

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

This Chapter reviews, in a historical perspective, our current understanding of the cell cycle control in terminally differentiated skeletal muscle cells. Attempts at inducing reentry into the cell cycle and proliferation of terminally differentiated muscle cells are reviewed in this context. The material is deliberately presented from the authors' point of view, providing nonetheless a broad coverage of the contributions of other research groups. Wherever possible, skeletal muscle cells are compared with other terminally differentiated cell types to highlight similarities and differences.
Introduction
According to a classic concept, a terminally differentiated (TD) cell is defined as one that, in the course of acquiring specialized functions, has irreversibly lost its ability to proliferate. This apparently simple definition stands on two legs, both of which are unsteady. On one side, it can be argued that all cells are somehow specialized, making the first leg vague and practically and theoretically useless. On the other side and most important, defining a TD cell based on its inability to proliferate means that the definition rests on negative evidence. The fact that a given cell type has never been shown to proliferate does not necessarily mean that it cannot. In fact, at least one cell type traditionally considered TD, the cardiomyocyte, has been shown to be capable of at least some proliferative activity (see Chapter 1). In addition, the “irreversible” loss of proliferative capacity has different characteristics, depending on the cell type considered. At one extreme, cells losing their nuclei during terminal differentiation, such as mammalian erythrocytes and keratinocytes, are obviously irreversibly growth arrested. At the other end, as already mentioned, adult cardiomyocytes are not completely incapable of proliferating even though they are remarkably resistant to attempts at inducing their proliferation in vitro and in vivo. TD mammalian skeletal muscle cells, the focus of this Chapter, are located in the middle of this range of proliferative potential. These cells have never been observed to divide spontaneously in vitro or in vivo. However, their cell cycle machinery is intact and can be reactivated in appropriate experimental conditions, as described here. The behavior of other TD cell types such as adipocytes and neurons will be briefly discussed in this Chapter, further documenting that the definition of terminal differentiation encompasses a variety of diverse states. What is really common to all of these cell types is that, in the adult animal, they are incapable of proliferating in a way that makes any significant contribution to the growth, maintenance, or repair of their respective tissues. Even this last notion is debatable in the case of cardiomyocytes, as argued in Chapter 1 and further discussed in Chapter 2.
Understanding the mechanisms that cause permanent loss of proliferative capacity in TD cells is a scientific challenge of fundamental interest and practical consequence. At the basic level, we are missing a description of the molecular mechanisms underlying the TD state, which is shared by the majority of the cells in an adult mammal. In part as a consequence of this lack of knowledge, we do not understand why evolution has favored this state in the building of higher multicellular animals. On the practical ground, possessing the ability to induce controlled, reversible proliferation of otherwise TD cells would have a potentially dramatic impact on the therapy of diseases and traumas of organs that are incapable of self-renewal through proliferation of their constituent TD cells. The most obvious example is perhaps that of the nervous system.
Skeletal muscle cells are a prototypic TD system. These cells are well suited for in vitro experimentation, as both primary cells and established cell lines can be easily cultured and induced to differentiate. In addition, the molecular mechanisms of their differentiation program are among the best understood. Figure 1 shows a schematic of muscle differentiation, as it is recapitulated in cell culture.1 Undifferentiated myoblasts can be propagated in vitro in growth factor-rich media. Growth factors promote myoblast proliferation and prevent their differentiation. When myoblasts are shifted to a mitogen-poor medium, they withdraw from the cell cycle and begin to express muscle-specific genes (biochemical differentiation). At this stage they are called myocytes, are still mononuclear and are already TD, as they cannot be induced to reinitiate the cell cycle by mitogenic stimulation. Myocytes then fuse to form large, multinucleated, syncytial myotubes. Typically, differentiation is complete in a few days.
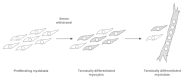
Figure 1
Skeletal muscle differentiation in vitro.
During the last ten years, we have been trying to understand the mechanisms that maintain the growth arrested or postmitotic state in TD skeletal muscle cells. The following is an account of what has been learned through the work of many laboratories, including ours.
Terminal Cell Cycle Withdrawal in Myotubes
It has long been known that expression of the myogenic program requires previous exit from the cell cycle. In 1987 the myogenic transcription factor MyoD was discovered.2 Astonishingly at the time, expression of MyoD was able to convert a variety of cell types into skeletal muscle cells capable of terminal differentiation.2 Identification of the other three members of the MyoD family followed in the next two years.3 During the initial characterization of the properties of MyoD, it had been noticed that its expression was somehow detrimental to growth. Indeed, in 1990 two groups reported that MyoD was capable of inducing growth arrest in normal and neoplastic cells alike, independent of its differentiation properties.4,5 The physiologic significance of the antiproliferative capacity of MyoD is not completely understood even nowadays, but the finding did prompt a search for the molecular mechanisms underlying this effect. In 1993 that pursuit led to the discovery that MyoD induces growth arrest through the obligatory cooperation with the then-emerging tumor suppressor and cell cycle regulator retinoblastoma protein (pRb).6 One year later it was demonstrated that terminal cell cycle withdrawal cannot take place in skeletal muscle cells in the absence of pRb.7 This finding was not substantiated at the time by the phenotype of Rb knockout mice,8-10 but was later fully confirmed by that of partially rescued animals.11 Thus, pRb plays a central role in the establishment of the postmitotic state in skeletal muscle, and cannot be replaced by other members of its family. pRb has similar functions in the establishment of the postmitotic state in neural and lens cells (reviewed in ref. 12). Although the role of pRb in initiating the postmitotic state is firmly established, less clear is its importance in maintaining terminal growth arrest. The selective inactivation of pRb in cells that have undergone terminal differentiation in its presence has not been attempted and in fact has not been possible until the recent development of conditional pRb knockout mice.13 Indeed, evidence exists that, although pRb is required for neuronal entry into the postmitotic state, inactivation of all pRb family members by an E1A mutant cannot reactivate the cell cycle in these cells once terminal growth arrest is established.14 If extended to other cell types, this result would lead to the conclusion that pRb is required only to initiate the postmitotic state, which is then maintained through other mechanisms.
