

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Summary
Genetic approaches complement functional approaches to the study of hereditary disease and have contributed substantially to our understanding of the biology of enterohepatic circulation in health and disease. The basic steps in genetic mapping of a disease gene are reviewed here. They include identification of the mode of inheritance; genetic mapping of the disease gene; identification and screening of candidate genes; and evaluation of the functional consequences of the mutation(s) identified. Ongoing advances in technology and analytic methods have increased the effectiveness and efficiency of genetic mapping approaches.
Introduction
Genetic and functional approaches are the two main ones used in identification of genes that, when mutated, cause disease. In genetic approaches to the study of hereditary disease, genetic mapping methods are employed to identify the position within the human genome of a genetic factor influencing development of a disorder. Then, the gene or genes within that region are screened for evidence of involvement in the disorder. In contrast, in functional approaches to study of genetic disease, ‘candidate genes’ that may be mutated in individuals with the disease under study are identified based on other information. This information can include the biology of the disorder and the known or predicted function of the protein encoded by the gene, as well as other knowledge about the biology of the gene or protein, such as the times during development and tissues in which the gene or protein is expressed and the subcellular or extracellular location of the protein.
In the past, genetic approaches to disease gene identification were often considered, with some validity, to be less direct and more laborious than functional approaches. However, with recent improvements in laboratory technology, analytic approaches, and available data (the latter including the nearly complete sequence of the human genome), genetic approaches can now be applied with steadily increasing efficiency to study of an ever-expanding set of diseases. In recent years, the application of genetic approaches has contributed greatly to our understanding of hereditary disorders, and this productive use of genetic approaches is likely to continue increasing.
Human genetics has changed dramatically during the past two decades, as the following two examples illustrate. In 1983, the initial localization of the gene mutated in Huntington disease (HD) to human chromosome 4p was one of the earliest successes in use of genetic mapping to genetically localize a previously unknown disease gene and involved a combination of very hard work and substantial luck. Selected members of two large pedigrees (including one with more than 3000 identified members) were genotyped, along with other unaffected relatives. Luck came into play in that one of the first 12 genetic markers (in this case, restriction fragment length polymorphisms analyzed using Southern hybridization techniques) tested was linked to HD.1 A decade and the work of many dozens of researchers affiliated with a number of collaborating and competing laboratories was required for the subsequent identification of the gene mutated in HD (i.e., the HD ‘disease gene’) by a collaborative group of 58 authors from six institutions.2
In 1994, a gene mutated in some cases of benign recurrent intrahepatic cholestasis (BRIC) was mapped to human chromosome 18q through study of four patients and six unaffected relatives.3 The following year, the progressive familial intrahepatic cholestasis type 1 (PFIC1) locus was mapped to the same region through study of two patients and their parents.4 It then took less than three years, and the work of many fewer individuals than for HD, to identify the gene mutated in these two disorders.5
Twenty years after the initial mapping of the HD gene, identification of disease genes for disorders with straightforward inheritance patterns (like HD) borders on the routine. Often, the time lapsing between initiation of a mapping project and identification of a disease gene is on the order of a few years; typically, such projects now require the concentrated efforts of only a few individuals to succeed.
One of the key advantages of genetic approaches over many functional ones is that genetic approaches depend very little upon knowledge about the underlying disease biology. Genetic approaches permit discovery of a disorder's genetic etiology, even if the identity of the disease gene could not have been predicted, given current understanding of cell, molecular, and disease biology. Therefore, genetic approaches allow a comprehensive screening of the possible genetic factors leading to development of disease and facilitate unexpected, unpredictable discoveries. This impressive power of genetic approaches has been one factor driving their increased use.
The identification of the BRIC/PFIC1 gene, ATP8B1 (also called FIC1, for familial intrahepatic cholestasis 1), illustrates this point. Prior to identification of ATP8B1, it was hypothesized that the BRIC/PFIC1 gene most likely encoded a canalicular bile acid transporter that was a member of the ATP-binding cassette (ABC) protein family.6 The reasonableness of this hypothesis was borne out when the gene mutated in a different form of PFIC was identified; the PFIC2 gene is indeed an ABC protein that transports bile acids across the canalicular membrane. In contrast, ATP8B1 is not an ABC transporter but belongs to a different family of membrane transporters, the P-type ATPases.7 ATP8B1 is expressed in a wide variety of tissues, suggesting that it plays a role important in many tissues, rather than being specifically a canalicular bile acid transporter.5 Within the liver, the protein is found in the canalicular membrane of hepatocytes, and in cholangiocytes.8,9 ATP8B1 may function in transport of aminophospholipids between membrane leaflets.8 Initial evaluation of a mouse model suggests that ATP8B1 may participate in regulation of intestinal bile salt absorption.10 Further studies to determine the function of ATP8B1 are currently under way and promise to shed new light on the biology of bile acid transport.
Genetic and functional approaches to the study of hereditary disease complement each other and can be applied together. Such an approach facilitated rapid identification of the PFIC2 gene; initially, the PFIC2 locus was genetically mapped to chromosome 2q24.11 An ABC transporter gene lay in the region and was indeed the PFIC2 gene, ABCB11 (also called BSEP, for bile salt export protein).12 If no such excellent candidate gene lay in the region, it likely would have taken longer to identify the disease gene.
In recent years, genetic approaches have been applied to the study of cholestatic disorders, with exciting results. In this chapter, the basic concepts in human genetics that are helpful for understanding of these and future advances will be reviewed, using as illustrations examples from hereditary cholestatic disease.
Our DNA: The Basics
The nucleus of each human somatic cell carries 46 chromosomes: two copies of each of the 22 autosomal chromosomes, and either two copies of the X chromosome (in females), or one copy each of the X and Y chromosomes (in males). Each cell also contains numerous mitochondria, which have their own ˜16 kb (kilobase pair) genome. A single copy of the human nuclear genome (one copy each of chromosomes 1-22, X and Y) is ˜3.2 Gigabase pairs in length and has been estimated to contain approximately 30,000–35,000 protein-coding genes; approximately 1.5% of our DNA is protein-coding sequence, hidden within a large excess of non-coding DNA.13,14 This non-coding DNA contains introns, as well as genes that produce non-coding RNAs, including transfer RNA, ribosomal RNA, small nuclear RNA, small nucleolar RNA, microRNAs, small interfering mRNAs, and small temporal RNAs.15,16 Non-coding DNA also contains numerous other functionally important sequences, including those necessary for maintenance of chromosome ends, proper segregation of chromosomes during cell division, and initiation and regulation of gene expression. Much of our genome has no currently known function; this sequence includes pseudogenes, and the roughly 48% of our DNA that consists of interspersed repetitive elements. This latter group includes many types of transposon-derived repeats, as well as simple sequence repeats.13
Review of a few terms may aid understanding of the discussion that follows. The term locus refers to the chromosomal location of a gene or DNA marker. A specific DNA sequence is polymorphic if it varies between individuals, and the different sequence variants are alleles. A haplotype is a set of specific co-inherited alleles; typically, these alleles are co-inherited because they are present at neighboring genetic markers. An individual carrying the same allele at both copies of a locus is homozygous for that allele, while someone carrying two different alleles is heterozygous. Genetic markers are specific DNA sequences that are polymorphic and employed in mapping of disease genes. When a single disorder can be caused by mutation in different genes, locus heterogeneity is present; low-gamma-glutamyl-transpeptidase (GGT) PFIC exhibits locus heterogeneity, since mutations in either ATP8B1, ABCB11, or one or more as-yet-unidentified genes can cause the disease.17 Allelic heterogeneity exists if different mutations in a single disease gene can cause the same disorder; many genetic disorders manifest allelic heterogeneity, complicating mutation screens.
