane current. This is because the lightsensitive current is cooperatively regulated by free [cGMP], obeying a Hill relation in which the equilibrium between the free cGMP and the channels is established very rapidly,
within less than 3 milliseconds.58,59 Therefore, the instantaneous magnitude of the lightsensitive membrane current reflects the free [cGMP] in the outer segment.
There were no significant differences in the dark current values of GCAPs/ and wildtype rods (ref. 57, Table 1). Assuming that the PDE basal activity hasn't changed in GCAPs/ mice, this result suggests that the dark [cGMP] is similar in GCAPs/ and wildtype rods, implying that both wildtype and GCAPs/ rods have similar dark (basal) levels of GC activity. Consistent with this result, no significant differences were observed in the total levels of cGMP in wildtype and GCAPs/ retinas, when total (bound and free) cGMP levels were determined in whole retinas by radioimmunoassay (data not shown).
In conclusion, removal of GCAPs had little effect on the size of the inward dark current (and therefore, basal GC activity) or the physiological levels of cGMP in resting photoreceptors. Because GCAPs have dual actions to activate and inhibit RetGCs, there would be a certain value of [Ca2+] for which the GC activity is the same in the presence or absence of GCAPs. This would be ˜400 nM for GCAP225,60 and ˜1 mM for GCAP1.29 The fact that the basal GC activity is similar in wildtype and GCAPs/ mice might be explained if at the free [Ca2+] of resting rods some of the GCAP molecules are locally activating RetGC activity while others are inhibiting it, so that the steady state activity is unchanged in the presence or the absence of GCAPs.
Effect of GCAPs Deletion on DarkAdapted Flash Responses
It has been known since the 1980s that slowing the rate of change of [Ca2+] by incorporation of Ca2+ buffers such as BAPTA has profound effects on the darkadapted response of photoreceptors.61,63 Ca2+feedback to the transduction cascade severely limits the response to light. But what is the effect of abolishing Ca2+ regulation of RetGCs in the darkadapted light response?
Figure 3 shows representative flash families from a GCAPs/ and a wildtype rod. For dim flashes of comparative strength (see figure legend) GCAPs/ responses rose for a longer time, to a larger peak amplitude than wildtype responses, and took longer to recover. The flash sensitivity (the amplitude of the linear range response divided by the strength of the flash) was about 6 times higher in GCAPs/ rods. Another index of sensitivity, the flash strength required to elicit a halfmaximal response (Io), was about 8fold lower in the GCAPs/ rods (ref. 57, Table 1). These results indicate that Ca2+ feedback to RetGCs via GCAP1 and/or GCAP2 greatly limits the flash sensitivity of wildtype darkadapted rods. These changes are also reflected in the single photon response (Fig. 4). Heterozygous knockout rods, expressing about half the normal levels of GCAP1 and GCAP2 (GCAPs+/) had flash sensitivities and response kinetics between those of knockout and wildtype rods (Fig. 4), indicating that GCAPs are not present in excess in wildtype rod outer segments.
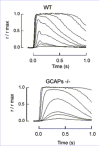
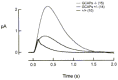
Figure 4 shows that Ca2+feedback to RetGCs is sensed at the single photon level. This fact implies that the local closure of cGMPchannels triggered by photoactivation of a single rhodopsin molecule is enough to cause a local drop in [Ca2+]that is sensed by RetGCs during the timecourse of the single photon response (on the order of a few hundred milliseconds). The speed of this Ca2+feedback loop might derive from the close proximity of the cGMPgated channels, the Na/Ca2+, K exchanger and the RetGCs at the rim of the discs. These proteins may be assembled in a macromolecular complex that may also include cGMPPDE and the ATPbinding cassette transporter (ABCR) by interactions with the multivalent glutamicacidrich proteins (GARPs).64 Fluctuations of [Ca2+]would be greatest near the plasma membrane at the site of Ca2+ entry and extrusion. By rapidly responding to local changes in [Ca2+] (through a high Hill coefficient1 and rapid conformational change65) GCAPs transmit a local drop in [Ca2+] during the photoresponse to a quick and robust stimulation of RetGC activity that limits the amplitude and shortens the duration of the light response.
Interestingly, the Ca2+ feedback to RetGCs that boosts cGMP synthesis imposes a constraint on the amplitude of the light response even in mouse models with defects in other biochemical processes governing inactivation. The loss of rhodopsin phosphorylation by RK in several mouse models,66,68 for instance, results in constant rhodopsin's catalytic activity, but causes only a 23 fold increase in single photon response amplitude, in contrast to the 5fold increase seen in GCAPs/ mice. These results indicate that the amplitude of the responses in all these mouse models is limited by replenishment of cGMP by stimulated RetGCs, and stress the strength of the feedback loop mediated by the GCAP proteins.
Effect of GCAPs Deletion on Light adaptation of Rod Photoreceptors
As mentioned above, [Ca2+] fluctuations are sensed at several enzymatic steps in the phototransduction cascade, and each of these steps could potentially contribute to the photoreceptor's ability to light adapt. Therefore, a complete understanding of the molecular mechanisms behind this important photoreceptor cell attribute requires a quantitative assessment of each of these steps. This can only be accomplished in a system where each of the pathways can be specifically disrupted without affecting Ca2+feedback to the other enzymatic reactions. As we have demonstrated, the GCAPs/ rod fulfills this requirement. Measurements of flash sensitivities of wildtype and GCAPs/ rods in the presence of background lights of increasing intensities showed the gradual desensitization obeying the WeberFechner law for wildtype rods (filled symbols, Fig. 5), but a much steeper decline of sensitivity in GCAPs/ rods (open symbols, Fig. 5). However, adaptation was not completely absent in GCAPs/ rods. The dependence of incremental sensitivity on background intensity for a GCAPs/ rod lacking any adaptation to background light was theoretically predicted by assuming an exponential saturation relation and making use of the GCAPs/ darkadapted response parameters (thin trace, Fig. 5). A comparison between the experimental results and this theoretical prediction indicated that GCAPs stimulation of GC activity accounts for only one half of the factor by which the rods' operating range of light intensities is extended. The remainder is mediated by other mechanisms (discussed in ref. 57).