In the second half of the nineties, another important theme emerged, that of cell cycle inhibitors. These proteins can be divided in two families: the INK4 family comprises four members, p15, p16, p18 and p19, which bind specifically the cdk4 and cdk6 cyclin-dependent kinases and prevent their binding to D-type cyclins. The second, Cip or Kip class includes three members, p21, p27 and p57. These proteins preferentially bind and inhibit assembled cyclin/cdk complexes, with broad specificity. All seven inhibitors were identified and cloned between 1993 and 1995.15 Naturally, people wondered whether these inhibitors are involved in the control of terminal growth arrest. Indeed, it was found that p21 increases during skeletal muscle differentiation.16 Later on, it was shown that MyoD is responsible for the transcriptional upregulation of the p21 promoter in the early muscle differentiation,17,18 although the mechanisms involved are unclear. These early findings were followed by a large number of studies, collectively strongly indicating that multiple cell cycle inhibitors are highly expressed during terminal muscle differentiation (see ref. 19 and references therein). Similar evidence was produced for terminal differentiation of other cell types, including for example keratinocytes.20 The phenotypes of mice knockout for individual cell cycle inhibitors did not appear to support an important role for these molecules in skeletal muscle differentiation, as these mice showed little or no muscle phenotype. However, when p21 and p57 knockout mice were crossed, their progeny showed a dramatic reduction in muscle mass due to overproliferation and death of myoblasts.21 In addition, the myotubes that did develop showed evidence of DNA synthesis and endoreplication. At first sight, the last finding would suggest that the cyclin-dependent kinase inhibitors are responsible for the maintenance of the postmitotic state that accompanies and defines terminal differentiation. However, as in the case of pRb, the experiment of withdrawing cell cycle inhibitors in already TD cells has not yet been performed. Thus, it may still be argued that these inhibitors are more important to keep in check cell cycle kinases at the inception of terminal growth arrest, rather than after its establishment.
One reason to be prudent about the role of cell cycle inhibitors in the maintenance of the postmitotic state comes from classic cell fusion studies performed in the late sixties and seventies. These experiments sought to determine whether the inability to reenter the cell cycle is a dominant or recessive property of TD cells. The essentially uniform conclusion was that it is recessive since, in a cell hybrid, a proliferating cell drives into the cell cycle a TD one, belonging to virtually any lineage including macrophages, neurons and chick (nucleated) erythrocytes.22 An apparent, partial exception to this rule is the finding that myocyte × fibroblast hybrids respond to mitogens undergoing DNA synthesis only for a limited time, after which the proliferating partner also becomes growth arrested.23 One possible view is that such hybrids initially conform to the rule stated above, but eventually the proliferating partner is induced to differentiate in trans by MyoD, thus ceasing to be the mitotic driving force of the heterokaryon. Whatever the interpretation of this specific experiment, the overwhelming evidence derived from cell hybrids suggests that the main reason why TD cells are mitotically arrested is not their expression of cell cycle inhibitors which, in a trans fashion, would halt the proliferating partner. Rather, the indication is that TD cells are growth arrested mainly because they lack factors necessary for entrance into or progression through the cell cycle. These factors can be provided in trans by a fused, proliferating cell. It is important to understand that this reasoning is based on indirect evidence and in any case it does not negate that cell cycle inhibitors contribute to the maintenance of the postmitotic state, as opposed to being their principal determinants.
Altogether, current wisdom ascribes in large part the maintenance of the postmitotic state in myotubes to the presence of high levels of pRb. In turn, pRb would not be inactivated through phosphorylation by cyclin-dependent kinases due to the presence of high levels of inhibitors. We have tried to argue that these assumptions must undergo more stringent experimental tests before being definitively accepted.