Steps in Genetic Mapping
Genetic mapping studies typically involve a series of steps: identification of the likely mode of inheritance of the disease; application of experimental and analytical methods to initially map the disease gene and then refine its location; identification of candidate genes; screening of candidate genes for mutation; and determination of the functional consequences of the mutation(s).
Mode of Inheritance
Understanding the mode of inheritance of a disease can focus genetic studies, permits selection of the most appropriate form of genetic analysis, and is important when counseling affected families. Additionally, many forms of genetic analysis require specification of the mode of inheritance of a disorder. The modes reported for cholestatic diseases to date are mostly autosomal recessive and autosomal dominant (Table 1), so these are discussed in some detail below. Basic features of several other modes are indicated in Table 2.
Table 1
Genetics of cholestatic disorders and disorders of bilirubin metabolism and transport: genes identified or mapped.
Table 2
Mendelian modes of inheritance: typical features.
For an autosomal recessive (AR) disorder, disease develops only if both copies of the ‘disease gene’ in an individual possess deleterious mutations. Generally, AR disorders are seen in one or more siblings in a family, but not in parents or children of patients. Heterozygous carriers are usually clinically normal but may exhibit biochemical abnormalities. The ratio of males to females affected is typically ˜1:1, and on average, one-fourth of the children of two heterozygous carrier parents will be affected. Consanguinity is seen more often in AR disease than in disorders with other modes of inheritance and has proven helpful in mapping of PFIC1, AR BRIC, PFIC2, LCS, and North American Indian childhood cirrhosis (NAIC). 3,4,11,12,38,39,42 In consanguineous families, affected children are typically homozygous for a disease mutation. Patients with an AR disease who carry two different disease mutations are compound heterozygotes.
In a simple autosomal dominant (AD) disorder, one defective copy of the disease gene is sufficient to produce disease. In a typical pedigree for an AD disease, individuals in multiple generations will be affected, no generations will be skipped, males and females are equally likely to be affected, and if affected, to transmit the disorder. In a family in which one parent has an AD disorder, 50% of the children, on average, will also have the disorder. Homozygotes, when they occur, are generally more severely affected than heterozygotes. A form of BRIC demonstrating AD inheritance has recently been described.43
As mitochondrial DNA is maternally transmitted, affected fathers do not transmit disorders with a mitochondrial mode of inheritance to their children. Of note with regard to mitochondrial inheritance is that many genetic diseases in which mitochondrial defects occur do not demonstrate mitochondrial inheritance. This is because many proteins essential for normal function of mitochondria are encoded by the nuclear genome. Navaho neurohepatopathy may be an example of such a disorder; it is AR, but appears to involve depletion of mitochondrial DNA.44
Chromosomal abnormalities are large enough to be visible using cytogenetic techniques and include changes in chromosome number (polyploidy, aneuploidy) or structure (i.e., translocations, deletions, inversions, duplications). Some such abnormalities can be genetically transmitted, although review of their inheritance patterns is beyond the scope of this chapter. Before embarking on extensive genetic mapping studies of a disorder, it is sensible to have karyotyping performed on some patients to ensure that they do not have a chromosomal abnormality. For example, early identification of a large deletion in a patient with Alagille syndrome (AGS) narrowed the focus of mapping studies for this disorder to a region on chromosome 20.45
Other features can influence the manifestations and apparent inheritance pattern of a genetic disorder. Disorders that present at different rates or with different symptoms in males versus females can be due to mutation in autosomal genes. Such disorders are sex-influenced or sex-limited; intrahepatic cholestasis of pregnancy (ICP) is a sex-limited disorder. Another important feature of a genetic disease is its penetrance—i.e., the proportion of people with a disease-causing genotype who actually develop the disease. Penetrance can be complete (i.e., all patients with a disease-causing genotype develop the disease) or incomplete, and also may be age-dependent; BRIC due to mutation in ATP8B1 exhibits incomplete, age-dependent penetrance.46
In a disorder with variable expressivity, patients differ in the severity and/or constellation of disease manifestations they suffer. Sometimes variable expressivity is due to mutations with effects of differing severity on protein function, but in other cases, substantial variability is seen even between patients possessing the same disease mutation. Both BRIC and Alagille syndrome have substantially variable expressivity.28,46
Several other features should be kept in mind when considering the pattern of inheritance of a genetic disorder:
- A new mutation may have occurred. For example, a new mutation for a dominant disease may occur in a parent, so that a child is affected, although the parent was not. If the possibility of new mutation is not considered, the disorder in that family might be thought recessive. In Alagille syndrome, as in many AD disorders that reduce reproductive success, a majority of the mutations are ‘de novo’.28 Such a new mutation can occur in a single germ cell in the parent, or the parent can be mosaic for it— i.e., a proportion of the parent's cells carry the mutation. In a dominant disorder, parental germline mosaicism for a new disease mutation means that multiple children in the family can suffer from an AD disorder, although neither parent is affected.
- Some disorders show anticipation, in which the age of onset decreases, and/or disease severity increases (on average), with each successive generation. Anticipation is seen most typically in disorders caused by expansion of a trinucleotide repeat, such as HD, as the repeat can further expand in successive generations.
- Imprinting is said to occur when inheritance of the same mutation has a different effect on the child, depending upon whether it was inherited from the mother or father.
Sometimes, a disorder's mode of inheritance can be evaluated statistically using a formal segregation analysis. In other cases, especially for rare disorders, and given today's typically smaller families, too few patients and family members are available to permit statistically definitive segregation analysis; nevertheless, it is often possible to identify the most likely mode of inheritance. Knowledge of the mode of inheritance of a disorder can help greatly in identifying the genetic etiology of the disease; for example, if a disorder demonstrates X-linked inheritance, a genetic screen need only be performed for the X-chromosome, rather than for the entire nuclear genome.