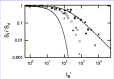
Thus, RetGCs activation by GCAPs in wildtype rods served to increase the incremental flash sensitivity in the presence of bright steady light by restoring current to rods that would otherwise have saturated. It is interesting to note that rod photoreceptors, specialized for the detection of dim illumination and extremely sensitive to light, could be considerably more sensitive to light in the absence of the GCAP proteins. However, in the absence of these proteins the range of light intensities in which the rods could operate would be severely limited.
Effect of Restoring GCAP2 in GCAPs/ Photoreceptors
Introduction
While the mechanism of modulation of RetGCs by GCAPs is now better understood, less is known about the significance of multiple RetGCs and GCAPs. In vitro, GCAPs display some preferences toward RetGCs. For example, GCAP1 stimulates RetGC1 more efficiently than RetGC2, while GCAP2 and GCAP3 effectively stimulate both RetGC1 and RetGC2. RetGC1 and GCAP1 appear to be important for cone and rod function in humans. Mutations in the RetGC1 gene have been linked to Leber's congenital amaurosis type I13 and to autosomal dominant conerod dystrophy (adCORD),69 and defects in the GCAP1 gene have been linked to adCORD.34,35,37 A naturally occurring null mutation in RetGC1 in chickens, which have conedominated retinas, leads to the loss of cones and rods.70 However, disruption of RetGC1 expression in mice leads to the degeneration of cones primarily,15 indicating that RetGC2 may substitute for the loss of RetGC1 in murine rods.
It is not clear why photoreceptors express two GCAP proteins with very similar biochemical behavior. The ratio of GCAP1 to GCAP2 in bovine retinas has been estimated at between 3:1 and 4:1 (reference 18, and references herein), and the GC activity stimulation attributable to GCAP2 has been estimated at about 30%.18 Based on this data, the question was raised of whether GCAP2 would have a physiological role in phototransduction.18 The compartmentalization of GCAP2 differs from that of GCAP1 in rods and cones of retinas from higher species.18,20 GCAP1 appears to be more abundantly expressed in the cone outer segments of human, monkey and bovine retinas, whereas GCAP2 immunoreactivity, even if weaker in the outer segments, is seen in the whole region of rods and cones.20
As a starting point to address functional differences and redundancy of GCAPs in the photoreceptors, we generated a transgenic mouse line that expressed GCAP2 in the GCAPs/ background.
GCAP2 is Functional in Rods and Partially Rescues the GCAP1/GCAP2 Knockout Phenotype
GCAP2 is first expressed in murine retinas at postnatal day 9, paralleling rod opsin expression (Fig. 6). Transgenic mice expressing GCAP2 in rod photoreceptors were generated by expressing the bovine GCAP2 cDNA under the control of the rod opsin promoter (Fig. 7A). Several lines were obtained, and a line in which heterologous GCAP2 expression was about 2fold higher than endogenous GCAP2 expression was selected for the study (Fig. 7B, see figure legend). This line was bred to GCAPs/ mice to establish a line in which GCAP2 was expressed in rod photoreceptors in the absence of GCAP1. Heterologous GCAP2 in the GCAPs/GCAP2+ line showed the same pattern of localization as endogenous GCAP2 in wildtype retinas (Fig. 7C); and in vitro GC assays using retinal homogenates showed that heterologous GCAP2 fully restored stimulation of RetGC activity at low [Ca2+] (Fig. 7D).


GCAPs/GCAP2+ transgenic mice were analyzed by single cell recordings. Although GCAP2 expression seemed to vary from cell to cell (see reference 57, summarized here in Fig. 8 legend), the mean single photon response from GCAPs/GCAP2+ rods was between those of wildtype and GCAPs/ responses (Fig. 8A). This result indicates that GCAP2 expression alone partially restores the dark flash sensitivity in GCAPs/ rods. Furthermore, a correlation between the dark flash sensitivity and the time course of the decline of the Na/Ca2+, K exchange current (thought to reflect the level of expression of GCAP2 in individual cells, see ref. 57), could be established for individual rods. In general, cells with time constants of the Na/Ca2+, Kexchanger close to that of wildtype rods (and presumably high GCAP2 expression) had nearly normal dark flash sensitivities. Cells with briefer exchanger time constants (and presumably lower GCAP2 expression) had higher flash sensitivities, approaching those of GCAPs/ (Fig. 8B). These results indicate that GCAP2 has the capacity to stimulate GC activity in vivo.
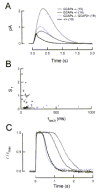
In addition, responses from GCAPs/GCAP2+ rods to saturating flashes showed that GCAP2 alone can maximally stimulate GC activity. After a bright flash, in which the [Ca2+] drops to the minimum level, the time at which a rod comes out of saturation is determined by the lighttriggered PDE activity (assumed to be the same in wildtype, GCAPs/ and GCAPs/GCAP2+ rods for a flash of the same strength), and the maximal cyclase activity. The similarity of wildtype and GCAPs/GCAP2+ responses to bright flashes indicated that GCAP2 alone was capable of producing maximal GC activation (Fig. 8C).
Taken together, these results show that GCAP2 is active in rod outer segments, partially rescuing the GCAPs/ phenotype, and that GCAP2 alone is capable of maximally activating RetGC activity under low intracellular [Ca2+].
