

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

U.S. Food and Drug Administration Regulation of Medical Devices
The U.S. Food and Drug Administration (FDA) regulates all medical devices sold in the United States. As depicted in Figure 3.1, there are a variety of possible paths that a medical device manufacturer may follow in order to obtain approval or clearance to market products in the U.S. Many of the simpler, Class I, devices are excepted from the premarket review process. Most of these devices raise few, if any, biocompatability issues. The more complex Class II and Class III devices frequently include materials that closely interact with the body. In these cases, biocompatability data can make up a significant portion of the submission. Understanding FDA's concerns regarding a particular biomaterial and its application will enhance the quality of the submission and likely accelerate the review process.
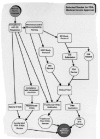
Figure 1
Selected routes for FDA medical device approval.
Table 1
International standards for medical devices.
Medical Device Amendments of 1976
The 1976 Medical Device Amendments to the 1938 Food, Drug and Cosmetic Act (FDCA) gave FDA the responsibility for regulation of medical devices sold in the U.S. Not all devices are regulated in the same manner. Class I devices such as eyeglasses, tooth brushes, scalpels, prosthetic heart valve sizers and stomach pH electrodes are, for the most part, exempt from premarket review by FDA, although they can be subject to some or all quality system (QS) regulation manufacturing and development controls.
Preclinical Testing and IDEs
Preclinical testing is an integral part of the product development process. If required, much of this testing must be completed and reports available, prior to the start of human testing. 510(k) notifications for Class II devices with direct patient contact or contact with the blood supply will contain biocompatability testing data, according to ISO 10993, in order to demonstrate that the risks posed by the new device are “substantially equivalent” to the risks posed by the predicate device. Preclinical testing can also include animal and bench studies to demonstrate the effectiveness of the new device, confirm electrical safety or investigate electromagnetic interference. Although it is often prudent to discuss preclinical testing plans with FDA, no prior approvals are necessary in order to perform this testing. Both the Investigational Device Exemption (IDE)1 and the PreMarket Approval (PMA)2 regulations require that nonclinical laboratory studies be performed under the Good Laboratory Practices (GLP) regulations in 21CFR 58,3 or that an explanation be provided if a study was not performed in accordance with the GLPs. Once preclinical testing has progressed to the appropriate stage, the reports are written, overall summaries of related tests compiled, and overall conclusions drawn. For relatively simple devices, conclusions of substantial equivalence may be drawn from preclinical testing alone. When complex devices are considered, preclinical testing serves as the foundation for more extensive clinical testing.
A relatively small number of 510(k) notifications, perhaps 5%,4 also contain human clinical data. As shown in Figure 3.1, there are two routes available in order to initiate a human clinical trial. Non Significant Risk studies involve devices that pose little risk to the study population and can be initiated after local Institutional Review Board (IRB) review, while Significant Risk studies require submission of an IDE and IRB approval prior to study initiation. Informed Consent must be obtained from all study subjects, regardless of the risk status of the device, unless the emergency use provisions of the IDE regulations are invoked.
An IDE application contains a description of the device, theory of operation, indication for use and method of manufacture. Available preclinical data, including biocompatability information is also included in the IDE. A clinical study protocol, clinical investigator information and a clinical monitoring SOP must also be submitted. If the manufacturer plans to charge for the investigational device, this must be stated in the IDE. FDA permits recovery of research and development costs but will not allow commercialization during the investigation. Once the IDE is received, FDA has 30 days to review the application. The FDA reviewer's main concern is to assure the level of risk incurred by the patient population is appropriate given the benefits of the investigational device. The IDE can be approved, rejected or FDA can ask for additional information before making a final determination. Typically, about 70%5 of IDEs are approved during the first round of review. Once the IDE is approved, the manufacturer and clinical investigators must comply with a wide variety of regulations which are described in 21CFR 812. Approval of an IDE does not imply that marketing approval via the 510(k) or PMA routes is assured. It only signifies that the clinical research does not pose unreasonable risks to the study population. The manufacturer must periodically update FDA regarding the progress of the investigation.
