

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Mitogen-Dependent and -Independent Phases of the Cell Cycle
Production of two cells from one requires duplication of all molecules and organelles that compose each cell. DNA does not duplicate throughout the cycle but only during several hours in S-phase (Fig. 1). During S-phase, the DNAs of the 100,000 genes located on 23 pairs (in humans) of chromosomes are replicated, each in a timely fashion. This requires the ordered assembly of many proteins at the origins of DNA replication to form a competent, pre-replicative chromosomal state. All eukaryotes use similar proteins to license replication origins.1 After formation, competent origins are activated by cyclin-dependent kinases. Once started, DNA replication must be finished. Therefore, extracellular signals such as growth factors (GF) must not and do not control S-phase progression. Although S-phase is independent of growth factors, massive DNA damage or deprivation of nucleotides forces a cell to be arrested in S-phase, but such arrest is usually accompanied by cell death.
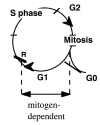
Figure 1
Mitogen-dependent and -independent phases of the cell cycle
After successful completion of DNA synthesis, cells enter G2 phase in preparation for mitosis. Protein kinases that are activated in G2 phase prevent re-replication of the DNA. Quiescent mammalian cells are usually diploid, so after DNA duplication a cell must divide. Therefore, control by growth factors is also unnecessary in the G2 phase. This is a time to check the internal signaling with events like DNA damage.
At the onset of mitosis, cyclin B/cdc2 phosphorylates laminin, a nuclear membrane protein, thus dissolving the nuclear envelope. In metaphase of mitosis, condensing of chromosomes associates with forming of mitotic spindles. The endoplasmic reticulum and the Golgi complex break down into small vesicles. During mitosis, chromosomes are condensed, proteins hyperphosphorylated and transcription and biosynthesis are reduced or absent. Progression of mitosis is determined by monitoring of the microtubule function to ensure chromosomal fidelity. External regulation by growth factors would be detrimental for a cell, and consequently, mitosis is a growth factor-independent phase of the cell cycle. In anaphase of mitosis the two sets of chromosomes are pulled apart. During telophase and cytokinesis the preceding fragmentation process of intercellular membranes is reversed. The nuclear envelope reappears around the chromosomes, endoplasmic reticulum is rebuilt and cytoplasmic microtubules reassemble. The transcription and biosynthetic functions of the cell are normalized and intracellular membrane traffic is resumed.2 As cells exit mitosis, the cell cycle is reset, allowing the establishment of a new, competent replication state in G1 phase (Fig. 1). Therefore, as the logic dictates, G1 phase is the only part of the cell cycle that can and must be growth factor-dependent.
The Restriction Point
The major regulatory events leading to proliferation occur in the G1 phase of the cell cycle. In vivo as well as in cell culture, most quiescent cells have a G1 DNA content. The growth of normal cells in culture is regulated by complex interactions between growth factors, cell density, and attachment to substrate.3 Growth factors are necessary to initiate and maintain the transition through G1 phase leading to S-phase. In normal cells, withdrawal of growth factors prevents the onset of S phase. The point at G1 at which commitment occurs and the cell no longer requires growth factors to complete the cell cycle has been termed the restriction (R) point.4 The R point has been temporally mapped at 2–3 hours prior to the onset of DNA synthesis.5 Once beyond the R point, or point of no return, cells are committed to DNA synthesis and they no longer require the extracellular growth factors during the remainder of the cell cycle. Transition of the restriction point was proposed to be determined by accumulation of a labile protein.4
In Search of Mediators of the Restriction Point
In 1974, comparison of growth of cancer and normal cells revealed that restriction point is lost in cancer.4,6 Identification of the R point provided the difference between normal and cancer cell cycles. But what is the biochemical nature of the point of no return? R-protein is a functionally short-lived (labile) regulatory protein, whose synthesis is sensitive to growth factors and needs to accumulate to a critical amount before a cell can pass the R point and proceed towards DNA synthesis.5 When this question had been asked, oncogenes and signal transduction pathways were not known. Many of the later discovered proteins were evaluated for meeting the criteria of the R-protein.
Search for proteins that are differentially expressed in the late G1 phase versus resting cells identified numerous proteins.7 In order to effectively identify and isolate the late G1-expressing genes, Liang and Pardee developed a method known as differential display.8 The differential display detects mRNA species that are different between sets of mammalian cells, permitting their recovery and cloning of the corresponding cDNAs. Differential display allows one to separate and clone individual mRNAs by PCR. The key element is to use a set of oligonucleotide primers, one being anchored to the polyadenylate tail of a subset of mRNAs, the other being short and arbitrary in sequence so that it anneals at different positions relative to the first primer. The mRNA subpopulations defined by these primer pairs were amplified after reverse transcription and resolved on a DNA sequencing gel. When multiple primer sets were used, reproducible patterns of amplified complementary DNA fragments were obtained that showed strong dependence on sequence specificity of either primer. Many late-G1-expressing genes were isolated by differential display.9 However, R-protein is also accumulated by the mechanism of protein stabilization, but protein stabilization cannot be detected by the differential display method.