Although GCAP2 was capable of maximally activating RetGC activity and restoring sensitivity, it was not sufficient to restore the normal kinetics of the photoresponse. Responses to subsaturating flashes lacked the rapid initial recovery that follows the peak of the response in wildtype rods.57 This result suggests that GCAP1 is responsible for the characteristic rapid recovery, indicating that GCAP1 might act faster in stimulating RetGCs. This functional difference might result from differences in the rates of Ca2+ binding and dissociation to each GCAP. The rate of Ca2+release by GCAP1 is very rapid (k1 = 72 s1; ref. 65) and would be consistent with an early role of GCAP1 in the recovery phase of the photoresponse, while the Ca2+dissociation rate for GCAP2 has not yet been determined. In addition, as the EC50s for the inhibitory effect of Ca2+ were estimated to be ˜580 nM for GCAP1 and ˜250 nM for GCAP2, GCAP1 would be expected to sense a decrease in [Ca2+] earlier than GCAP2. Alternatively, this difference in kinetics may be imparted by the topology of GCAPs and RetGCs near the plasma membrane where [Ca2+] changes first occur. As mentioned above, the proximity of cGMPgated channels, the Na/Ca2+, K exchanger and RetGCs at the rim of the disc near the plasma membrane may play a role in the rapid feedback to resynthesize cGMP. In this regard, the concentration of each GCAP near this complex would affect the speed of the feedback. Future experiments will be needed to determine the mechanisms that underlie the differential effects of GCAP1 and GCAP2 on the time course of the flash response.
GCAPs and Disease: Mouse Models Expressing Forms of GCAPs with Impaired Ca2+Binding Properties
Three mutations in the gene encoding GCAP1 have been linked to autosomal dominant cone dystrophy, a disease characterized by the loss of color vision and central visual acuity with retention of peripheral sight.34,35,37 The first mutation characterized, Y99C,34 impairs Ca2+ binding at EF3 and has been shown to alter Ca2+ sensitivity of GCAP1 in in vitro assays. The mutant protein causes normal stimulation of RetGC1 at low [Ca2+], but this stimulation persists as the [Ca2+] increases even above 1 mM,30,38 leading to constitutive activation of the cyclase over the physiological range of [Ca2+]. Another mutation, causing an E155G substitution within the EF4 domain, was shown to alter Ca2+ sensitivity of GCAP1 in the same way.37 Both Y99CGCAP1 and E155GGCAP1 stimulate RetGCs constitutively in the presence of wildtype GCAP1, consistent with the dominant phenotype of the disease. A model has been proposed in which constitutive synthesis of cGMP in vivo would increase the steadystate level of cGMP and lead to cell death. Increased cGMP levels have been associated with cell death in a number of animal models with mutations in the genes encoding cGMPPDE subunits.71,73
Consistent with this interpretation, substitutions at Arg838 in RetGC1 that have been associated with autosomal dominant cone rod dystrophy (adCORD) result in constitutive synthesis of cGMP in vitro, even at the highest physiological [Ca2+] concentrations.31,36 As in the case of GCAP1 mutations, the Arg838substituted RetGC1 mutants exhibit a dominant biochemical phenotype that would explain the characteristics of the disease. Arg838 was proposed to be critical for maintaining the normal structure of the ahelical coiledcoil domain, which could regulate the strength of the dimerization of the catalytic domains and consequently the Ca2+sensitivity.31
Despite the logic of the predicted association between elevated levels of cGMP and photoreceptor cell death in these cone or conerod dystrophies, some questions remain. A third mutation in GCAP1, leading to a P50L substitution, exhibits a marked variability in phenotype, ranging from mild abnormalities of macular function to severe conerod dystrophy.35 Contrary to the other mutants, however, P50LGCAP1 activates RetGC with the same Ca2+sensitivity as the wildtype protein in vitro.74,75 Interestingly, P50LGCAP1 showed a reduced thermal stability when compared to wildtype GCAP1, observed both by circular dicroism and by in vitro GC assays.75 Expression of a thermally unstable GCAP1 in human photoreceptors, therefore, may be the underlying cause of this dominantly inherited disease.
To investigate how mutations affecting Ca2+ binding in GCAPs may lead to retinal degeneration in vivo, a mutant form of GCAP2 with inactivated EF hands (E80Q/E116Q/D158N; or EF(2;3;4) GCAP2, ref. 23), which leads to constitutive activation of RetGCs in vitro over the whole physiological range of [Ca2+],23 was expressed in the photoreceptor cells of transgenic mice. Because this GCAP2 mutant shows a biochemical behavior similar to that of Y99CGCAP1,30,38 this transgenic animal model is expected to show a similar phenotype to the human disease.
GCAP2(EF2;3;4) Expression in Transgenic Mice Causes Retinal Dysplasia and Retinal Degeneration
Four independent transgenic lines were established that express the bovine GCAP2(E80Q/E116Q/D158N) cDNA under the control of the mouse rod opsin promoter. Two of the lines expressed the mutant protein to higher than the endogenous GCAP2 levels (lines A and B), whereas the other two expressed mutant GCAP2 to lower than the endogenous levels (lines C and D) (A. Mendez, A. Dizhoor and J. Chen, unpublished results). Lines could be ordered by the level of expression of the transgene as follows: line B > line A > lines C and D.