Response of Myotubes to Growth Factors
The fact that myotubes, as TD cells, do not synthesize DNA in response to mitogens does not necessarily imply that they are absolutely refractory to mitogenic stimuli. Indeed, already in 1986, it has been found that growth factors can induce accumulation of c-myc mRNA in TD myotubes, indicating that these cells can sense and respond to at least some growth factors.23A
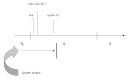
Several years ago we set out to assess whether and to what extent TD myotubes can react to various proliferation-promoting stimuli. Although scientists in the cell cycle field knew that TD cells cannot be reactivated by retroviral oncogenes, that was largely unwritten knowledge and it was difficult to find precise assessments in the scientific literature. We transfected into myotubes derived from primary mouse satellite cells (myoblasts) a number of retroviral oncogenes and/or cell cycle regulators, alone or in combinations (Table 1). Perhaps not surprisingly, none of the transfections brought about DNA synthesis in myotubes.24 We then asked what kind of response serum stimulation can induce in myotubes. Here came a surprise: as judged by the expression of a number of cell cycle markers (Fig. 2), serum-stimulated myotubes were able to leave G0 and progress through G1, at least up to a mid-G1 point that seemed to coincide approximately with the upregulation of the cyclin D1 gene. As shown in Figure 2, the early serum response of myotubes is practically indistinguishable from that of starved, quiescent myoblasts that are fully reactivated by the addition of serum. However, whereas quiescent myoblasts proceeded to upregulate late-G1 genes such as cyclin E, PCNA, B-myb and cyclin A, serum-stimulated myotubes did not appear to go beyond the cyclin D1 stage. These results were important for several reasons. They confirmed that myotubes are able to sense growth factors and transduce mitogenic signals to their nuclei. Furthermore, they refuted the intuitive idea that myotubes cannot proliferate because of their inability to escape from G0 phase. Rather, they suggested that one block prohibiting DNA synthesis in myotubes lies in mid-G1 (Fig. 3).
Table 1
Genes whose expression is unable to reactivate the cell cycle in mouse myotubes.

The E1A Oncogene Reactivates the Cell Cycle in Myotubes
Meanwhile, we were exploring ways to force TD myotubes to fully reenter the cell cycle. We started from reports which had long indicated that some DNA-tumor-virus oncogenes can reactivate the cell cycle in TD myotubes. As early as 1967, it was reported that infection of myoblasts with polyoma or SV40 viruses and subsequent induction of differentiation yielded myotubes that reentered the cell cycle, synthesizing DNA and undergoing mitosis.25,26 However, since the polyoma and SV40 viruses are unable to infect non-dividing cells, the infections had to be performed on myoblasts. This leaves the possibility open that the T antigens might be able to induce DNA synthesis in myotubes only if expressed, though at low levels, before differentiation takes place, but not afterwards. Indeed, it has been reported that injection of polyomavirus into myotubes does not induce DNA synthesis, despite T antigen expression.27 Similarly, SV40 Large T antigen seems to be able to affect the cell cycle only in proliferating, but not in TD cardiomyocytes (see Chapter 2). SV40-mediated reactivation of myotubes was taken up again by Endo and Nadal-Ginard who, in 1989, reported the generation of a C2C12 myoblast cell line expressing a temperature-sensitive mutant of T antigen under the control of an inducible promoter.28 This cell line can undergo differentiation in the functional absence of T antigen and subsequently undergoes DNA synthesis and mitosis when oncogene expression is induced at the permissive temperature. This system was potentially exposed to the same criticism as those involving viruses: one could not rule out the possibility that some T antigen might be expressed and functional before the onset of differentiation. However, the inducible cell line was much more amenable to cellular, biochemical and molecular analyses than the acutely infected, primary cells, witness several studies published thereafter (see Chapter 5 and references therein).
We reasoned that if some DNA-tumor-virus oncogenes truly had the ability to reactivate the cell cycle in TD cells, this capacity might be shared by another oncogene belonging to the same functional class, adenoviral E1A. Adenoviruses possess the invaluable ability to infect a wide variety of cells across species, irrespective of their proliferation status. Thus, we simply asked whether a wild-type adenovirus could infect TD myotubes and express E1A. If so, whether E1A would then be able to reactivate the cell cycle in these cells. The answers to all of these questions turned out to be positive.29,30 It was quickly found that the adenovirus mutant dl520, expressing the 12S but not the 13S transcript of the E1A gene, is far more efficient and less toxic than the wild-type virus.30 Thus, virtually all subsequent experiments with non-mutant E1A were performed with the dl520 virus. Depending on the experimental settings, E1A is able to drive up to 100% of the myotubes in an infected culture into S phase and a substantial proportion of them into, through and beyond mitosis (Fig. 4).30 As we found subsequently, most E1A-reactivated myotubes go through the first cell cycle and die in the course of the second one by apoptosis.31 Apoptosis can be delayed by coexpressing the antiapoptotic adenoviral gene E1B.31 Whether it can be completely prevented is not yet known and awaits further experimentation. We could show that E1A can reactivate other TD cell types, including adipocytes30 and neurons (Fig. 5). This provided a good degree of generality to the conclusion that adenovirus-mediated expression of E1A is a powerful and convenient means to reactivate the cell cycle in TD cells. Indeed, the method was shortly adopted in the heart field and E1A was proven capable of efficiently reactivating TD cardiomyocytes.32,33 However, differences emerged. Unlike myotubes, adipocytes and neurons, cardiomyocytes are not brought into mitosis by E1A, but accumulate in G2. Furthermore, at variance with myotubes,34 the ability of E1A to bind the “pocket proteins” of the pRb family was not required for cardiomyocyte reactivation. These differences and others that will be highlighted later support the notion that the specific mechanisms holding TD cells arrested vary in different tissues.