510(k) and PMA Marketing Applications
More than 99% of all the medical devices that FDA reviews in a given year are cleared for marketing through the 510(k) PreMarket Notification process. (510(k) refers to Section 510(k) of the FDCA.) The goal of the process is to demonstrate to FDA that the new device is “substantially equivalent” to a “predicate device” which was already on the U.S. market prior to the May 28, 1976 effective date of the Medical Device Amendments. Historically, FDA has interpreted substantial equivalence in a broad sense. For an electronic device with no direct patient contact, a table containing a point-by-point comparison of key engineering features of the device is often sufficient. Class II devices such as some implants, many catheters, electrocardiograph systems, and ultrasound imaging systems are all included in an intermediate risk category where 510(k) notifications and adherence to FDA Quality system regulations are required. Under the Medical Device Amendments of 1976, the submitter of a 510(k) notification could market a device 90 days after submitting the notification to FDA. As we shall see later in this chapter, the Safe Medical devices Act of 1990 made several significant changes to the 510(k) process.
Approximately 40 medical devices are approved for marketing in the U.S. through the PMA process each year. All PMA devices are Class III. The PMA process is used for devices that cannot be shown to be “substantially equivalent” to a suitable predicate device. This commonly occurs because the device utilizes a novel technology or material, or is intended for a novel indication for use. The goal of the PMA process is for the manufacturer to provide “reasonable assurance” that the new device is safe and effective. This is a higher standard than the 510(k)'s “substantial equivalence” and nearly always requires manufacturers to perform human clinical trials. Biomaterial and biocompatability concerns, as with 510(k)s, are related to the type of device, materials used and the indication for use, rather than the application type. PMA review times generally extend from 9–18 months.
Quality System Regulation Requirements
The Good Manufacturing Practices regulations for medical devices became effective in 1978 and were the subject of a major revision, effective mid 1997. As a result of the revision, the regulation has been renamed the Quality system Regulation.6 The purpose of this regulation is to assure that manufacturers maintain a system that can reliably design, produce, control, install and service medical devices. It is based on the international ISO 9000 standard with revisions to accommodate U.S. legal requirements. The FDA district offices, in collaboration with headquarters personnel, are responsible for enforcing these regulations by sending FDA investigators to inspect medical device manufacturers. FDA actions when noncompiling systems are identified range from documentation on the FDA form 483, warning letters and in rare instances, seizures, injunctions, and civil penalties.
It is the manufacturer's responsibility to assure that the device described in the 510(k) notification or PMA is the device actually being produced. While some changes in manufacturing methodology and some substitutions of materials can be accomplished without prior FDA review,7 the Quality system Regulation provides for controls that, among other functions, controls for changes in materials and production methods. Before any change is implemented, the manufacturer must consider if a new 510(k) notification or a PMA supplement is necessary and document that review. The Quality systems Regulations contain many other provisions, most not directly related to biomaterials. The reader is referred to the regulation and various FDA guidance documents for further information.6,8,9
Safe Medical Devices Act of 1990
The Safe Medical devices Act of 1990 (SMDA) [http://www.fda.gov/cdrh/ode/655.pdf] modified the 1976 Medical Device Amendments in many key areas. SMDA permits applicants to utilize any device that has been cleared through the 510(k) process as a predicate device. It is no longer necessary to select a device that was on the U.S. market prior to May 28, 1976. With regard to PMAs, SMDA makes advisory panel review optional so that the panels can focus on the most innovative technology. SMDA also contains the “four of a kind” rule which could permit submitters of the fourth PMA for a particular device to reference data submitted by the first three applicants. These data, could potentially, contain biocompatability information. As with several other provisions of this law, FDA has not drafted enabling regulations for this section and it is not currently being used. Another provision of SMDA gives FDA the ability to regulate the design process for medical devices. After considerable public comment, FDA published final regulations, which implement this provision and significantly revise all the manufacturing regulations. These changes are discussed in the following section.
Design Controls and Design History Files
The Safe Medical devices Act of 1990 (SMDA), for the first time, gave FDA the authority to regulate the design process for medical devices. Up until this time FDA investigators were limited to the examination of production and quality control records and could not review research and development documentation. The design control regulations are part of the Quality systems regulation which was fully implemented in mid-1998.