Complex disorders are those which do not exhibit simple Mendelian inheritance patterns but are multifactorial (i.e., influenced by multiple genetic and environmental factors). Such disorders may be oligogenic (influenced by a small number of genetic loci) or polygenic (influenced by many loci). Given an adequate study sample, the extent to which a trait is inherited can be estimated. For example, in one study, sisters and mothers of women with ICP were found to have ˜12 times greater risk of developing ICP than were women in the general popu- lation;47 however, both genetic and environmental factors could contribute to this increased relative risk. A number of susceptibility loci may exist for a disorder; a susceptibility locus is one at which mutation increases the risk of developing the disease but does not lead inevitably to disease. Modifier loci may influence the phenotype of a disorder; in cystic fibrosis (CF), a modifier locus for meconium ileus has been mapped.41
Localizing a Disease Gene: Genetic Markers
A number of experimental and analytic approaches can be used to map a disease gene. Experimentally, these approaches rely upon genotyping of genetic markers, although the number and type of markers vary. The genetic markers most frequently used in mapping studies to date are polymorphic simple sequence repeats (SSRs). These are widely distributed, short, tandemly repeated sequences. The most commonly employed SSRs include dinucleotide and tetranucleotide repeats. The number of copies of the repeat unit varies between alleles, so alleles differ in length. The inheritance pattern can be assessed by amplifying the repeat from genomic DNA using the polymerase chain reaction (PCR), with unique primers flanking the repeat, and then electrophoresing the PCR products to separate the alleles by size. Over 8,000 polymorphic SSRs have been positioned on a comprehensive genetic map, for an average density of 1 SSR every ˜400 kb;48 a typical genome screen might involve typing of 200–800 of these markers. A major advantage of SSRs for genetic mapping is that they are highly polymorphic and consequently, very informative in genetic analyses. SSRs have been successfully employed in genetic mapping of PFIC1, AR BRIC, PFIC2, LCS, and NAIC, amongst other disorders. 3,4,11,38,39
The use of single nucleotide polymorphisms (SNPs; genomic sites at which a single base varies between alleles) has been increasing. An advantage of SNPs is that they are extremely common; 2 chromosomes differ from each other at ˜1 bp in 1,300.13,14 Also, SNPs have a lower mutation rate than do SSRs. Numerous SNP genotyping technologies are at various stages of development; these include methods employing electrophoresis, oligonucleotide microarrays, mass spectrometry, fluorescent microtiter plate reading, or flow cytometry, among others. Some of these methods are amenable to extremely high-throughput genotyping, compared to what can be achieved with SSRs.49 A disadvantage of SNPs relative to SSRs is that SNPs are less polymorphic, since most SNPs have only 2 alleles.
Genotyping data can be interpreted using various statistical or empiric forms of analysis; what is most appropriate in a specific situation depends upon characteristics of the disorder, the type and size of sample available, and characteristics of the population(s) from which the sample is derived. Genetic mapping data can be evaluated using linkage analysis and/or population genetic mapping.
Localizing a Disease Gene Part 1: Linkage Analysis
Linkage analysis is a powerful, family-based approach to disease mapping. It makes use of the fact that specific copies of the genomic region containing the disease gene are co-inherited with the disease within a family; this reflects lack of recombination between the disease mutation and neighboring genetic markers, due to their close proximity. Within a family, individuals who share a disease will typically share alleles at markers near the disease gene ( Fig. 1). The particular alleles co-inherited with the disease often differ between families, reflecting allelic heterogeneity or ancestral genetic recombination events. Results of linkage analysis are reported as LOD scores representing the relative likelihood that a disease locus and a genetic marker are genetically linked (with a recombination fraction theta), rather than that they are genetically unlinked. A LOD score of at least +3.3 is typically considered evidence of linkage from a genome-wide screen. A LOD score of −2 or below excludes disease linkage to a region. Linkage analysis permitted mapping of the CF gene.5052
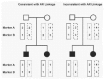
Standard parametric linkage analysis requires specification of a genetic model, including the mode of inheritance, penetrance, and frequency of the disease in the population, and often also necessitates estimation of marker allele frequencies in the population. For disorders with a poorly understood genetic model, other mapping approaches are more appropriate. Additionally, locus heterogeneity within the collection of families studied complicates standard linkage analysis and, if not properly taken into account, can lead to incorrect interpretation of results. Finally, extremely complex pedigrees present computational difficulties for linkage analysis.
Homozygosity mapping is a particularly powerful form of parametric linkage analysis applicable to recessive conditions in consanguinous families. In such a family, patient(s) are likely to be homozygous by descent for a single disease mutation, and for alleles at nearby genetic markers, i.e., both the mutation and marker alleles were inherited from a single ancestor shared by their mother and father. Homozygosity mapping identifies segments of homozygous DNA in patients. Such an approach was employed to map the loci for Wilson disease (WD) and PFIC2.11,53,54
When the genetic model of a disorder cannot be determined, modified forms of linkage analysis may be used. When penetrance of a disorder is unknown, an affecteds-only parametric linkage analysis can be performed, in which the phenotype of unaffected individuals is considered unknown. This approach results in some loss of statistical power. If the mode of inheritance of a disorder is also unknown, a nonparametric linkage approach, such as affected sib pair analysis, can be performed. Regions that are shared by affected siblings or other relatives more often than expected by chance are identified; such approaches are useful in mapping susceptibility loci for complex traits.
Localizing a Disease Gene Part 2: Population Genetic Mapping
Population genetic mapping can also be employed to map disease genes. In this approach, the genome is screened in patients from a single population to identify chromosomal segments that the patients share identical by descent (IBD). The concept behind this approach is that members of a population are relatives, even if their exact relationships are unknown. Distantly related patients may share the same disease mutation, and the same version of the chromosomal sequence surrounding the mutation, inherited from a common ancestor, especially if the disease is rare (Fig. 2). Due to genetic recombination, the greater the number of generations that have passed since the introduction of the mutation into the population, the smaller this shared region will be. The presence of a shared haplotype, or set of specific co-inherited alleles, at several consecutive genetic markers indicates IBD sharing of such an ancestral region. Such sharing reflects the presence of linkage disequilibrium (LD, i.e., a non-random association) between a disease and particular genetic marker alleles.
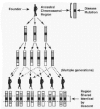
Population genetic mapping approaches can be applied in circumstances where successful linkage analysis is difficult or impossible. In population genetic mapping, mode of inheritance and penetrance of the disorder need not be specified, and allelic and/or locus heterogeneity may be present without resulting in erroneous interpretation of results (although it does reduce statistical power). Also, precise family relationships need not be known, and it is not necessary to identify large pedigrees with multiple patients; DNA need only be obtained from single patients (and if possible, their parents). Additionally, population genetic approaches often permit further refinement of a disease gene's localization than is possible with linkage analysis.