Transgenic mice from lines A and B showed a progressive retinal degeneration, the time course and severity of which correlated with the level of expression of the transgene (Fig. 9). Mice from line B, the line with the highest level of expression, manifested a retinal dysplasia as early as 20d of age. Initially, a loss of the integrity of the outer limiting membrane was seen, with occasional invaginations of the outer nuclear layer into the inner retina (Fig. 10B). At this early age, however, retinas maintained the 1012 normal rows of nuclei at the outer nuclear layer, and about the normal rod outer segment and inner segment length. By 30d of age, disorganization and shortening of the outer segments was apparent, and the outer nuclear layer showed whirls and rosettes over the whole retinal section in some cases. By 2.5 months of age, the outer nuclear later was reduced to 45 rows of nuclei (Fig. 9, line B). Mice from line A, which expressed the mutant form of GCAP2 to approximately four times the endogenous GCAP2 levels, showed a slower progression of the disease. Disorganization of the outer limiting membrane and occasional rosette formation were seen early in time in some retinal sections, but the outer nuclear layer thickness was still 89 rows by 2.5 months of age (Fig. 9). Mice from lines C and D showed normal retinal morphology up to one year of age. In contrast to bGCAP2(E80Q/E116Q/D158N), expression of wildtype bGCAP2 to twofold the endogenous levels did not cause retinal degeneration or signs of rosette formation in a control line of transgenic mice (data not shown), suggesting that the degeneration observed in mice expressing bGCAP2(E80Q/E116Q/D158N) most probably results from the physical properties of this particular mutant form of GCAP2 that does not bind Ca2+.


The Severity of Retinal Degeneration Correlates with Transgene Expression, but not with the Levels of Total cGMP
Historically, the hypothesis that elevated levels of cGMP are toxic for the photoreceptor cell originated from the rd/rd mouse model, in which autosomal recessive mutations in the gene encoding the beta subunit of cGMPPDE result in massive photoreceptor cell death.72,76,77 In rd/rd mouse retinas, the deficiency of cGMPPDE activity results in 45 fold higher total cGMP levels than normal at 1415 postnatal days. This peak of [cGMP] preceeds the fast degeneration of all the rod photoreceptors that occurs in the third postnatal week.77,78 Other animal models of retinal degeneration with imbalances in cGMP metabolism, due to a deficiency in cGMPPDE (e.g., the Irish Setter dog, ref. 71) or in RetGC1 (the rd/rd chicken, ref. 70) show either abnormally high or abnormally low total [cGMP] in the retina. In these animal models deregulation of cGMP metabolism is reflected in changes in the total [cGMP] in the photoreceptors that preceeds photoreceptor cell death.
If the loss of photoreceptor cells in transgenic mouse retinas expressing bGCAP2(E80Q/E116Q/D158N) was due to constitutive activation of the cyclase in rods, one reasonable expectation would be that an increase in the levels of total cGMP might precede the onset of the degeneration. Another predictable outcome would be that the time course and severity of the degeneration should be affected by the lighting conditions in which the mice are reared, accelerating or worsening when mice are raised in constant darkness, and slowing down when mice are exposed to constant light. This latter prediction is based on the fact that PDE activity increases with background light intensity. Therefore, constitutive activation of the cyclase should translate to the higher increase in the steadystate levels of cGMP when mice are raised under constant darkness, when only basal PDE activity is counteracting cGMP synthesis. However, in the presence of background light, PDE activity can be expected to overcome the cyclase activity.
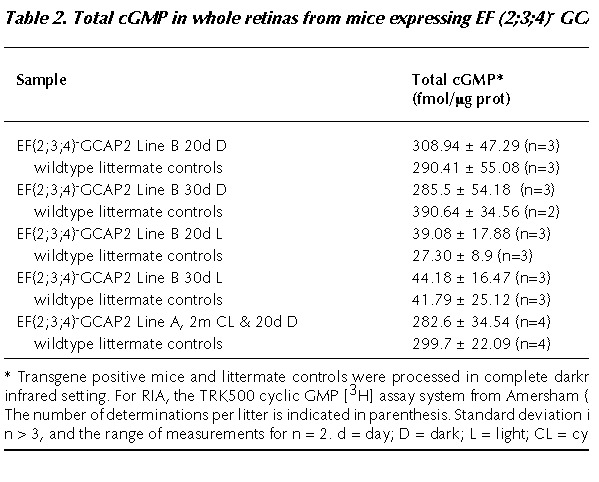
The first prediction was tested by measuring total cGMP in freshly dissected retinas by radioimmunoassay (RIA), at time points that precede the retinal degeneration in each transgenic line. For mice from line B (showing the earliest and more severe phenotype), the selected time points were 20 and 30 postnatal days, as in this time window the disorganization of the outer nuclear layer becomes apparent (Fig. 10B), and loss of outer nuclei rows begins to be measurable (Fig. 10A). Mice from line B were raised under constant darkness and sacrificed at 20 or 30 postnatal days. Whole litters were processed at each timepoint, so that transgenepositive mice could be directly compared to transgenenegative littermate controls. One eye of each mouse was fixed for morphological analysis, and the corresponding eye was used for cGMP measurement by RIA. In this manner, individual comparisons could be established between cGMP levels and retinal morphology. For RIA, mice were processed and samples handled in complete darkness.
As shown in Table 2, no significant differences were observed between transgenepositive and transgenenegative control mice at 20d when these mice were raised in constant darkness. At this age, transgene positive retinas showed normal outer nuclear layer thickness (Fig. 10A), and inner and outer segment length (data not shown), indicating that retinas were not degenerated. However, it should be pointed out that disorganization of the outer limiting membrane and rosette formation were already incipient in some retinas. These signs of the pathology, shown in Figure 10B in the 20d lightrearedmouse retina, were present in some of the transgenic retinas and absent in others within the same litter. Nevertheless, the degree of disorganization of the outer nuclear layer did not correlate with higher total cGMP levels in the corresponding eye (e.g., mice showing the more disorganized retinas did not show the higher cGMP values).
At 30 days, the levels of total cGMP were actually lower in the transgenepositive mice than in their littermate controls, most probably reflecting the loss of a percentage of rod photoreceptors (see measurement of rod outer nuclear layer thickness at 30d, Fig.10A).
Similarly, no differences in total cGMP could be observed between transgenepositive and littermate control mice of line A, when mice were raised in cyclic light for 2 months, and dark-adapted for 20d prior to the experiment (Table 2); nor in mice from lines C or D at later ages (data not shown).