The regulation requires each manufacturer of Class II and Class III devices to implement design controls. These controls consist of a development plan applicable to the development of all new products and a Design History File (DHF) where these activities are documented. These controls are intended to regulate the design process for specific medical devices, not basic research such as the formulation and testing of novel biomaterials. A specific example may serve to clarify this distinction. If a firm is developing a polymer that may have applications for a wide variety of devices that contact the circulatory system and have other applications outside of the medical device industry, then, design controls would not apply to the development of the polymer. Once the development process for the polymer has been completed and a device manufacturer obtains supplies of this material for evaluation in a particular device, then the evaluation process is regulated under design controls. The evaluation process for biomaterials including engineering studies for functionality, durability, biocompatability, and manufacturability would all be described in the DHF. Once a specific polymer has been chosen, the device manufacturer and polymer manufacturer must agree upon specifications. The regulation recommends that the device manufacturer obtain agreement from the polymer supplier that they will receive advance notification if changes are made to the polymer manufacturing process or raw materials. The polymer specification becomes part of both the Device Master Record and the Design History File. The key sections of the DHF are described below:
Design Input: Initial specifications and requirements for the device are described here. The needs of both the device user and the patient must be considered. The intended use of the device should also be included. The material that appears in the first version of this section does not have to be final and complete, but a mechanism must be in place to update the section. This section must be reviewed (signed and dated) by an appropriate individual at proper intervals.
Design Output: This section contains the results of the design effort and may include various engineering analyses as well as biocompatability data. The important feature here is that results of the design effort be quantifiable and meet or exceed previously determined specifications. It is important that all relevant design outputs be identified and included in this analysis. As with the design input section, this section must be reviewed prior to release and the reviewer's signature and date must appear on the approval page.
Design Review: A formal design review needs to occur according to a master schedule during the design process. The review team must include representatives from each functional area responsible for design activities, additional specialists, if necessary, and one individual not directly involved in the phase of the design process under review. Decisions regarding the replacement of one biomaterial with another are often made at these meetings, once the relevant data are examined. The Design history File (DHF) must document names of the participants of the design review, the version of the design reviewed, the date of the review and the results of the review.
Design Verification: This process confirms that the design output meets the specifications of the design input. The DHF must document the version of the design, verification method, date of verification and the individuals participating in the verification.
Design Validation: The device design must be validated using initial production material, not prototype units. The purpose of the validation is to ensure that the device functions according to predefined user requirements and intended uses. Devices are tested under actual or simulated use conditions. Biomaterials/biocompatability concerns, if applicable, must be addressed during this process. The entire validation process must be fully documented in the DHF.
Design Transfer: Procedures must be in place to assure that the design is correctly translated into production specifications. Raw material specifications, storage conditions, and process parameters are key biomaterials issues here.
Design Changes: A change control system must be in place. This system must identify and document all changes to the device, materials and/or production methodology.
Design History File: This document must be prepared for each type of device as mentioned in the preceding paragraphs. The DHF can either be a collection of the actual documents or an index listing storage locations for those documents.
A much more detailed description of these requirements may be found in FDA guidance documents8,9 and independently published sources.10
The FDA Modernization Act of 1997 (FDAMA97)
This wide-ranging law [http://www.fda.gov/cdrh/modact/modern.html] adds many features to the medical device regulatory landscape. One provision, Section 216 will permit FDA to use biocompatability data submitted in one PMA when reviewing another PMA, once the original data is at least six years old. There are also several provisions that detail various types of meetings that sponsors of IDEs or PMAs can request with FDA. This law also formalizes FDA's reliance on compliance with recognized national and international standards to expedite the 510(k) review process.
FDA Regulation of Biomaterials
The Tripartite Agreement11
This agreement between the U.S., Canada, and the UK described a common view for the assessment of biocompatability for polymeric materials [http://www.fda.gov/cdrh/g87-1.html]. FDA officially adopted this agreement in April of 1987, and it was in effect until superseded by ISO 10993 in July of 1995. The key element to the Tripartite Agreement is a matrix that categorizes devices according to type of body contact, such as external devices on intact skin to internal devices in contact with blood; and according to the duration of contact. Duration of contact is categorized as transient (<5 minutes), short term (5 minutes–29 days), and long term (>29 days). Eleven tests were specified for possible inclusion in a testing program depending on the indications for use of the device. As the complexity of biomaterials technology increased and the types of biocompatability assays proliferated, FDA testing requirements strayed further from the program specified in this agreement. In addition, this agreement specifically relates to polymeric biomaterials and was never intended to address concerns for other classes of biomaterials. An April 1993 FDA guidance document provided industry with a more detailed explanation of some of the Tripartite Agreement's provisions but made no attempt to change any of them.