We then sought to investigate the molecular mechanisms underlying E1A-mediated myotube reactivation. To this aim, we subjected adenovirus-infected myotubes to time-course Northern analyses similar to those shown in Figure 2. The resulting picture was virtually the reverse of that obtained by serum stimulation. The early genes activated by serum, including c-fos, c-myc, Id-1 and cyclin D1, were sharply downregulated by E1A as soon as it began to accumulate in the infected myotubes. Conversely, the late G1 genes that were refractory to serum, such as cyclin E, PCNA, B-myb, cyclin A, were strongly upregulated by E1A. This led us to the conclusion that E1A does not force the previously described mid-G1 block of myotubes, but rather bypasses it by acting directly at the G1/S boundary (Fig. 6).
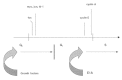
Figure 6
The cell cycle in myotubes (II).
A Second Barrier
Based on current cell cycle wisdom, we predicted that E1A should be substituted for by E2F overexpression. At the time (1995), most of the cell cycle activity of E1A was believed to be mediated by its ability to bind pRb and/or the other pocket proteins and release E2F from their control.35 In its turn, E2F would activate the genes effecting DNA synthesis. According to this model, which is still held largely true, overexpressing unbound E2F is equivalent to releasing it from pocket protein control and should bring cells into S phase. This model has been confirmed by the ability of E2F-1 overexpression to induce DNA synthesis in fibroblasts in a variety of circumstances.36,37 Even in TD cardiomyocytes, E2F-1 was indeed able to fully replace E1A and bring about both DNA synthesis and suppression of tissue-specific transcription.38 However, in agreement with the results of E2F-1 transfection (Table 1), Puri et al showed that neither E2F-1 nor E2F-4 overexpression can induce DNA synthesis in TD myotubes.39 Our group demonstrated that this is not due to lack of transcriptional activity of exogenous E2Fs in myotubes and concluded that E1A, in order to reactivate myotubes, must exert other activities beyond freeing E2F.40 We believe these conclusions to be correct despite the report that E2F-1 triggers DNA synthesis in L6 myotubes,41 as L6 cells are an atypical myoblast cell line that does not express MyoD,42 downregulates pRb during differentiation43 and shows a partially transformed phenotype.44 In addition, E2F-1 has been reported to reactivate TD neurons derived from the P19 embryonal carcinoma cell line,45 while E2F-1 and E2F-2 have a limited but significant capacity to induce DNA synthesis in primary adult rat sensory neurons.46 The competence of E2F to reactivate cardiac myocytes and neurons but not myotubes underscores once more the diversity of the control of the postmitotic state in different TD cell types.
Another means to force cells through the G1/S boundary is activating the cyclin E/cdk2 kinase. This kinase is capable of promoting the transition into S phase even in the absence of E2F activity,47 in a way acting as a parallel control to the pRb-E2F pathway.48 However, activation of the endogenous cyclin E/cdk2 kinase in myotubes through expression of an E1A mutant, does not lead to DNA synthesis despite significant pRb phosphorylation.49 Furthermore, recombinant adenovirus-mediated overexpression of both cyclin E and cdk2 in myotubes achieved kinase activity levels far above physiology and induced pRb phosphorylation but was still unable to trigger DNA replication.50
Altogether, these results indicate that the control of the transition into S phase in myotubes is different from that of non-TD cells and involves what could be thought of as a secondary block at the G1/S boundary (Fig. 7).
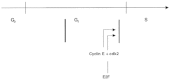
Figure 7
The cell cycle in myotubes (III). The putative secondary block is represented by a double, thin line close to the G1/S boundary.
Reactivation of Myotubes by Expression of Cellular Genes
At least in one sense, oncogene-mediated myotube reactivation is unsatisfactory. Since E1A acts downstream of the primary, mid-G1 block of myotubes, it provides little information as to the nature of such block. Thus, it was important to address this problem by other means.
One clue came from the analysis of serum-stimulated myotubes. As already described, the last cell cycle event activated by serum we detected was cyclin D1 accumulation (Fig. 2). Since it is known that cyclin D-dependent kinase activity is indispensable for cell cycle progression in normal cells,51 we wondered whether the lack of this activity in myotubes might explain, at least in part, their inability to proliferate. Initial experiments confirmed that cdk4, one of the kinase partners of cyclin D1, is constitutively expressed in myotubes. In addition, the cyclin D1 protein could be induced by serum to accumulate at levels similar to those of proliferating myoblasts. However, immunoprecipitations for cdk4 or cyclin D1 could detect no measurable kinase activity in serum-stimulated myotubes. We then used recombinant adenoviruses to express cyclin D1 and/or cdk4 in myotubes.50 While either factor alone induced no DNA synthesis in myotubes, the two proteins together, in the presence of serum, efficiently reactivated up to 85% of the infected myotubes (Fig. 8). In order to achieve this feat, it was necessary to express very high levels of the two proteins. However, maximal myotube reactivation was obtained by expressing them just as much as necessary to attain a level of kinase activity very similar to that found in proliferating myoblasts. TD 3T3-L1 adipocytes and, to a limited extent, P19-derived neurons were also reactivated by cyclin D1/cdk4 expression.