A disadvantage of population genetic studies is that, if the patients are very distantly related, evidence of LD may only be detectable over a very small genomic region, necessitating screening of a large sample with densely spaced genetic markers. This potential problem can be minimized through careful choice of study populations. Recently founded genetically isolated populations are ideal for population genetic mapping, as the size of the genomic region(s) in LD with the disease is likely to be relatively large, and more easily detectable. If such a population was founded by a relatively small number of individuals, the risk of extensive allelic and/or locus heterogeneity, which reduce power to detect a disease locus, is decreased.
Population genetic mapping was successfully employed in the mapping of BRIC, PFIC1, and LCS.3,4,38 A modification of this approach was used to map the NAIC locus; a genome screen was performed on three pools of DNA, one each from patients, unaffected siblings, and parents. Markers with enrichment of an allele in the patient pool, as compared to the other two pools, were identified. Those regions were then further characterized to identify the NAIC region.39
Typically, association studies are used to evaluate candidate genes (or regions) in less consanguineous populations. DNA from a set of patients is collected and data from a polymorphism of interest is generated and statistically evaluated to determine whether one of the alleles is overrepresented in patients as compared to controls. Large patient samples are usually needed for such studies, and patients and controls must be carefully matched. False-positive results can be caused by factors such as undetected population stratification in the sample used, as well as effects of natural selection. Some association tests, such as the transmission disequilibrium test and the more recently developed genomic control and structured association tests, account for population stratification.55 In Finnish women, an association study using markers spanning chromosome 2 identified a region that may contain a susceptibility locus for ICP;40 another study of this same population suggested that a polymorphism in the gene encoding the angiotensin-converting enzyme (not located on chromosome 2) may be associated with ICP.56 Due to the high rate of false-positive results obtained in association studies, confirmation of these results will be important.
Localizing a Disease Gene Part 3: Fine-Mapping
Often the initial mapping of a disease locus to a region is too imprecise to permit immediate identification of the actual disease gene, particularly if the region contains many genes, and none of them are especially promising from a functional standpoint. The location of a disease gene is genetically further refined through study of additional patients, and/or use of additional genetic markers. Often a combination of analytic approaches and populations is used for study of a single disease, as one approach or population may be most useful for initial mapping of a disease locus, and another for the refinement of localization.
To initially localize ATP8B1, for example, haplotypes shared by distantly related patients were identified. 3,4 The position of the locus was further refined using linkage analysis in additional families and finally, detailed haplotype analysis of a larger set of PFIC1 and BRIC patients from multiple populations.42,57 This analysis permitted identification of shared disease haplotypes, and one disease-associated deletion, and localized the disease gene to a ˜1 cM (centimorgan) interval, greatly facilitating identification of ATP8B1.
Candidate Gene Identification
Once a disease gene has been mapped, genes within the candidate region must be identified. In the past, this required much laborious and clever laboratory work, but it has become vastly easier. With the availability of the human genome sequence, a substantially complete inventory of the genes in a given region can be obtained without performing any experiments. At a public or private website devoted to the human genome (e.g., http://www.ensembl.org/ Homo_sapiens/, http://www.genome.ucsc.edu/goldenPAth/help/hgTracksHelp.html, http:// www.ncbi.nlm.nih.gov/, or http://www.ncbi.nlm.nih.gov/genome/guide/human/ ), the genomic region of interest can be selected, and a list of known or predicted genes in the region displayed. Sequence from the region can be used to scan databases of transcribed sequences. To find genes that have not yet been identified through laboratory experiment, sequence from the region can be evaluated using computer programs designed to identify genes within genomic sequence. Nevertheless, difficulties remain. There are still gaps and errors in the genome sequence. Also, the process of computationally predicting the presence and structure of genes, given DNA sequence, is imperfect. Particularly in the case of genes that do not demonstrate substantial sequence homology to known genes, it is possible that a gene might be present, but not predicted to exist.
Usually, one or more promising candidate genes are identified in the region. When multiple genes are present, those screened first for mutation are usually those that data suggest are the most promising candidates, given the known biology of the disease. A gene that encodes a functionally uncharacterized protein can be evaluated as a candidate using information such as whether it has homology to proteins that have known functions consistent with the disease phenotype, and/or whether it is expressed in the tissue(s) most affected by the disease. Genetic and functional approaches to disease gene identification often meet at this point.
Genetic Polymorphisms and Mutations: Types and Identification
The categories of mutation that can occur in our DNA are summarized in Table 3. For most disorders, the most common category of disease mutation identified is the single base pair substitution, in which a single base pair of DNA is replaced with another base pair. Deletions, inversions and insertions (including duplications and repeat expansions) also occur.
Table 3
Categories of mutation.
Deleterious mutations are changes in the DNA sequence that lead to development of, or increased susceptibility to, disease. Neutral polymorphisms or normal variants are DNA sequence changes that have no apparent functional significance. When it is unclear whether a sequence change has any functional consequence, it can conservatively be referred to as a variant.
A number of methods for identifying disease mutations are available. The types and proportion of total mutations detected depends on the method used. Most commonly used methods involve as an initial step PCR amplification of the coding sequence of the candidate gene; to screen for regulatory mutations, the gene's promoter should also be identified and screened. Genes can be screened for mutation using RNA or genomic DNA. Screening of genomic DNA usually requires more effort, as each exon of the gene is amplified by PCR, then analyzed. Where RNA from tissue in which the candidate gene is expressed is available, the transcript can be amplified from it. Unfortunately, patient RNA from relevant tissues is often not readily available. For example, ATP8B1 has 27 coding exons, and screening of the genomic DNA encoding it requires generation and evaluation of 24 separate PCR products (in three cases, small introns enable two exons to be included in a single PCR product). The coding portion of the ATP8B1 transcript is 3.8 kb in length, so when patient RNA from a tissue in which ATP8B1 is expressed is available, the coding portion of the gene can be amplified in substantially fewer PCR reactions.
Once PCR products have been generated, they can be analyzed using one of several methods; the ‘gold standard’ method is DNA sequencing. However, generation and analysis of DNA sequence can be comparatively expensive and time-consuming, so other methods are sometimes used for screening, especially when a large number of samples is involved. Also, heterozygous mutations may occasionally go unnoticed, due to base-calling errors; sequencing both strands of DNA can minimize this problem.
Numerous other approaches to identification of mutations through analysis of PCR products have been developed. One commonly used approach is single strand conformation polymorphism analysis (SSCP); in this approach PCR products are denatured and then electrophoresed through a non-denaturing gel. Mutations may alter the conformation, and thus the mobility, of the single-stranded PCR product; however, some mutations are typically not detected. Three other approaches, denaturing gradient gel electrophoresis (DGGE), mismatch cleavage, and denaturing high performance liquid chromatography (DHPLC), use various techniques to detect differences between homoduplex and heteroduplex DNA molecules. Heteroduplexes are double-stranded DNA molecules in which the sequence differs between the two strands. They are formed by denaturation and reannealing of a PCR product; as a patient may be homozygous for a mutation, the mutation detection rate for these approaches is highest if the patient PCR product is mixed with a PCR product from a control sample prior to denaturation, to ensure that heteroduplexes as well as homoduplexes will form. DHPLC in particular lends itself to automation, and reportedly detects over 95% of mutations; however, the necessary equipment is expensive.58 In general, these techniques identify samples that possess sequence changes, and then the region containing the sequence change is sequenced from that sample to precisely characterize the mutation. The presence of a frequent neutral polymorphism may necessitate a lot of extra sequencing. In the future, mutation screening may increasingly be performed using oligonucleotide microarrays; these techniques are still being perfected.