These data show that even when the experimental conditions were optimized to identify changes in the levels of total cGMP, no major differences in the total [cGMP] could be detected in transgenepositive versus control mouse retinas.
The second prediction was tested by raising mice from line B under constant light exposure (1,500 lux), and analyzing their retinal morphology at 20, 30, and 45 postnatal days. The outer nuclear layer thickness was measured at four points along the retinal section. No difference was observed between transgene positive mice raised in constant darkness or raised in constant light at 20 or 45 postnatal days (Fig. 10A). In addition, no differences in retinal morphology were observed when 2month cycliclightreared mice from line A were placed for 20d in constant light, or under constant light exposure (data not shown).
Taken together, these results show that overexpression of bGCAP2(E80Q/E116Q/D158N) in transgenic mice did not lead to major changes in the retinal content of cGMP, or differences in retinal morphology depending on the lightrearing conditions.
Several scenarios would explain these results. First, that bGCAP2(E80Q/E116Q/D158N) causes constitutive activation of RetGC activity resulting in the 512 fold increase in cGMP synthesis in the dark state as predicted from in vitro studies; but that the cGMP hydrolysis rate by basal PDE is accelerated to adjust to the new darkness equilibrium. In this case, a change in the cGMP turnover rate wouldn't necessarily be reflected in bulk [cGMP], but could result in small changes in free [cGMP]. Since the lightsensitive current is cooperatively regulated by free [cGMP] with a Hill coefficient of ˜3, a small increase in free [cGMP] would result in a much bigger difference in the lightsensitive conductance and therefore in Ca2+ influx, which could be toxic for the cell (see refs. 79 and 80, and references herein).
A second possibility is that bGCAP2(E80Q/E116Q/D158N) is affecting some other cellular processes unrelated to cGMP synthesis, e.g., cell adhesion at the outer limiting membrane, by interacting with an as yet unidentified target. Finally, the possibility cannot be excluded that bGCAP2(E80Q/E116Q/D158N) in transgenic lines kills the cells nonspecifically due to an overexpression artifact.
Future experiments will be oriented toward the expression of this mutant form of GCAP2 in the GCAPs/ background to address whether these mutant proteins localize to rod outer segments, and whether they are active in vivo. The question remains open, therefore, as to whether similar mutations affecting Ca2+ binding in the GCAP1 protein cause the loss of cones through constitutive activation of RetGC, or whether other mechanisms are involved.
Conclusion
In vivo, the GCAP proteins mediate a Ca2+feedback loop to RetGCs that greatly limits the amplitude and duration of the photoresponse of darkadapted rods, but also serves to extend the rod's operational range of light intensities during background light adaptation. The biophysical properties of GCAPs (their kon and koff for Ca2+, their Ca2+sensitivity and the cooperativity of GC activation by Ca2+) confer speed and robustness to this feedback loop. During the photoresponse, when there is a decrease in the free [cGMP] and some cGMPchannels close, the GCAPs sense the resulting decrease in [Ca2+] very quickly, transducing it to a robust stimulation of RetGC activity that restores free cGMP and accelerates recovery. During background adaptation, this feedback loop prevents rod's saturation by restoring current to rods that are exposed to ambient illumination. In vivo, GCAP2 is functional in rod outer segments, and has the capacity to maximally stimulate RetGC activity when [Ca2+] drops to a minimum. However, responses to dim flashes of rods expressing GCAP2 alone lack the rapid initial decline that follows the peak in wildtype responses, suggesting that GCAP1 may function earlier in the photoresponseand be responsible for this early phase of the recovery, and GCAP2 regulation of GCs may proceed more slowly and/or require a higher intensity of light. Future experiments are needed to confirm this subtle difference in GCAP1 and GCAP2 kinetic behavior. Finally, the in vivo mechanism by which mutant forms of GCAP1 lead to cone photoreceptor death remains to be determined.
Acknowledgements
The electrophysiological analysis of GCAPs/ and GCAPs/GCAP2+ mice was done by Dr. M. E. Burns in Dr. D. A. Baylor's laboratory at Stanford University. Financial support to J. C. was provided by the National Eye Institute (NEI) (EY 12703).
References
- 1.
- Koch KW, Stryer L. Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature. 1988;334:64–66. [PubMed: 2455233]
- 2.
- Koutalos Y, Nakatani K, Tamura T. et al. Characterization of guanylate cyclase activity in single retinal rod outer segments. J Gen Physiol. 1995;106:863–890. [PMC free article: PMC2229293] [PubMed: 8648296]
- 3.
- Koutalos Y, Yau KW. Regulation of sensitivity in vertebrate rod photoreceptors by calcium. Trends Neurosci. 1996;19:73–81. [PubMed: 8820871]
- 4.
- Gorczyca WA, Polans AS, Surgucheva IG. et al. Guanylyl cyclase activating protein. A calciumsensitive regulator of phototransduction. J Biol Chem. 1995;270:22029–22036. [PubMed: 7665624]
- 5.
- Dizhoor AM, Olshevskaya EV, Henzel WJ. et al. Cloning, sequencing, and expression of a 24kDa Ca(2+)binding protein activating photoreceptor guanylyl cyclase. J Biol Chem. 1995; 270:25200–25206. [PubMed: 7559656]
- 6.
- Palczewski K, Subbaraya I, Gorczyca WA. et al. Molecular cloning and characterization of retinal photoreceptor guanylyl cyclaseactivating protein. Neuron. 1994;13:395–404. [PubMed: 7520254]
- 7.
- Shyjan AW, de Sauvage FJ, Gillett NA. et al. Molecular cloning of a retinaspecific membrane guanylyl cyclase. Neuron. 1992; 9:727–737. [PubMed: 1356371]
- 8.