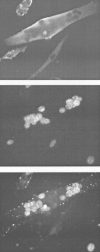
These results indicate that the main block preventing myotubes, and possibly other TD cells, from entering S phase rests in their inability to activate the cyclin D-associated kinase activity. It is important to note that, when reactivated by cyclin D1/cdk4 overexpression, myotubes ignore the late G1 block already described (Fig. 7). This suggests that the more “physiological” reactivation afforded through restoration of the cyclin D1-associated kinase activity is able to properly activate the cell cycle machinery in a way that removes the block.
A Third Block?
Somewhat surprisingly, cyclin D1/cdk4-reactivated myotubes very rarely undergo mitosis. Cytofluorimetric analysis of cyclin D1/cdk4-reactivated TD myocytes shows that they accumulate in G2 indefinitely. This raises the question whether a third block exists in myotubes preventing them from dividing even after DNA replication. Several hypotheses can be made as to the causes of such arrest, which is not observed in myotubes reactivated by E1A.30,31 It might be a last line of defense preventing myotubes from undergoing catastrophic cell division. The more “physiological” cell cycle reentry mediated by expression of cyclin D1 and cdk4 in myotubes might unveil a G2 block that is nullified and thus hidden by E1A. In fact, as already described, myocardiocytes, even when reactivated by E1A, accumulate in G2.32,33 A G2 block also takes place in skeletal myoblasts derived from Rb knockout mice. Although these cells never definitively withdraw from the cell cycle during differentiation,7,52 they rarely undergo mitosis.52 Interestingly, in vivo invasion of muscle fibers by the parasite Trichinella spiralis induces DNA replication in the fiber nuclei. In this natural instance too, mitosis does not occur.53 These examples would suggest that a constitutive G2 block exists in at least some TD cell types, safeguarding them from potentially disruptive mitoses. Alternatively, these observations are compatible with a possible activation of the G2 checkpoint by DNA damage consequent to forced cell cycle reentry. The block due to activation of the G2 checkpoint is partially mediated by p21,54 which is often expressed at high levels in the reactivated myotubes.34,50 In addition, in the case of cyclin D1/cdk4-induced cell cycle reentry, the deregulated expression of cdk4 kinase activity might alter the control of the late cell cycle. Further work is necessary to evaluate the constitutive or reactive nature of the G2 block and its molecular basis. At any rate, not all TD cells reactivated by cyclin D1/cdk4 fail to enter mitosis. At least adipocytes frequently undergo M phase in response to expression of these proteins,50 providing one more example of the differences among diverse TD cell types.
Suppression of Muscle-Specific Gene Expression
Cell cycle reactivation in myotubes is accompanied by suppression of the muscle differentiation program, as myotubes reactivated by either E1A or cyclin D1/cdk4 lose muscle-specific gene expression.34,50 Since skeletal muscle differentiation and proliferation are generally incompatible, it may be argued that forcing myotubes to reenter the cell cycle entails suppressing tissue-specific gene expression. This is certainly true in part, since cell cycle reentry involves pRb phosphorylation. As active, hypophosphorylated pRb contributes substantially to muscle differentiation, it has been proposed that pRb acts as a switch determining whether a myoblast should continue to proliferate (in the presence of phosphorylated pRb) or differentiate (when pRb is dephosphorylated).6 However, all myotube-reactivating genes so far identified (E1A, cyclin D1, cdk4) also block differentiation, in part independently of their cell cycle activity, when expressed in myoblasts.34,55–57 Thus, whether suppression of differentiation is a necessary consequence of cell cycle reactivation, or is accidentally brought about by the specific means so far employed to achieve such reactivation remains to be established.
A Graphic Summary
Our current working model is summarized in Figure 9. The three blocks to cell cycle progression discussed above are represented, together with the variable ability of different mitogenic stimuli to take TD myotubes through none, one or more of them. As to their nature, the mid-G1 block can likely be ascribed to the presence of high levels of various cell cycle inhibitors in myotubes.19 Accordingly, overexpression of cyclin D1 and cdk4 would allow serum-stimulated myotubes to pass this block by simply titering away the inhibitors. Such an explanation does not easily account for the second, late-G1 block, which persists even in the face of very high levels of exogenous cyclin E/cdk2 kinase activity.50 The characteristics of this barrier must be further investigated. The uncertain nature of the third block, already discussed, is indicated by a dotted line. The first block arrests the initial cell cycle progression elicited by serum growth factors, as one necessary mediator of their action is the cyclin D-associated kinase activity. Retroviral oncogenes, whose activity in broad terms mimics the presence of growth factors, possibly hit the same barrier. E1A and T antigen are not arrested by any of the blocks and are capable of pushing myotubes through one or more31 cell cycles. However, the reactivation mediated by these oncogenes results in eventual apoptotic cell death.31,58 E2F or cyclin E/cdk2 expression cannot force the second block, while cyclin D1 and cdk4 cannot take myotubes beyond G2.
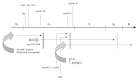
Figure 9
The cell cycle in myotubes: a graphic summary. A possible third block in G2 phase is represented by a dotted line.