Although PCR-based methods of mutation detection are efficient and require only small quantities of patient DNA or RNA, some types of mutation are difficult or impossible to detect using them. Therefore, other methods still play a role. Use of karyotyping and forms of fluorescence in situ hybridization (FISH) allow detection of changes in chromosome number, as well as large deletions or other rearrangements. Preparation of standard genomic Southern blots, followed by hybridization with probes from the candidate region, and/or pulsed field gel electrophoresis (PFGE) of genomic DNA digested with restriction enzymes that yield large fragments, followed by Southern blotting and hybridization, can permit detection of deletions and rearrangements of intermediate size. Although comparatively large amounts of DNA and labor are required for such studies, they can be worthwhile, particularly in patients in whom mutations remain unidentified after use of other screening methods.
Regardless of the method used for mutation detection, findings in patients should be compared with those in a control sample (ideally ethnically matched, although this is not always possible), to help distinguish disease-causing mutations from neutral polymorphisms. If a particular disease mutation is found to occur frequently in patients, a specific assay for its efficient detection can be developed.
Functional Consequences of Mutation
There are a variety of ways in which a disease mutation in a gene can ultimately affect the function of the encoded protein; for example a mutation may affect transcription, mRNA stability, translation, or protein stability, localization, or function. The functional consequences of a mutation can often be predicted based upon the sequence change induced; nevertheless, functional studies are extremely valuable in confirming, refining, or changing these predictions.
- Transcription: a mutation may occur in a promoter or enhancer element of a gene, and prevent transcription of the gene, or alter the levels, timing, and/or tissue distribution of expression. An example of such a mutation was recently described in the Wilson's Disease (WD) gene, ATP7B; a 15-bp deletion in the promoter region of the gene was present on 60.5% of Sardinian WD chromosomes, and reduced transcriptional activity by 75% in expression assays.59 Mutations can also change splicing patterns of a gene.
- mRNA stability and translation: Another way in which a mutation may effect mRNA levels is by decreasing the stability of the transcript. Mutation in a polyadenylation site may prevent polyadenylation of the transcript, and lead to decreased stability of the mRNA, and/or inhibition of its translation. The presence of a sequence change that leads to premature termination of translation (such as a nonsense mutation, or an insertion, deletion, or splicing mutation that leads to a frameshift) can also lead to mRNA degradation through nonsense-mediated mRNA decay.60 Rarely, the initiation codon may be mutated.
- Protein stability or localization: a mutation may have no effect on transcript levels, but alter the stability or localization of the protein. A mutation causing abnormal folding of a protein may result in its ubiquitination and subsequent degradation. Coding sequence mutations may alter localization signals, or prevent post-translational modification, necessary for delivery of a protein to its correct location. Several examples of mutations leading to liver disease through such mechanisms are known. For instance, the common WD mutation, H1069Q, appears to cause a protein-folding defect resulting in degradation of the protein.61 Similarly, the common ABCC7 mutation in CF patients, deltaF508, causes ABCC7 to be incompletely glycosylated, mislocalized, and degraded.62,63 Two JAG1 mutations found in AGS lead to abnormal glycosylation and mislocalization of JAG1.64
- Protein function: normal levels of a protein may be produced, and the protein may be delivered to its proper location, but a change in the amino acid sequence of the protein may prevent it from functioning properly; its function may be partially or completely destroyed. For example, two ABCC7 mutations found in CF patients have been shown not to prevent normal protein localization, but to disrupt protein function.62,65 Occasionally, mutations may instead cause a ‘gain of function’, in that the protein has higher activity than normal, or functions in ways or circumstances different from those of the normal protein. Dominant negative mutations are loss-of-function mutations in which, in heterozygous individuals, the mutated protein interferes with the function of the normal versions of the protein; generally, this effect occurs with multimeric proteins.
Genetics of Cholestatic Disorders
Application of genetic approaches has greatly increased our understanding of the molecular biology of cholestasis. Through use of genetic and functional approaches, the genes mutated in a number of disorders have been identified; others have been genetically mapped (Table 1; a helpful source of more information and references on these disorders is the Online Mendelian Inheritance in Man database at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM). There remains much knowledge to be gained through further genetic study of cholestatic disorders. For example, a recent report indicates that at least one additional low-gamma-GT PFIC gene remains to be identified; in some families segregating low-GGT PFIC, the disease does not map to either ATP8B1 or to ABCB11.17 Mutation of ABCB11 in a BRIC patient has recently been reported, and a third BRIC locus waits to be identified, as a family in which BRIC demonstrates an autosomal dominant mode of inheritance unlinked to either ATP8B1 or to ABCB11 has been described.20,43 It is clear that many cholestatic disorders exhibit extensive allelic and/or locus heterogeneity, and this is both a challenge with respect to identifying the genetic etiologies of these disorders, as well as a rich source of information about the liver and the enterohepatic circulation in health and disease. Given the rapid pace and high efficiency with which genetic research can now move, we will soon be learning much that is new about the molecular basis of cholestasis.
This new knowledge will eventually extend beyond increased understanding of rare cholestatic diseases; common genetic polymorphisms associated with increased or decreased susceptibility to relatively common, less obviously ‘genetic’ disorders will likely be identified, as will factors influencing disease severity, rate of progession, age of onset, and other similar variables. Work in this area is already beginning, with the mapping of a modifier locus for meconium ileus in CF and of potential susceptibility loci for ICP in the Finnish population.40,41,56 It is likely that some of the sequence variants eventually found to influence more common hepatic disorders will occur in genes that encode proteins involved in hepatic transport and metabolism; these genes may include those initially functionally linked to cholestasis through their identification as disease genes for rare disorders.