- Lowe DG, Dizhoor AM, Liu K. et al. Cloning and expression of a second photoreceptorspecific membrane retinal guanylyl cyclase (RetGC), RetGC2. Proc Natl Acad Sci USA. 1995;92:5535–5539. [PMC free article: PMC41730] [PubMed: 7777544]
- 9.
- Liu X, Seno K, Nishizawa Y. et al. Ultrastructural localization of retinal guanylate cyclase in human and monkey retinas. Exp Eye Res. 1994;59:761–768. [PubMed: 7698269]
- 10.
- Dizhoor AM, Lowe DG, Olshevskaya EV. et al. The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron. 1994;12:1345–1352. [PubMed: 7912093]
- 11.
- Koch KW. Purification and identification of photoreceptor guanylate cyclase. J Biol Chem. 1991;266:8634–8637. [PubMed: 1673683]
- 12.
- Hallett MA, Delaat JL, Arikawa K. et al. Distribution of guanylate cyclase within photoreceptor outer segments. J Cell Sci. 1996;109:1803–1812. [PubMed: 8832403]
- 13.
- Perrault I, Rozet JM, Calvas P. et al. Retinalspecific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat Genet. 1996;14:461–464. [PubMed: 8944027]
- 14.
- Yang RB, Garbers DL. Two eye guanylyl cyclases are expressed in the same photoreceptor cells and form homomers in preference to heteromers. J Biol Chem. 1997;272:13738–13742. [PubMed: 9153227]
- 15.
- Yang RB, Robinson SW, Xiong WH. et al. Disruption of a retinal guanylyl cyclase gene leads to conespecific dystrophy and paradoxical rod behavior. J Neurosci. 1999;19:5889–5897. [PMC free article: PMC6783089] [PubMed: 10407028]
- 16.
- Haeseleer F, Sokal I, Li N. et al. Molecular characterization of a third member of the guanylyl cyclaseactivating protein subfamily. J Biol Chem. 1999;274:6526–6535. [PubMed: 10037746]
- 17.
- Howes K, Bronson JD, Dang YL. et al. Gene array and expression of mouse retina guanylate cyclase activating proteins 1 and 2. Invest Ophthalmol Vis Sci. 1998;39:867–875. [PubMed: 9579466]
- 18.
- OttoBruc A, Fariss RN, Haeseleer F. et al. Localization of guanylate cyclaseactivating protein 2 in mammalian retinas. Proc Natl Acad Sci USA. 1997;94:4727–4732. [PMC free article: PMC20792] [PubMed: 9114059]
- 19.
- Cuenca N, Lopez S, Howes K. et al. The localization of guanylyl cyclaseactivating proteins in the mammalian retina. Invest Ophthalmol Vis Sci. 1998;39:1243–1250. [PubMed: 9620085]
- 20.
- Kachi S, Nishizawa Y, Olshevskaya E. et al. Detailed localization of photoreceptor guanylate cyclase activating protein1 and 2 in mammalian retinas using light and electron microscopy. Exp Eye Res. 1999;68:465–473. [PubMed: 10192804]
- 21.
- Imanishi Y, Li N, Sowa ME. et al. Characterization of retinal guanylate cyclaseactivating protein 3 (GCAP3) from zebrafish to man. Eur J Neurosci. 2002;15:63–78. [PMC free article: PMC1363676] [PubMed: 11860507]
- 22.
- Duda T, Goraczniak R, Surgucheva I. et al. Calcium Modulation of Bovine Photoreceptor Guanylate Cyclase. Biochemistry. 1996; 35:8478–8482. [PubMed: 8679607]
- 23.
- Dizhoor AM, Hurley JB. Inactivation of EFhands makes GCAP2 (p24) a constitutive activator of photoreceptor guanylyl cyclase by preventing a Ca2+induced “activatortoinhibitor” transition. J Biol Chem. 1996;271:19346–19350. [PubMed: 8702620]
- 24.
- Laura RP, Dizhoor AM, Hurley JB. The membrane guanylyl cyclase, retinal guanylyl cyclase1, is activated through its intracellular domain. J Biol Chem. 1996;271:11646–11651. [PubMed: 8662612]
- 25.
- Laura RP, Hurley JB. The kinase homology domain of retinal guanylyl cyclases 1 and 2 specifies the affinity and cooperativity of interaction with guanylyl cyclase activating protein2. Biochemistry. 1998;37:11264–11271. [PubMed: 9698373]
- 26.
- Krylov DM, Niemi GA, Dizhoor AM. et al. Mapping sites in Guanylyl Cyclase Activating Protein1 required for regulation of photoreceptor membrane Guanylyl Cyclases. J Biol Chem. 1999; 274:10833–10839. [PubMed: 10196159]
- 27.
- Krylov DM, Hurley JB. Identification of proximate regions in a complex of retinal guanylyl cyclase 1 and guanylyl cyclaseactivating protein1 by a novel mass spectrometrybased method. J Biol Chem. 2001;276:30648–30654. [PubMed: 11387342]
- 28.
- Olshevskaya EV, Boikov S, Ermilov A. et al. Mapping functional domains of the guanylate cyclase regulator protein, GCAP2. J Biol Chem. 1999;274:10823–10832. [PubMed: 10196158]
- 29.
- RudnickaNawrot M, Surgucheva I, Hulmes JD. et al. Changes in biological activity and folding of guanylate cyclaseactivating protein 1 as a function of calcium. Biochemistry. 1998;37:248–257. [PubMed: 9425045]
- 30.
- Dizhoor AM, Boikov SG, Olshevskaya EV. Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP1. Possible role in causing human autosomal dominant cone degeneration. J Biol Chem. 1998;273:17311–17314. [PubMed: 9651312]
- 31.
- Ramamurthy V, Tucker C, Wilkie SE. et al. Interactions within the coiledcoil domain of RetGC1 guanylyl cyclase are optimized for regulation rather than for high affinity. J Biol Chem. 2001;276:26218–26229. [PubMed: 11306565]
- 32.