Conclusions and Perspectives
In this Chapter we have tried to summarize what has been learned in the past fifteen years on the mechanisms that arrest the cell cycle in skeletal muscle cells and, more generally, in TD cells. We have consciously presented the evolution of the field as seen from our particular vantage point. However, we have made an effort to acknowledge and report comprehensively the many other contributions to this field made by other laboratories.
We hope to have contributed to clarify that the “conventional” cell cycle knowledge, mostly derived from studies on fibroblasts, does not necessarily apply to TD cells, a fact which is perhaps insufficiently perceived by people working outside the reactivation and regeneration fields.
In reviewing the efforts to achieve sustained proliferation of TD cells, one must admit that all attempts focused on the cell cycle machinery have been frustrating, resulting in partial cell cycle reentry or true proliferation followed by cell death. Hope comes from studies on regeneration and development. A very recent paper reports that expression of the mouse homeobox gene msx1 can induce segmentation of TD C2C12 murine myotubes and indefinite proliferation of the derived mononucleated cells.59 Even more exciting, the resulting cells appear to dedifferentiate to a stage compatible with redifferentiation into diverse cell types, including muscle itself. If confirmed, this report might mark a watershed in our field. It is probably the first indication that a developmental regulator can act on “adult” cells in a way that presumably reflects its functions in development. Moreover, for the first time TD cells have been induced to proliferate escaping cell death. Virtually nothing is known about the mechanisms mediating the cell cycle and dedifferentiation effects of msx1 and much research will be required to work them out. However, the spin-offs of this discovery might pave the way to achieving tissue or even organ regeneration in human beings. It is really an exciting time to be in this field.
References
- 1.
- Okazaki K, Holtzer H. Myogenesis: fusion, myosin synthesis, and the mitotic cycle. Proc Natl Acad Sci USA. 1966;56:1484–1490. [PMC free article: PMC220007] [PubMed: 5339623]
- 2.
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. [PubMed: 3690668]
- 3.
- Weintraub H, Davis R, Tapscott S. et al. The myoD gene family: nodal point during specification of the muscle cell lineage. Science. 1991;251:761–766. [PubMed: 1846704]
- 4.
- Sorrentino V, Pepperkok R, Davis RL. et al. Cell proliferation inhibited by MyoD1 independently of myogenic differentiation. Nature. 1990;345:813–815. [PubMed: 2359457]
- 5.
- Crescenzi M, Fleming TP, Lassar AB. et al. MyoD induces growth arrest independent of differentiation in normal and transformed cells. Proc Natl Acad Sci USA. 1990;87(21):8442–8446. [PMC free article: PMC54972] [PubMed: 2236052]
- 6.
- Gu W, Schneider JW, Condorelli G. et al. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. [PubMed: 8381715]
- 7.
- Schneider JW, Gu W, Zhu L. et al. Reversal of terminal differentiation mediated by p107 in Rb−/− muscle cells. Science. 1994;264:1467–1471. [PubMed: 8197461]
- 8.
- Lee EY, Chang CY, Hu N. et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992; 359(6393):288–294. [PubMed: 1406932]
- 9.
- Jacks T, Fazeli A, Schmitt EM. et al. Effects of an Rb mutation in the mouse. Nature. 1992;359(6393):295–300. [PubMed: 1406933]
- 10.
- Clarke AR, Maandag ER, van Roon M. et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359(6393):328–330. [PubMed: 1406937]
- 11.
- Zacksenhaus E, Jiang Z, Chung D. et al. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10(23):3051–3064. [PubMed: 8957005]
- 12.
- Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene. 1999;18(55):7873–7882. [PubMed: 10630640]
- 13.
- Vooijs M, van der Valk M, te Riele H. et al. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene. 1998;17(1):1–12. [PubMed: 9671308]
- 14.
- Slack RS, El-Bizri H, Wong J. et al. A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol. 1998;140(6):1497–1509. [PMC free article: PMC2132670] [PubMed: 9508781]
- 15.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512. [PubMed: 10385618]
- 16.
- Steinman RA, Hoffman B, Iro A. et al. Induction of p21 (WAF-1/CIP1) during differentiation. Oncogene. 1994;9:3389–3396. [PubMed: 7936667]
- 17.
- Halevy O, Novitch BG, Spicer DB. et al. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. [PubMed: 7863327]
- 18.
- Guo K, Wang J, Andres V. et al. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol. 1995;15(7):3823–3829. [PMC free article: PMC230621] [PubMed: 7791789]
- 19.
- Zabludoff SD, Csete M, Wagner R. et al. p27Kip1 is expressed transiently in developing myotomes and enhances myogenesis. Cell Growth Differ. 1998;9(1):1–11. [PubMed: 9438383]
- 20.
- Missero C, Calautti E, Eckner R. et al. Involvement of the cell-cycle inhibitor Cip1/WAF1 and the E1A-associated p300 protein in terminal differentiation. Proc Natl Acad Sci USA. 1995;92(12):5451–5455. [PMC free article: PMC41712] [PubMed: 7777529]
- 21.
- Zhang P, Wong C, Liu D. et al. p21(CIP1) and p57(KIP2) control muscle differentiation at the myogenin step. Genes Dev. 1999;13(2):213–224. [PMC free article: PMC316389] [PubMed: 9925645]
- 22.