Association studies of candidate genes have already revealed genetic factors that potentially influence development of a number of hepatic disorders. In primary biliary cirrhosis (PBC), alleles have been identified that appear associated with increased or decreased disease susceptibility or rate of disease progression; these polymorphisms include ones in the major histocompatibility locus (MHC), as well as one in the cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) gene, one in the interleukin 1 gene, another in the vitamin D receptor, and a 4-SNP haplotype in the mannose-binding lectin (MBL) gene. 6671 Variation in susceptibility to primary sclerosing cholangitis (PSC), alcoholic liver disease, and chronic viral hepatitis may also be associated with alleles of polymorphisms in the MHC. 7277 PSC susceptibility also appears associated with a functional polymorphism in the stromelysin (MMP-3) gene;78 other genetic associations to development of severe alcoholic liver disease have also been identified, including an association with a common amino acid change in manganese superoxide dismutase, one with a variant in the promoter of the CD14 endotoxin receptor, and a third with variants in the interleukin-1beta gene.7981 In chronic hepatitis C, progression of liver disease may be associ- ated with particular variants in the transforming growth factor beta 1 and angiotensin II genes.82 In evaluating these types of study, it is important to remember that false-positive evidence for association is sometimes obtained, and replication is necessary to confirm results. Also, due to the existence of linkage disequilibrium (LD) between closely linked genetic markers, evidence for association between a disease and a polymorphic allele does not necessarily indicate that that allele itself is the functional cause of the association; another nearby genetic variant in LD with the polymorphism studied may be the functional cause of the association, i.e., it may affect production or function of a protein, and thus influence disease development. Understandably, many genetic association studies of common liver diseases have focused on evaluation of genes encoding proteins involved in function of the immune system. It will be interesting to see whether associations between sequence variants in hepatic transporter genes and particular disease features, such as ability to maintain adequate hepatic function despite liver damage, may also be found.
Conclusions and Outlook
In the field of human genetics, rapid progress has occurred in development of technology and amassing of data including sequencing of the human genome, characterization of expressed sequence tags and identification of SNPs. This progress has set the stage for broader application of genetic techniques. Identification of genes mutated in disorders demonstrating simple Mendelian modes of inheritance is now relatively straightforward. Genetic mapping of genes mutated in disorders with more complex genetic etiology, as well as of modifier genes of ‘simple traits’, is increasingly feasible. In the future, many already identified DNA sequence variants will likely be shown to influence disease susceptibility and severity for both common and rare disorders. This knowledge will permit better prediction of disease prognosis in individuals, and also, more targeted preventive interventions.
Another area in which research is rapidly expanding is that of pharmacogenetics, the study of genetic factors influencing drug response. Systematic study of known genetic variations that have no currently identified functional consequences will likely lead to identification of a subset that impact response to medications. Genetic factors leading to a greater risk of deleterious side effects, as well as factors influencing efficacy and optimal dosing, may be identified, dramatically altering medical practice.
Developments in cDNA expression array technology are beginning to yield large quantities of information on gene expression patterns in health and disease. Such studies complement genetic approaches and have parallels to sequencing of the human genome, in that they can enable creation of a vast repository of ‘functional’ data that can be systematically mined to identify genes encoding proteins likely to play a key role in disease processes.
References
- 1.
- Gusella JF, Wexler NS, Conneally PM. et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature. 1983;306:234–238. [PubMed: 6316146]
- 2.
- Huntington's Disease Collaborative Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's Disease chromosomes. Cell. 1993;72:971–983. [PubMed: 8458085]
- 3.
- Houwen RH, Baharloo S, Blankenship K. et al. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nat Genet. 1994;8:380–6. [PubMed: 7894490]
- 4.
- Carlton VE, Knisely AS, Freimer NB. Mapping of a locus for progressive familial intrahepatic cholestasis (Byler disease) to 18q21-q22, the benign recurrent intrahepatic cholestasis region. Hum Mol Genet. 1995;4:1049–53. [PubMed: 7655458]
- 5.
- Bull LN, van Eijk MJ, Pawlikowska L. et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–24. [PubMed: 9500542]
- 6.
- Sela-Herman S, Bull L, Lomri N. et al. In search of a gene for hereditary cholestasis. Biochem Mol Med. 1996;59:98–103. [PubMed: 8986630]
- 7.
- Tang X, Halleck MS, Schlegel RA. et al. A subfamily of P-type ATPases with aminophospholipid transporting activity [published erratum appears in Science 1996 Dec 6;274(5293):1597] Science. 1996;272:1495–7. [PubMed: 8633245]
- 8.
- Ujhazy P, Ortiz D, Misra S. et al. Familial intrahepatic cholestasis 1: Studies of localization and function. Hepatology. 2001;34:768–775. [PubMed: 11584374]
- 9.
- Eppens EF, van Mil SWC, de Vree JM. et al. FIC1, the protein affected in two forms of hereditary cholestasis, is localized in the cholangiocyte and the canalicular membrane of the hepatocyte. J Hepatol. 2001;35:436–443. [PubMed: 11682026]
- 10.
- Pawlikowska L, Ottenhoff R, Looije N. et al. FIC1 mutant mice have a defect in the regulation of intestinal bile salt absorption Hepatology 200134240AAbstr.
- 11.
- Strautnieks SS, Kagalwalla AF, Tanner MS. et al. Identification of a locus for progressive familial intrahepatic cholestasis PFIC2 on chromosome 2q24. Am J Hum Genet. 1997;61:630–3. [PMC free article: PMC1715942] [PubMed: 9326328]
- 12.
- Strautnieks SS, Bull LN, Knisely AS. et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–8. [PubMed: 9806540]
- 13.
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. [PubMed: 11237011]
- 14.
- Venter JC, Adams MD, Myers EW. et al. The sequence of the human genome. Science. 2001;291:1304–1351. [PubMed: 11181995]
- 15.
- Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919–929. [PubMed: 11733745]
- 16.
- Moss EG. RNA interference: It's a small RNA world. Curr Biol. 2001;11:R772–R775. [PubMed: 11591334]
- 17.
- Strautnieks S, Byrne J, Knisely AS. et al. There must be a third locus for low GGT PFIC Hepatology 200134240AAbstr.
- 18.
- Klomp LW, Bull LN, Knisely AS. et al. A missense mutation in FIC1 is associated with greenland familial cholestasis. Hepatology. 2000;32:1337–41. [PubMed: 11093741]
- 19.
- Jansen PLM, Strautnieks SS, Jacquemin E. et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–1379. [PubMed: 10579978]
- 20.
- Kullak-Ublick GA, Kerb R, Mullhaupt B. et al. A novel R432T mutation in the bile salt export pump gene (BSEP;ABCB11) is associated with recurrent intrahepatic cholestasis in an adolescent patient Hepatology 200134216AAbstr.
- 21.
- Deleuze JF, Jacquemin E, Dubuisson C. et al. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology. 1996;23:904–8. [PubMed: 8666348]
- 22.
- de Vree JM, Jacquemin E, Sturm E. et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998;95:282–7. [PMC free article: PMC18201] [PubMed: 9419367]
- 23.
- Jacquemin E, de Vree JM, Cresteil D. et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120:1448–1458. [PubMed: 11313315]
- 24.
- Jacquemin E, Cresteil D, Manouvrier S. et al. Heterozygous non-sense mutation of the MDR3 gene in familial intrahepatic cholestasis of pregnancy [letter] Lancet. 1999;353:210–1. [PubMed: 9923886]
- 25.