- Olshevskaya EV, Ermilov AN, Dizhoor AM. Dimerization of guanylyl cyclaseactivating protein and a mechanism of photoreceptor guanylyl cyclase activation. J Biol Chem. 1999;274:25583–25587. [PubMed: 10464292]
- 33.
- Yu H, Olshevskaya E, Duda T. et al. Activation of retinal guanylyl cyclase1 by Ca2+binding proteins involves its dimerization. J Biol Chem. 1999;4:15547–15555. [PubMed: 10336449]
- 34.
- Payne AM, Downes SM, Bessant DAR. et al. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998;7:273–277. [PubMed: 9425234]
- 35.
- Downes SM, Holder GE, Fitzke FW. et al. Autosomal dominant cone and conerod dystrophy with mutations in the guanylate cyclase activator 1A geneencoding guanylate cyclase activating protein1. Arch Ophthalmol. 2001;119:96–105. [PubMed: 11146732]
- 36.
- Wilkie SE, Newbold RJ, Deery E. et al. Functional characterization of missense mutations at codon 838 in retinal guanylate cyclase correlates with disease severity in patients with autosomal dominant conerod dystrophy. Hum Mol Genet. 2000; 9:3065–3073. [PubMed: 11115851]
- 37.
- Wilkie SE, Li Y, Deery EC. et al. Identification and functional consequences of a new mutation (E155G) in the gene for GCAP1 that causes autosomal dominant cone dystrophy. Am J Hum Genet. 2001;69:471–480. [PMC free article: PMC1235478] [PubMed: 11484154]
- 38.
- Sokal I, Li N, Surgucheva I. et al. GCAP1(Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol Cell. 1998;2:129–133. [PubMed: 9702199]
- 39.
- Surguchov A, Bronson JD, Banerjee P. et al. The human GCAP1 and GCAP2 genes are arranged in a tailtotail array on the short arm of chromosome 6 (p21.1). Genomics. 1997;39:312–322. [PubMed: 9119368]
- 40.
- Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol (Lond). 1979;288:613–634. [PMC free article: PMC1281447] [PubMed: 112243]
- 41.
- Leskov IB, Klenchin VA, Handy JW. et al. The gain of rod phototransduction: reconciliation of biochemical and electrophysiological measurements. Neuron. 2000;27:525–537. [PubMed: 11055435]
- 42.
- Matthews HR, Murphy RL, Fain GL. et al. Photoreceptor light adaptation is mediated by cytoplasmic calcium concentration. Nature. 1988;334:67–69. [PubMed: 2455234]
- 43.
- Nakatani K, Yau KW. Calcium and light adaptation in retinal rods and cones. Nature. 1988;334:69–71. [PubMed: 3386743]
- 44.
- Pepperberg DR, Cornwall MC, Kahlert M. et al. Lightdependent delay in the falling phase of the retinal rod photoresponse. Vis Neurosci. 1992;8:918. [PubMed: 1739680]
- 45.
- Rispoli G, Sather WA, Detwiler PB. Visual transduction in dialysed detached rod outer segments from lizard retina. J Physiol (Lond). 1993;465:513–537. [PMC free article: PMC1175444] [PubMed: 8229848]
- 46.
- Koutalos Y, Nakatani K, Yau KW. The cGMPphosphodiesterase and its contribution to sensitivity regulation in retinal rods. J Gen Physiol. 1995;106:891–921. [PMC free article: PMC2229286] [PubMed: 8648297]
- 47.
- Matthews HR. Actions of Ca2+ on an early stage in phototransduction revealed by the dynamic fall in Ca2+ concentration during the bright flash response. J Gen Physiol. 1997;109:141–146. [PMC free article: PMC2220062] [PubMed: 9041444]
- 48.
- Kawamura S, Murakami M. Calciumdependent regulation of cyclic GMP phosphodiesterase by a protein from frog retinal rods. Nature. 1991;349:420–423. [PubMed: 1846944]
- 49.
- Kawamura S. Rhodopsin phosphorylation as a mechanism of cyclic GMP phosphodiesterase regulation by Smodulin. Nature. 1993; 362:855–857. [PubMed: 8386803]
- 50.
- Klenchin VA, Calvert PD, Bownds MD. Inhibition of rhodopsin kinase by recoverin. Further evidence for a negative feedback system in phototransduction. J Biol Chem. 1995;270:16147–16152. [PubMed: 7608179]
- 51.
- Hsu YT, Molday RS. Modulation of the cGMPgated channel of rod photoreceptor cells by calmodulin. Nature. 1993;361:76–79. [PubMed: 7678445]
- 52.
- Hsu YT, Molday RS. Interaction of calmodulin with the cyclic GMPgated channel of rod photoreceptor cells. Modulation of activity, affinity purification, and localization. J Biol Chem. 1994; 269:29765–29770. [PubMed: 7525588]
- 53.
- Nakatani K, Koutalos Y, Yau KW. Ca2+ modulation of the cGMPgated channel of bullfrog retinal rod photoreceptors. J Physiol (Lond). 1995;484:69–76. [PMC free article: PMC1157922] [PubMed: 7541463]
- 54.
- Lolley RN, Racz E. Calcium modulation of cGMP synthesis in rat visual cells. Vision Res. 1982;22:1481–1486. [PubMed: 6305024]
- 55.
- Kawamura S, Murakami M. Regulation of cGMP levels by guanylate cyclase in truncated frog rod outer segments. J Gen Physiol. 1989;94:649–668. [PMC free article: PMC2228963] [PubMed: 2575652]
- 56.
- Hu G, Jang GF, Cowan CW. et al. Phosphorylation of RGS91 by an endogenous protein kinase in rod outer segments. J Biol Chem. 2001; 276:22287–22295. [PMC free article: PMC1364467] [PubMed: 11292825]
- 57.