- Ringertz NR, Savage RE. Cell HybridsNew York, San Francisco, London: Academic Press, 1976. [PMC free article: PMC287625]
- 23.
- Clegg CH, Hauschka SD. Heterokaryon analysis of muscle differentiation: Regulation of the postmitotic state. J Cell Biol. 1987;105:937–947. [PMC free article: PMC2114765] [PubMed: 3624312]
- 23A.
- Endo T, Nadal-Genard B. Transcriptional and posttranscriptional control of c-myc during myogenesis: its mRNA remains inducible in differentiated cells and does not suppress the differentiated phenotype. Mol Cell Biol. 1986;6(5):1412–1424. [PMC free article: PMC367665] [PubMed: 2431278]
- 24.
- Tiainen M, Pajalunga D, Ferrantelli F. et al. Terminally differentiated skeletal myotubes are not confined in G0, but can enter G1 upon growth factor stimulation. Cell Growth Diff. 1996;7:1039–1050. [PubMed: 8853900]
- 25.
- Yaffe D, Gershon D. Multinucleated muscle fibres: induction of DNA synthesis and mitosis by polyoma virus infection. Nature. 1967;215:421–424. [PubMed: 4293732]
- 26.
- Fogel M, Defendi V. Infection of muscle cultures from various species with oncogenic DNA viruses (SV40 and polyoma). Proc Natl Acad Sci USA. 1967;58:967–973. [PMC free article: PMC335733] [PubMed: 4293268]
- 27.
- Gruen R, Graessmann M, Graessmann A. et al. Infection of human cells with polyoma virus. Virology. 1974;58:290–293. [PubMed: 4362550]
- 28.
- Endo T, Nadal-Ginard B. SV40 large T antigen induces reentry of terminally differentiated myotubes into the cell cycle In: Kedes LH, Stockdale FE, eds. Cellular and Molecular Biology of Muscle Development New York: Alan R. Liss, Inc., 198995–104.
- 29.
- Crescenzi M, Soddu S, Sacchi A. et al. Adenovirus infection induces reentry into the cell cycle of terminally differentiated skeletal muscle cells. Ann NY Acad Sci. 1995;752:9–18. [PubMed: 7755299]
- 30.
- Crescenzi M, Soddu S, Tato' F. Mitotic cycle reactivation in terminally differentiated cells by adenovirus infection. J Cell Physiol. 1995;162:26–35. [PubMed: 7814449]
- 31.
- Latella L, Sacchi A, Crescenzi M. Long-term fate of terminally differentiated skeletal muscle cells following E1A-initiated cell cycle reactivation. Cell Death Differ. 2000;7(2):145–154. [PubMed: 10713729]
- 32.
- Kirshenbaum LA, Schneider MD. Adenovirus E1A represses cardiac gene transcription and reactivates DNA synthesis in ventricular myocytes, via alternative pocket protein- and p300-binding domains. J Biol Chem. 1995;270(14):7791–7794. [PubMed: 7713869]
- 33.
- Liu Y, Kitsis RN. Induction of DNA synthesis and apoptosis in cardiac myocytes by E1A oncoprotein. J Cell Biol. 1996;133(2):325–334. [PMC free article: PMC2120791] [PubMed: 8609165]
- 34.
- Tiainen M, Spitkovsky D, Jansen-Dürr P. et al. Expression of E1A in terminally differentiated muscle cells reactivates the cell cycle and suppresses tissue-specific genes by separable mechanisms. Mol Cell Biol. 1996;16(10):5302–5312. [PMC free article: PMC231529] [PubMed: 8816442]
- 35.
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. [PubMed: 7736585]
- 36.
- Johnson DG, Schwarz JK, Cress DW. et al. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature. 1993;365:349–352. [PubMed: 8377827]
- 37.
- Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12(15):2245–2262. [PubMed: 9694791]
- 38.
- Kirshenbaum LA, Abdellatif M, Chakraborty S. et al. Human E2F-1 reactivates cell cycle progression in ventricular myocytes and represses cardiac gene transcription. Dev Biol. 1996;179:402–411. [PubMed: 8903355]
- 39.
- Puri PL, Cimino L, Fulco M. et al. Regulation of E2F4 mitogenic activity during terminal differentiation by its heterodimerization partners for nuclear translocation. Cancer Res. 1998;58(7):1325–1331. [PubMed: 9537223]
- 40.
- Pajalunga D, Tognozzi D, Tiainen M. et al. E2F activates late-G1 events but cannot replace E1A in inducing S phase in terminally differentiated skeletal muscle cells. Oncogene. 1999;18(36):5054–5062. [PubMed: 10490842]
- 41.
- Gill RM, Hamel PA. Subcellular compartmentalization of E2F family members is required for maintenance of the postmitotic state in terminally differentiated muscle. J Cell Biol. 2000;148(6):1187–1201. [PMC free article: PMC2174298] [PubMed: 10725332]
- 42.
- Braun T, Bober E, Arnold HH. Inhibition of muscle differentiation by the adenovirus E1a protein: repression of the transcriptional activating function of the HLH protein Myf-5. Genes Dev. 1992;6(5):888–902. [PubMed: 1315706]
- 43.