- Dixon PH, Weerasekera N, Linton KJ. et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genet. 2000;9:1209–1217. [PubMed: 10767346]
- 26.
- Li L, Krantz ID, Deng Y. et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–51. [PubMed: 9207788]
- 27.
- Oda T, Elkahloun AG, Pike BL. et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–42. [PubMed: 9207787]
- 28.
- Spinner NB, Colliton RP, Crosnier C. et al. Jagged1 mutations in Alagille syndrome. Hum Mutat. 2001;17:18–33. [PubMed: 11139239]
- 29.
- Wada M, Toh S, Taniguchi K. et al. Mutations in the canalicular multispecific organic anion transporter (cMOAT) gene, a novel ABC transporter, in patients with hyperbilirubinemia II/Dubin-Johnson syndrome. Hum Mol Genet. 1998;7:203–207. [PubMed: 9425227]
- 30.
- Ritter JK, Yeatman MT, Kaiser C. et al. A phenylalanine codon deletion of the UGT1 gene complex locus of a Crigler-Najjar type I patient generates a pH-sensitive bilirubin UDP-glucuronosyltransferase. J Biol Chem. 1993;268:23573–23579. [PubMed: 8226884]
- 31.
- Kadakol A, Ghosh SS, Sappal BS. et al. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum Mutat. 2000;16:297–306. [PubMed: 11013440]
- 32.
- Setchell KD, Schwarz M, O'Connell NC. et al. Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7-alpha-hydorxylase gene causes severe neonatal liver disease. J Clin Invest. 1998;102:1690–1703. [PMC free article: PMC509117] [PubMed: 9802883]
- 33.
- Bull PC, Thomas GR, Rommens JM. et al. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. [PubMed: 8298639]
- 34.
- Tanzi RE, Petrukhin K, Chernov I. et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. [PubMed: 8298641]
- 35.
- Riordan JR, Rommens JM, Kerem B. et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. [PubMed: 2475911]
- 36.
- Rommens JM, Iannuzzi MC, Kerem B. et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. [PubMed: 2772657]
- 37.
- Kerem B, Rommens JM, Buchanan JA. et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. [PubMed: 2570460]
- 38.
- Bull LN, Roche E, Song EJ. et al. Mapping of the locus for cholestasis-lymphedema syndrome (Aagenaes syndrome) to a 6.6-cM interval on chromosome 15q. Am J Hum Genet. 2000;67:994–9. [PMC free article: PMC1287903] [PubMed: 10968776]
- 39.
- Betard C, Rasquin-Weber A, Brewer C. et al. Localization of a recessive gene for North American Indian childhood cirrhosis to chromosome region 16q22-and identification of a shared haplotype. Am J Hum Genet. 2000;67:222–228. [PMC free article: PMC1287080] [PubMed: 10820129]
- 40.
- Heinonen ST, Eloranta ML, Heiskanen JTM. et al. Maternal susceptibility locus for obstetric cholestasis maps to chromosome region 2p13 in Finnish patients. Scand J Gastroenterol. 2001;36:766–770. [PubMed: 11444477]
- 41.
- Zielenski J, Corey M, Rozmahel R. et al. Detection of a cystic fibrosis modifier locus for meconium ileus on human chromosome 19q13. Nat Genet. 1999;22:128–129. [PubMed: 10369249]
- 42.
- Bull LN, Juijn JA, Liao M. et al. Fine-resolution mapping by haplotype evaluation: the examples of PFIC1 and BRIC. Hum Genet. 1999;104:241–8. [PubMed: 10323248]
- 43.
- Floreani A, Molaro M, Mottes M. et al. Autosomal dominant benign recurrent intrahepatic cholestasis (BRIC) unlinked to 18q21 and 2q24. Am J Med Genet. 2000;95:450–453. [PubMed: 11146465]
- 44.
- Vu TH, Tanji K, Holve SA. et al. Navajo neurohepatopathy: a mitochondrial DNA depletion syndrome? Hepatology. 2001;34:116–120. [PubMed: 11431741]
- 45.
- Byrne JL, Harrod MJ, Friedman JM. et al. Del(20p) with manifestations of arteriohepatic dysplasia. Am J Med Genet. 1986;24:673–678. [PubMed: 3740100]
- 46.
- van Mil SWC, Klomp LW, Bull LN. et al. FIC1 Disease: A spectrum of intrahepatic cholestatic disorders. Semin Liver Dis. 2001;21:535–544. [PubMed: 11745041]
- 47.
- Eloranta MJ, Heinonen S, Mononen T. et al. Risk of obstetric cholestasis in sisters of index patients. Clin Genet. 2001;60:42–45. [PubMed: 11531968]
- 48.
- Broman KW, Murray JC, Sheffield VC. et al. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet. 1998;63:861–9. [PMC free article: PMC1377399] [PubMed: 9718341]
- 49.
- Gut IG. Automation in genotyping of single nucleotide polymorphisms. Hum Mutat. 2001;17:475–492. [PubMed: 11385706]
- 50.
- Tsui LC, Buchwald M, Barker D. et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science. 1985;230:1054–1057. [PubMed: 2997931]
- 51.
- White R, Woodward S, Leppert M. et al. A closely linked genetic marker for cystic fibrosis. Nature. 1985;318:382–384. [PubMed: 3906407]
- 52.
- Wainwright BJ, Scambler PJ, Schmidtke J. et al. Localization of cystic fibrosis locus to human chromosome 7cen-q22. Nature. 1985;318:384–385. [PubMed: 2999612]
- 53.
- Frydman M, Bonne-Tamir B, Farrer LA. et al. Assignment of the gene for Wilson disease to chromosome 13: linkage to the esterase D locus. Proc Natl Acad Sci USA. 1985;82:1819–1821. [PMC free article: PMC397364] [PubMed: 3856863]
- 54.
- Bonne-Tamir B, Farrer LA, Frydman M. et al. Evidence for linkage between Wilson disease and esterase D in three kindreds: detection of linkage for an autosomal recessive disorder by the family study method. Genet Epidemiol. 1986;3:201–209. [PubMed: 3459695]
- 55.
- Devlin B, Roeder K, Bacanu SA. Unbiased methods for population-based association studies. Genet Epidemiol. 2001;21:273–284. [PubMed: 11754464]
- 56.
- Heiskanen JTM, Pirskanen MM, Hiltunen MJ. et al. Insertion-deletion polymorphism in the gene for angiotensin-converting enzyme is associated with obstetric cholestasis but not with preeclampsia. Am J Obstet Gynecol. 2001;185:600–603. [PubMed: 11568784]
- 57.
- Sinke RJ, Carlton VE, Juijn JA. et al. Benign recurrent intrahepatic cholestasis (BRIC): evidence of genetic heterogeneity and delimitation of the BRIC locus to a 7-cM interval between D18S69 and D18S64. Hum Genet. 1997;100:382–7. [PubMed: 9272159]
- 58.