ISO 1099312
The International Organization for Standardization's Technical Committee 194, comprised of members from the European Committee for Standardization (CEN) and the U.S.-based Association for the Advancement of Medical Instrumentation (AAMI), produced the ISO 10993 standard “Biological Evaluation of Medical devices Part 1: Evaluation and Testing” in 1994. FDA issued a memo in 1995 [http://www.fda.gov/cdrh/g951.html] that made compliance with a modified version of ISO 10993 mandatory after July 1, 1995. This document contains two tables (Refer to Figs. 3.2 and 3.3) and a flow chart (Refer to Fig. 3.4) to assist manufacturers in determining the appropriate testing plan for their device. Additional modifications to the ISO 10993 matrix which specifically relate to immunotoxicity were announced by FDA in the Immunotoxicity Testing Guidance dated May 6, 1999 [http://www.fda.gov/cdrh/ost/ostggp/immunotox.pdf]. The FDA modified ISO 10993 testing matrix (refer to Fig. 3.2 and 3.3) in several specific areas.13,14 First, for surface devices contacting mucosal membranes, acute and chronic toxicology testing along with implantation testing is expected in most cases. Second, for externally communicating devices, those that contact tissue or bone are often expected to include irritation, systemic toxicology, subchronic and chronic toxicology testing. Another important feature of the ISO 10993 standard that FDA accepted is a change in the time periods for the three exposure categories. The ISO 10993 matrix uses the contact terms “limited “ (>24 hours), “prolonged” (24 hours to 30 days), and permanent (>30 days). The most important change here is that the limited term extends to <24 hours, rather than the Tripartite's <5 minutes. This permits a greater number of devices to be assessed under the simpler “limited” requirements. In documentation that accompanies the testing matrix, FDA makes the point that test selection should be based upon sound scientific reasoning. Specific biomaterials utilized for particular indications may require more or less testing than the matrix specifies. Additional, more specialized tests may also be necessary. It should be kept in mind that samples of devices used for this testing should resemble the actual marketed product as closely as possible. Devices sold as sterile should be sterilized using the method that will be used for commercial production prior to biocompatability testing. In cases where aging may affect biocompatability, devices should be aged to the end of their shelf life before testing. Organizations with internal biocompatability experts can often make these judgments independently, while other organizations frequently enlist the assistance of outside consultants and scientists at contract testing laboratories. Contacts with FDA prior to embarking on a time-consuming and expensive testing program are also a prudent course of action.
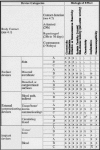
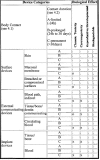
Figure 3
Supplementary evaluation tests for consideration; X = ISO Evaluation tests for consideration; O = Additional Tests which may be applicable; *See Figure 3.2 for initial evaluation tests.
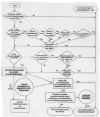
Figure 4
Biocompatibility flow chart for the selection of toxicity tests for 510(k)s.
Table 2
Principles for the evaluation of medical devices*.
FDA Product Specific Guidance Documents
FDA's Center for Devices and Radiological Health (CDRH) publishes a wide variety of guidance documents to assist medical device manufacturers in determining CDRH expectations for data in marketing applications. The three best sources for obtaining any of these documents are the FDA itself. FDA maintains a World-Wide Web Site at [http://www.fda.gov] which has extensive material from CDRH. The Division of Small Manufacturer's Assistance (DSMA), at CDRH, also maintains a Facts-On-Demand service at (800) 899-0381 (in USA) or (301) 827-0100 (international). The user calls this number from a touch-tone telephone and follows the automated instructions to receive a lengthy index of documents via FAX. Once the index is available, guidance documents can be ordered with a second telephone call. Finally, DSMA can be contacted directly at (800) 638-2041 (in USA) or (301) 443-6597 (international). A wide variety of private organizations provide FDA documentation for a fee. These organizations include newsletter publishers and CD-ROM publishers.
Table 3
Paradigm for biological evaluation and risk assessment of medical devices.
Proper Use of Guidance Documents
Guidance documents do not carry the force of law. They describe one way that industry can follow to meet FDA expectations. There may very well be many other equally acceptable paths that could be followed. When a device has one or more unique characteristics, it becomes increasingly likely that the manufacturer will need to deviate from the path described in the guidance document. These deviations should be for sound scientific reasons. In many cases, manufacturers will contact knowledgeable reviewers within the Office of Device Evaluation to obtain concurrence.