- Zentella A, Massague' J. TGF-β and myogenesis: Interference with cell cycle G1 phase progression may lead to differentiation In: Rifkind RA, ed. The Pharmacology of Cell Differentiation Amsterdam: Excerpta Medica, 199345–53.
- 44.
- Bignami M, Dogliotti E, Benigni R. et al. Expression of transformation-associated traits in the myogenic cell lines L6 and L8. Exp Cell Res. 1982;137:239–248. [PubMed: 6276204]
- 45.
- Azuma-Hara M, Taniura H, Uetsuki T. et al. Regulation and deregulation of E2F1 in postmitotic neurons differentiated from embryonal carcinoma P19 cells. Exp Cell Res. 1999;251(2):442–451. [PubMed: 10471329]
- 46.
- Smith DS, Leone G, DeGregori J. et al. Induction of DNA replication in adult rat neurons by deregulation of the retinoblastoma/E2F G1 cell cycle pathway. Cell Growth Differ. 2000;11(12):625–633. [PubMed: 11149597]
- 47.
- Lukas J, Herzinger T, Hansen K. et al. Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev. 1997;11:1479–1492. [PubMed: 9192874]
- 48.
- Santoni-Rugiu E, Falck J, Mailand N. et al. Involvement of Myc activity in a G(1)/S-promoting mechanism parallel to the pRb/E2F pathway. Mol Cell Biol. 2000;20(10):3497–3509. [PMC free article: PMC85642] [PubMed: 10779339]
- 49.
- Mal A, Chattopadhyay D, Ghosh MK. et al. p21 and retinoblastoma protein control the absence of DNA replication in terminally differentiated muscle cells. J Cell Biol. 2000;149(2):281–292. [PMC free article: PMC2175169] [PubMed: 10769022]
- 50.
- Latella L, Sacco A, Pajalunga D. et al. Reconstitution of cyclin D1-associated kinase activity drives terminally differentiated cells into the cell cycle. Mol Cell Biol. 2001;21(16):5631–5643. [PMC free article: PMC87284] [PubMed: 11463844]
- 51.
- Baldin V, Lukas J, Marcote MJ. et al. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7(5):812–821. [PubMed: 8491378]
- 52.
- Novitch BG, Mulligan GJ, Jacks T. et al. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J Cell Biol. 1996;135(2):441–456. [PMC free article: PMC2121049] [PubMed: 8896600]
- 53.
- Jasmer DP. Trichinella spiralis-infected skeletal muscle cells arrest in G2/M and cease muscle gene expression. J Cell Biol. 1993;121(4):785–793. [PMC free article: PMC2119794] [PubMed: 8491772]
- 54.
- Bunz F, Dutriaux A, Lengauer C. et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282(5393):1497–1501. [PubMed: 9822382]
- 55.
- Webster KA, Muscat GE, Kedes L. Adenovirus E1A products suppress myogenic differentiation and inhibit transcription from muscle-specific promoters. Nature. 1988;332:553–557. [PubMed: 2965790]
- 56.
- Skapek SS, Rhee J, Spicer DB. et al. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science. 1995;267:1022–1024. [PubMed: 7863328]
- 57.
- Zhang JM, Wei Q, Zhao X. et al. Coupling of the cell cycle and myogenesis through the cyclin D1-dependent interaction of MyoD with cdk4. EMBO J. 1999;18(4):926–933. [PMC free article: PMC1171185] [PubMed: 10022835]
- 58.
- Endo T, Nadal-Ginard B. Reversal of myogenic terminal differentiation by SV40 large T antigen results in mitosis and apoptosis. J Cell Sci. 1998;111(Pt 8):1081–1093. [PubMed: 9512504]
- 59.
- Odelberg SJ, Kollhoff A, Keating MT. Dedifferentiation of mammalian myotubes induced by msx1. Cell. 2000;103(7):1099–1109. [PubMed: 11163185]
- Introduction
- Terminal Cell Cycle Withdrawal in Myotubes
- Response of Myotubes to Growth Factors
- The E1A Oncogene Reactivates the Cell Cycle in Myotubes
- A Second Barrier
- Reactivation of Myotubes by Expression of Cellular Genes
- A Third Block?
- Suppression of Muscle-Specific Gene Expression
- A Graphic Summary
- Conclusions and Perspectives
- References
- Cell Cycle Reactivation in Skeletal Muscle and Other Terminally Differentiated C...Cell Cycle Reactivation in Skeletal Muscle and Other Terminally Differentiated Cells - Madame Curie Bioscience Database
- A Conceptual Framework - Madame Curie Bioscience DatabaseA Conceptual Framework - Madame Curie Bioscience Database
- Biocompatibility of Polyurethanes - Madame Curie Bioscience DatabaseBiocompatibility of Polyurethanes - Madame Curie Bioscience Database
- Ceramide in the Regulation of Neuronal Development: Two Faces of a Lipid - Madam...Ceramide in the Regulation of Neuronal Development: Two Faces of a Lipid - Madame Curie Bioscience Database
- Learning from Deficiency: Gene Targeting of Caspases - Madame Curie Bioscience D...Learning from Deficiency: Gene Targeting of Caspases - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...