- Mendez A, Burns ME, Sokal I. et al. Role of guanylate cyclaseactivating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci USA. 2001;98:9948–9953. [PMC free article: PMC55558] [PubMed: 11493703]
- 58.
- Cobbs WH, Pugh EN Jr. Kinetics and components of the flash photocurrent of isolated retinal rods of the larval salamander, Ambystoma tigrinum. J Physiol (Lond). 1987;394:529–572. [PMC free article: PMC1191975] [PubMed: 2832596]
- 59.
- Karpen JW, Zimmerman AL, Stryer L. et al. Gating kinetics of the cyclicGMPactivated channel of retinal rods: flash photolysis and voltagejump studies. Proc Natl Acad Sci USA. 1988;85:1287–1291. [PMC free article: PMC279752] [PubMed: 2448798]
- 60.
- Dizhoor AM, Hurley JB. Regulation of photoreceptor membrane guanylyl cyclases by guanylyl cyclase activator proteins. Methods. 1999;19:521–531. [PubMed: 10581151]
- 61.
- Lamb TD, Matthews HR, Torre V. Incorporation of calcium buffers into salamander retinal rods: a rejection of the calcium hypothesis of phototransduction. J Physiol (Lond). 1986;372:315–349. [PMC free article: PMC1192765] [PubMed: 3088263]
- 62.
- Matthews HR, Torre V, Lamb TD. Effects on the photoresponse of calcium buffers and cyclic GMP incorporated into the cytoplasm of retinal rods. Nature. 1985;313:582–585. [PubMed: 2578629]
- 63.
- Nikonov S, Engheta N, Pugh EN Jr. Kinetics of recovery of the darkadapted salamander rod photoresponse. J Gen Physiol. 1998;111:737. [PMC free article: PMC1887775] [PubMed: 9417132]
- 64.
- Korschen HG, Beyermann M, Muller F. et al. Interaction of glutamicacidrich proteins with the cGMP signalling pathway in rod photoreceptors. Nature. 1999;400:761–766. [PubMed: 10466724]
- 65.
- Sokal I, OttoBruc AE, Surgucheva I. et al. Conformational changes in guanylyl cyclaseactivating protein 1 (GCAP1) and its tryptophan mutants as a function of calcium concentration. J Biol Chem. 1999;274:19829–19837. [PubMed: 10391927]
- 66.
- Chen J, Makino CL, Peachey NS. et al. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOHterminal truncation mutant. Science. 1995;267:374–377. [PubMed: 7824934]
- 67.
- Chen CK, Burns ME, Spencer M. et al. Abnormal photoresponses and lightinduced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci USA. 1999;96:3718–3722. [PMC free article: PMC22360] [PubMed: 10097103]
- 68.
- Mendez A, Burns ME, Roca A. et al. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. [PubMed: 11086991]
- 69.
- Kelsell RE, GregoryEvans K, Payne AM. et al. Mutations in the retinal guanylate cyclase (RetGC1) gene in dominant conerod dystrophy. Hum Mol Genet. 1998;7:1179–1184. [PubMed: 9618177]
- 70.
- SempleRowland SL, Lee NR, Van Hooser JP. et al. A null mutation in the photoreceptor guanylate cyclase gene causes the retinal degeneration chicken phenotype. Proc Natl Acad Sci USA. 1998; 95:1271–1276. [PMC free article: PMC18742] [PubMed: 9448321]
- 71.
- Suber ML, Pittler SJ, Qin N. et al. Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase betasubunit gene. Proc Natl Acad Sci USA. 1993; 90:3968–3972. [PMC free article: PMC46427] [PubMed: 8387203]
- 72.
- Bowes C, Li T, Frankel WN. et al. Localization of a retroviral element within the rd gene coding for the beta subunit of cGMP phosphodiesterase. Proc Natl Acad Sci USA. 1993;90:2955–2959. [PMC free article: PMC46215] [PubMed: 8385352]
- 73.
- Tsang SH, Burns ME, Calvert PD. et al. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282:117–121. [PubMed: 9756475]
- 74.
- Sokal I, Li N, Verlinde C. et al. Ca2+binding proteins in the retina: from discovery to etiology of human disease. Biochim Biophys Acta. 2000;1498:233–251. [PubMed: 11108966]
- 75.
- Newbold RJ, Deery EC, Walker CE. et al. The destabilization of human GCAP1 by a proline to leucine mutation might cause conerod dystrophy. Hum Mol Genet. 2001;10:47–54. [PubMed: 11136713]
- 76.
- Pittler SB, Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase betasubunit gene of the rd mouse. Proc Natl Acad Sci USA. 1991;88:8322–8326. [PMC free article: PMC52500] [PubMed: 1656438]
- 77.
- Farber DB, Lolley RN. Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974;186:449–451. [PubMed: 4369896]
- 78.
- Farber DB. From mice to men: the cyclic GMP phosphodiesterase gene in vision and disease. The Proctor Lecture. Invest Ophthalmol Vis Sci. 1995;36:263–275. [PubMed: 7843898]
- 79.
- Fain GL, Lisman JE. Light, Ca2+, and Photoreceptor Death: New Evidence for the EquivalentLight Hypothesis from Arrestin Knockout Mice. Invest Ophthalmol Vis Sci. 1999;40:2770–2772. [PubMed: 10549634]
- 80.
- Palczewski K, Polans AS, Baehr W. et al. Ca(2+)binding proteins in the retina: structure, function, and the etiology of human visual diseases. Bioessays. 2000;22:337–350. [PubMed: 10723031]
Publication Details
Author Information and Affiliations
Authors
Ana Mendez and Jeannie Chen.Copyright
Publisher
Landes Bioscience, Austin (TX)
NLM Citation
Mendez A, Chen J. Mouse Models to Study GCAP Functions in Intact Photoreceptors. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.