Biocompatibility Testing Program Examples
In the real world, it is difficult for any guidance document or international standard to anticipate all the issues that may arise during the device development process. Device developers must utilize sound professional engineering, scientific, medical, and regulatory judgment when planning a project. In some cases, going beyond the tests specified in a standard or guidance document may make both good scientific and business sense. In other cases, specific tests may be avoided if they are not scientifically justified or if the relevant data already exist. The following section contains examples of how two products that required biocompatability testing were handled. In the first case, a complicated product, used to support life, was subjected to extensive testing. The second case describes a less critical device where much of the data were obtained from previous testing programs.
Example 1: A Membrane Oxygenator
Membrane oxygenators serve as a patient's lungs when the patient is undergoing a cardiopulmonary bypass procedure. Blood flows out of the patient, into the oxygenator where carbon dioxide is removed and oxygen is added. Clearly, this device plays a critical role in keeping the patient alive. Although FDA has proposed reclassifying this device from Class III down to Class II, it currently remains a Class III 510(k) device. In January of 2000, FDA issued a revised guidance document, “Guidance for Cardiopulmonary Bypass Oxygenators 510(k) submissions” <http://www.fda.gov/cdrh/ode/1361.pdf> which provides very detailed descriptions of the biocompatability and functional testing expected for this type of device. In this particular example, the manufacturer has significantly redesigned the oxygenator, incorporating a variety of design, material, and manufacturing changes.
ISO 10993 classifies this device as limited contact duration (less than 24 hours), externally communicating and circulating blood path contact. Therefore, referring to the Figure 3.2, the following tests are necessary:
Cytotoxicity, Sensitization, Irritation, Acute Systemic Toxicity, Genotoxicity and Hemocompatibility
The guidance document adds that the devices that are tested must be representative of actual production lots, utilizing the same design, materials, assembly, and packaging procedures. FDA also states that the devices should be subjected to shipping tests and aged under real-time or accelerated conditions to simulate the full shelf life prior to beginning testing. The manufacturer must also demonstrate that oxygenator materials are compatible with anesthetic agents and medications commonly added to the blood of bypass patients. These expectations are part of the ISO 10993 standard. This is an example of a case where it is important to refer to current guidance documents and/or contact a knowledgeable FDA reviewer in order to confirm that biocompatability testing plans are appropriate. This is especially true if the oxygenator contains any materials that are new to FDA or have not previously been used in this type of device.
Example 2: A skin electrode for monitoring cutaneous electrical activity
This electrode is used to acquire an electrical signal that is further processed to generate diagnostic information. A 510(k) for this Class II device had previously been cleared, but included electrodes that could only be used for a few hours. The manufacturer resourced the electrode from another supplier. The new electrode could be used for not more than 24 hours. In this case, the new electrode had already been cleared for use with a therapeutic medical device, so FDA was already familiar with product, although not with its application to this particular indication. This is an especially important point. Whenever one makes regulatory judgements on a medical device, the indication for use must be clearly stated and carefully considered. A relatively small biocompatability testing program may be entirely appropriate for a device used for one indication for use, but completely inadequate for the same device or material if it is used for another, more critical indication. In this case, both indications involved placing the electrode over intact skin, so, much of the data previously gathered for the original indication of the new electrode remained applicable for its new indication. Understanding this permitted the diagnostic device manufacturer to save time and money by requesting that the electrode manufacturer provide copies of these reports so that the diagnostic device manufacturer could include them in the 510(k) Notification. Alternately, the electrode manufacturer could include these reports in a Device Master File (MAF) that is filed with FDA. The electrode manufacturer then provides its customers with letters authorizing them to reference the data in the MAF. The customers do not have access to these reports, so confidentiality is preserved. The tests included in the 510(k) were Cytotoxicity, Sensitization, Irritation and Acute Systemic Toxicity.
Even though no new biocompatability testing was required in this case, other preclinical testing was necessary. As part of the Design Control process, the diagnostic device manufacturer needed to assure that the electrical characteristics of the system were within limits during the entire recommended use period. Shelf life was another issue that required testing. The therapeutic indication utilized quite different electrical characteristics than the diagnostic indication, so the testing conducted for the original shelf life contained quite different test methods and specifications. Because of this, even though the packaging remained the same, the shelf life study was repeated using entirely different test methodology and specifications.