- Xiao W, Oefner PJ. Denaturing high-performance liquid chromatography: a review. Hum Mutat. 2001;17:439–474. [PubMed: 11385705]
- 59.
- Loudianos G, Dessi V, Lovicu M. et al. Molecular characterization of Wilson disease in the Sardinian population-evidence of a founder effect. Hum Mutat. 1999;14:294–303. [PubMed: 10502776]
- 60.
- Byers PH. Killing the messenger: new insights into nonsense-mediated mRNA decay. J Clin Invest. 2002;109:3–6. [PMC free article: PMC150830] [PubMed: 11781342]
- 61.
- Payne AS, Kelly EJ, Gitlin JD. Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc Natl Acad Sci USA. 1998;95:10854–10859. [PMC free article: PMC27985] [PubMed: 9724794]
- 62.
- Cheng SH, Gregory RJ, Marshall J. et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. [PubMed: 1699669]
- 63.
- Jensen TJ, Loo MA, Pind S. et al. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. [PubMed: 7553864]
- 64.
- Morrissette JJD, Colliton RP, Spinner NB. Defective intracellular transport and processing of Jag1 missense mutations in Alagille syndrome. Hum Mol Genet. 2001;10:405–413. [PubMed: 11157803]
- 65.
- Logan J, Hiestand D, Daram P. et al. Cystic fibrosis transmembrane conductance regulator mutations that disrupt nucelotide binding. J Clin Invest. 1994;94:228–236. [PMC free article: PMC296301] [PubMed: 7518829]
- 66.
- Donaldson PT. TNF gene polymorphisms in primary biliary cirrhosis: a critical appraisal. J Hepatol. 1999;31:366–368. [PubMed: 10453954]
- 67.
- Wilson AG. Genetics of tumour necrosis factor (TNF) in autoimmune liver diseases: red hot or red herring? J Hepatol. 1999;30:331–333. [PubMed: 10068116]
- 68.
- Agarwal K, Jones DEJ, Daly AK. et al. CTLA-4 gene polymorphism confers susceptibility to primary biliary cirrhosis.J Hepatol 200032538–541. [PubMed: 10782900]
- 69.
- Donaldson P, Agarwal K, Craggs A. et al. HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: associations with disease progression and disease susceptibility. Gut. 2001;48:397–402. [PMC free article: PMC1760119] [PubMed: 11171832]
- 70.
- Vogel A, Strassburg CP, Manns MP. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis and autoimmune hepatitis. Hepatology. 2002;35:126–131. [PubMed: 11786968]
- 71.
- Matsushita M, Miyakawa H, Tanaka A. et al. Single nucleotide polymorphisms of the mannose-binding lectin are associated with susceptibility to primary biliary cirrhosis. J Autoimmun. 2001;17:251–257. [PubMed: 11712863]
- 72.
- Wiencke K, Spurkland A, Schrumpf E. et al. Primary sclerosing cholangitis is associated to an extended B8-DR3 haplotype including particular MICA and MICB alleles. Hepatology. 2001;34:625–630. [PubMed: 11584356]
- 73.
- Norris S, Kondeatis E, Collins RS. et al. Mapping MHC-encoded susceptibility and resistance in primary sclerosing cholangitis: the role of MICA polymorphism. Gastroenterology. 2001;120:1475–1482. [PubMed: 11313318]
- 74.
- Bernal W, Moloney M, Underhill J. et al. Association of tumor necrosis factor polymorphism with primary sclerosing cholangitis. J Hepatol. 1999;30:237–241. [PubMed: 10068102]
- 75.
- Spurkland A, Saarinen S, Boberg KM. et al. HLA class II haplotypes in primary sclerosing cholangitis patients from five European populations. Tissue Antigens. 1999;53:459–469. [PubMed: 10372541]
- 76.
- Grove J, Daly AK, Bassendine MF. et al. Association of a tumor necrosis factor promoter polymorphism with susceptibility to alcoholic steatohepatitis. Hepatology. 1997;26:143–146. [PubMed: 9214463]
- 77.
- Thio CL, Thomas DL, Carrington M. Chronic viral hepatitis and the human genome. Hepatology. 2000;31:819–827. [PubMed: 10733534]
- 78.
- Satsangi J, Chapman RW, Haldar N. et al. A functional polymorphism of the stromelysin gene (MMP-3) influences susceptibility to primary sclerosing cholangitis. Gastroenterology. 2001;121:124–130. [PubMed: 11438501]
- 79.
- Degoul F, Sutton A, Mansouri A. et al. Homozygosity for alanine in the mitochondrial targeting sequence of superoxide dismutase and risk for severe alcoholic liver disease. Gastroenterology. 2001;120:1468–1474. [PubMed: 11313317]
- 80.
- Jarvelainen HA, Orpana A, Perola M. et al. Promoter polymorphism of the CD14 endotoxin receptor gene as a risk factor for alcoholic liver disease. Hepatology. 2001;33:1148–1153. [PubMed: 11343243]
- 81.
- Takamatsu M, Yamauchi M, Maezawa Y. et al. Genetic polymorphisms of interleukin-1beta in association with the development of alcoholic liver disease in Japanese patients. Am J Gastroenterol. 2000;95:1305–1311. [PubMed: 10811344]
- 82.
- Powell EE, Edwards-Smith CJ, Hay JL. et al. Host genetic factors influence disease progression in chronic hepatitis C. Hepatology. 2000;31:828–833. [PubMed: 10733535]
- 83.
- Chagnon P, Michaud J, Mitchell G. et al. A missense mutation (R565W) in Cirhin (FLJ14728) in North American Indian Childhood Cirrhosis. Am J Hum Genet. 2002;71:1443–1449. [PMC free article: PMC378590] [PubMed: 12417987]
- Genetics, Mutations, and Polymorphisms - Madame Curie Bioscience DatabaseGenetics, Mutations, and Polymorphisms - Madame Curie Bioscience Database
- Tumor-Initiating Cells, Cancer Metastasis and Therapeutic Implications - Madame ...Tumor-Initiating Cells, Cancer Metastasis and Therapeutic Implications - Madame Curie Bioscience Database
- Dynamic Kinetic Modelling of Mitochondrial Energy Metabolism - Madame Curie Bios...Dynamic Kinetic Modelling of Mitochondrial Energy Metabolism - Madame Curie Bioscience Database
- Calcium Channel Block and Inactivation: Insights from Structure-Activity Studies...Calcium Channel Block and Inactivation: Insights from Structure-Activity Studies - Madame Curie Bioscience Database
- Scaling Laws in the Functional Content of Genomes: Fundamental Constants of Evol...Scaling Laws in the Functional Content of Genomes: Fundamental Constants of Evolution? - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...