FDA Regulation of the Biomaterials Testing Process
In the previous sections FDA data requirements have been discussed. We have answered the question, “What tests do we need to conduct?” In this section, we address the question, “How is the recommended testing conducted?”
Description of GLP Regulations
Safety testing data such as biocompatability data are vital components to the marketing application. In order to assure that these data actually represent the true experimental results, FDA regulates safety testing with the Good Laboratory Practices (GLP) regulations (21CFR 58).3 The regulation requires documented controls for the regulation of organization and personnel, facilities, equipment, testing facilities operation, test and control articles and study protocols. One key provision of the regulation is the establishment of a Quality Control Unit (QAU) which periodically and independently monitors GLP-regulated studies. Just as FDA investigators can inspect manufacturer's facilities, GLP laboratory facilities are also subject to on-site inspections by FDA. When choosing a contract testing laboratory it is important to select one with a good compliance history. In extreme cases, FDA has refused to review data from a particular laboratory because of a high level of noncompliance with the GLP regulations. Careful selection of contract testing laboratories, periodic auditing and maintenance of open communication channels will reduce the likelihood of compliance issues.
Examples of GLP Noncompliance Taken From FDA Warning Letters
From time to time, inspections of GLP-regulated testing laboratories result in the issuance of a warning letter to laboratory management detailing apparently serious violations of regulations. These publicly available documents list the major violations found and may include sanctions against the laboratory including disallowal of study data. One must always bear in mind that warning letters are written to prove a point and additional data may change initial FDA conclusions. The following excerpts from FDA warning letters, for medical device studies, serve to illustrate the GLP compliance areas that FDA considers important.
The Quality Assurance Unit
“Failure to establish a Quality Assurance Unit to monitor each nonclinical study and to assure that the facilities, equipment, personnel, methods, practices, records, and controls are in conformance with applicable regulations as required in 21 CFR 58.29(b), 58.31, and 58.35. The testing facility management failed to assure that:
- the facility has an impartial and independent Quality Assurance Unit (QAU);
- test articles, study documents, raw data, and specimens are maintained in accordance with 21 CFR 58.190;
- all study personnel are knowledgeable of their responsibilities;
- deviations from these regulations were corrected and documentation of the corrections was maintained...
The Quality Assurance Unit failed to have a master schedule sheet in accordance with 21 CFR 58.35(b)(1).”
Training for Study Personnel
“Failure to have documentation to show that each individual participating or auditing this study had received any training in GLP's or that this facility provided GLP training as required by 21 CFR 58.29(a)(b).
The testing facility management failed to assure that the personnel followed the current written standard operating procedures entitled, “[purged text] Biomedical Research Standard Operating Procedures,” dated September 30, 1997. For example, in Study [purged word] the testing facility failed to have current training summaries for each person involved in the study and documentation defining the critical phases and associated QAU audit schedule.”
SOPs for Study Activities
“Failure to have written standard operating procedures established, during the period of this study, e.g., animal room preparation, test systems observations, data handling, storage and retrieval, laboratory test and the housing, feeding, handling and care of the animals as required by 21 CFR 58.81(b)(1–12).
There were no written procedures available for the receipt, security, storage, maintenance, disposition, or inventory control of test articles; for laboratory tests, such as blood chemistry, urinalysis, and histological analysis; for specimen collection and labeling requirements; for histopathology; and for specimen archiving.”
Study Protocol
“Protocol was inadequate in that it did not clearly indicate all methods for conduct of the study as follows: (a) the model number of the control leads was not stated; the source of the dogs was not identified; the description of the diet was not included and did not address interfering contaminants; study methods to be used were not described, 21 CFR 58.120.
You failed to include in the final report the statistical methods for analyzing the data; the source of the dogs and the location of where the calibration and maintenance records for the test equipment are to be stored as required by 21 CFR 58.185.
Failure to have written documentation as required by 21 CFR 58.90(c) to show that using dog No. 714, on a previous study using a J lead and a ventricular lead, would not interfere with the dog's heart accepting the control lead.
Failure to maintain records as required by 21 CFR 58.130(e). For example: records not dated or initialed; raw data not recorded; changes not properly documented; records not recorded in ink; and records do not explain why some dogs had two sets of data or partial data collected on the same day, dog Nos. 714, 792, 745.
Specimens were not identified in a manner to preclude error in the recording of data. For example, specimens from Study [purged word] were labeled [purged text] with no other information to correlate specimens to test systems.
All data generated during the conduct of these nonclinical laboratory studies were not prepared and documented as required by this regulation. In Study [purged word] inappropriate changes were noted in data entries, such as obscuring entries in animal care charts and other records. The reasons for these changes were not documented. Also, some records associated with the study were not dated and signed by the person responsible for direct data input.”
Key GLP Compliance Points to Consider When Reviewing GLP Studies
As the points included above illustrate, documentation is a key component of all GLP systems. All tasks required by regulations must be documented or FDA will assume that they were not performed.
The Quality Assurance Unit (QAU) receives a great deal of attention during an FDA inspection as the first few items in this list indicate. QAU personnel must be trained, have adequate time and be knowledgeable enough to perform their review functions assuring that study protocols and laboratory SOPs are followed. There must be enough people in the QAU so that each study can be controlled in an adequate manner. The Standard Operating Procedures (SOPs) for the QAU must describe an appropriate QA system in a sufficient level of detail. Audit reports and logs must also be carefully maintained. When testing follows internationally recognized standards, the laboratory must have current copies of those standards, and their procedures must accurately reflect requirements in the standards.
Study protocols must be carefully drafted to fully describe the research effort, all test article used, number and types of animals used and their diet and all other related study procedures. It is especially important in surgical implant studies to carefully describe the location and method for placing the device into position.
Data processing and data management usually occur during or at the end of most studies and can sometimes be given less attention than the portions of the study protocol that directly relate to handling or observation of the device. No matter how well these earlier parts of the study are executed, the raw data only gain full credibility after the analysis phase of the study. These concerns begin when raw data are initially entered into study notebooks or electronic files and continue when data are placed into databases for statistical analysis.
General laboratory systems are also regulated. In order to generate credible data, laboratory equipment from electronic balances to spectrophotometers must be properly maintained and regularly calibrated. Of course, all these activities must be documented.
Validation of laboratory methods and equipment can often become an issue with GLP studies. Assays performed exactly as they are specified in the USP or standard AOAC methods do not need to be validated. If, on the other hand, an assay from the research literature is utilized, or a special purpose assay is developed, then validation becomes necessary. The validation process provides assurance that the assay accurately and reproducibly measures the parameter of interest under all expected conditions and using defined test equipment.15–17 Of course, there must also be assurance that the parameter measured does indeed reflect the condition monitored. If the analysis method should change, revalidation may be necessary. Calibration, as mentioned above, is an important part of laboratory operations, but it is not a substitute for validation. A properly calibrated instrument can generate highly accurate, but irrelevant, data if the overall assay method is flawed.
Electronic data capture, analysis and storage is becoming widespread in all areas of biomaterials testing. These types of software offer analytical capabilities that were previously unavailable and save considerable quantities of time. As with assay methodology, software that manipulates data must be validated. Often, vendors of software specifically designed for the GLP environment will perform validations and have a mechanism in place to make the results available to regulatory authorities, if requested. Software which is well known to the regulators such as Lotus 1-2-3 or Excel do not, in general, need to be validated. However, if a researcher writes a macro using a spreadsheet, the user-defined macro must be validated to assure that it processed data in the expected manner.
The Federal Register Notices can be found on [http://www.access.gpo.gov/su_docs/aces/aces140.html]
Electronic data storage presents other challenges. Well run paper based systems provide for audit trails when data must be corrected. These notations enable a reviewer to immediately learn who made the change, when it was made and what the previous value was. This audit trail capability must be present in electronic systems. The system must be validated to assure that data cannot be changed with an audit trail being created and that all other data remains undisturbed. System security issues such as assuring that only authorized personnel have data modification privileges must also be validated. The FDA Electronic Signature Regulation (21CFR11) contains the regulatory requirements for these systems. Electronic data management systems can save enormous quantities of time in the testing lab; however, measures must be take to assure that they perform only as intended.
Conclusion
Biomaterials involve a wide variety of regulatory issues throughout the product life cycle. Recent changes in regulations have focused an increased level of attention on the design phase of the device development process. This attention originates both from the U.S. FDA, in the form of the Quality system Regulations, and internationally from the ISO 10993 standards. Awareness of regulatory requirements and informal expectations early in the planning process can speed the entire development process, allowing the general population rapid access to innovative technology.
Acknowledgment
The author wishes to thank Ms. Angela Rogers, Regulatory Associate II, PAREXEL International Corp., San Diego, CA for her kind assistance with this manuscript.
References
- 1.
- Food and Drug Administration Investigational Device Exemption Regulation, 21CFR 812 April 1, 2001 .
- 2.
- Food and Drug Administration PreMarket Approval Regulation,21CFR 814 April 1,2001 .
- 3.
- Food and Drug Administration Good Laboratory Practices Regulation, 21CFR 58 April 1,2001 .
- 4.
- Food and Drug Administration Financial Disclosure by Clinical Investigators, Federal Register February 2,199863(251):72171–72181. [PubMed: 10344800]
- 5.
- Food and Drug Administration Center for Devices and Radiologic Health, Office of Device Evaluation Fiscal Year 1998 Annual Report,1999. [http://www
.fda.gov/cdrh/ode/annrp298.pdf] - 6.
- Food and Drug Administration Quality System Regulation, 21CFR April1999820.
- 7.
- Food and Drug Administration Center for Devices and Radiologic Health, Office of Device Evaluation,Deciding When to Submit a 510(k) for a Change to an Existing Device January 10,1997[http://www
.fda.gov/cdrh/ode/510kmod.pdf] - 8.
- Food and Drug Administration Design Control Guidance for Medical Device Manufacturers, Federal Register March199661(58):12075–12076.
- 9.
- Food and Drug Administration Do It By Design; an Introduction to Human Factors in Medical devices Federal Register March199661(58):12075–12076.
- 10.
- Vercimak Sutton C, DeSain Meeting GMP and ISO 9001 Expectations for Product Development PAREXEL International Corp.,1996[http://38
.15.21.244/products /books/meeting.html] [PMC free article: PMC172907] - 11.
- Food and Drug Administration General Program Memorandum #87-1, Tripartite Biocompatability Guidance April 24,1987 .
- 12.
- International Organization for Standardization. Biological Safety Testing for Medical Devices, DIS 10993. 1993
- 13.
- Food and Drug Administration General Program Memorandum #G95-1, Use of International Standard ISO-10993, “Biological Evaluation of Medical Devices Part 1: Evaluation and Testing May 1,1995http://www
.fda.gov/cdrh/g951.html. - 14.
- Food and Drug Administration Draft Immunotoxicity Testing Framework October 21,1996 .
- 15.
- Food and Drug Administration Federal Register, International Conference on Harmonisation; Guideline on Validation of Analytical Procedures: Definitions and Terminology Availability, March 1,199560(40):11259–11262. The Federal Register Notices can be found on [http:// www.access.gpo.gov/su_docs/aces/aces140.html]
- 16.
- Food and Drug Administration Federal Register, International Conference on Harmonisation; Draft Guideline on the Validation of Analytical Procedures: Methodology Availability, March 7,199661(46):9315–9319. The Federal Register Notices can be found on [http://www
.access.gpo .gov/su_docs/aces/aces140.html] - 17.
- Food and Drug Administration Center for Drugs and Biologics and Center for Devices and Radiologic Health, Guideline on General Principles of Process Validation May1987.
- PubMedLinks to PubMed
- Regulation of Medical Devices - Madame Curie Bioscience DatabaseRegulation of Medical Devices - Madame Curie Bioscience Database
- Calreticulin in Cytotoxic Lymphocyte-Mediated Cytotoxicity - Madame Curie Biosci...Calreticulin in Cytotoxic Lymphocyte-Mediated Cytotoxicity - Madame Curie Bioscience Database
- Predicting the Spread of a Transgene in African Malaria Vector Populations: Curr...Predicting the Spread of a Transgene in African Malaria Vector Populations: Current Knowledge and Limitations - Madame Curie Bioscience Database
- Coronin 1 in Innate Immunity - Madame Curie Bioscience DatabaseCoronin 1 in Innate Immunity - Madame Curie Bioscience Database
- CTLA-4 in Type 1 Diabetes Mellitus - Madame Curie Bioscience DatabaseCTLA-4 in Type 1 Diabetes Mellitus - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...