R-protein is a labile protein which is induced, stabilized and accumulated in response to growth factors leading to growth factor independence. The discovery of G1-phase cyclins (D and E) was an important breakthrough. Cyclin D1, a labile nuclear protein, accumulates following growth factor stimulation.10–15 It is commonly overexpressed in human cancer. Cyclin E was discovered in the course of a screen for human complementary DNAs that rescue a deficiency of G1 cyclin function in budding yeast. The amounts of both the cyclin E protein and an associated protein kinase activity (cyclin E-Cdk2 complexes) fluctuated periodically through the human cell cycle; both were maximal in late G1 and early S phases.16 In 1993, Pardee and his colleagues suggested that the molecular basis of the R-protein could be production of cyclin E17,18 because cyclins satisfy all of the criteria for the R-protein, which includes a late G1-phase increase, a delay of appearance after inhibition of protein synthesis in nontransformed cells, and a faster recovery in transformed cells.
Cyclins: From Mitogen Signaling to the Restriction Point
The progression from one phase to another is driven by enzymes named cyclin-dependent kinases (CDK). Their activators, the cyclins, however, are unstable and are “cycling” during the cell cycle.19 Thus, cyclins control the activities of cyclin-dependent protein kinases (CDKs) and play a key role in cell cycle regulation. As cells proceed through the cycle, four major cyclins are produced sequentially (D, E, A, and B), and they activate CDKs (Fig. 2). B-type cyclins associate with p34cdc2 to trigger mitosis. Progression through S phase requires cyclin A, presumably in association with p33cdk2. Cyclins D and E drive a cell into S-phase. The three D-type cyclins (cyclins D1, D2, D3) are very similar but they share very little homology with cyclin E. During cell cycle progression, D cyclins start accumulating at mid-G1, whereas cyclin E appears later, just prior to the G1/S transition.
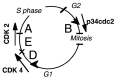
Figure 2
Cyclin-dependent kinases (CDKs) and cyclins
Mitogen-dependent progression through G1 phase is mediated by induction of the cyclin D family. Growth factors regulate cyclin D1 by four simultaneous mechanisms (Fig. 3): (i) transcriptional induction, (ii) stabilization of the cyclin D protein, (iii) its translocation to the nucleus, and (iv) assembly with their catalytic partners, CDK-4 and CDK-6.14,15 The promoters of D-type cyclins respond to a variety of mitogen-activated signals such as Ras, β-catenin-Tcf/LEF pathways.13,20 The transcriptional induction of cyclin D1 by growth factors is dependent on the Ras/Raf-1/Mek/ ERK pathway.21–23 However, cyclin D1 protein is rapidly degraded and therefore it has a short half-life. It is important to emphasize that not only cyclin D and E but other proteins that regulate or affect the restriction point can be increased by protein stabilization. Normally, wt p53 is rapidly degraded by the proteasome, but it is accumulated after DNA damage, almost exclusively by protein stabilization. Similarly the CDK inhibitors p27 and p21, as well as cyclin E and the E2F-1 transcription factor, are degraded by the proteasome.24–28 Cyclin D1 turnover is governed by ubiquitination and proteasomal degradation, which are stimulated by cyclin D1 phosphorylation on threonine 286.29 This phosphorylation can be inhibited by signaling through a pathway that sequentially involves Ras, phosphatidylinositol-3-OH kinase (PI3K), and protein kinase B (Akt). Thus, turnover of cyclin D1, like its assembly, is mitogen dependent.30 Nuclear localization and assembly of newly synthesized cyclin D1 with CDK-4 is also GF-dependent.31
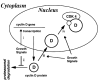
Figure 3
Cyclins D are growth factor sensors. Expression of cyclins D, their nuclear location, stability, and their assembly with CDK4 and CDK6 into active kinase complexes are regulated by growth factors
After active cyclin D-dependent kinase is assembled in the nucleus, it phosphorylates the retinoblastoma (Rb) protein,32 preventing its binding to E2F thus activating E2F-1 mediated transcription.33 The E2F transcription factor activates genes whose products are involved in nucleotide metabolism and DNA synthesis.34 For understanding the restriction point, it is important to emphasize that E2F transactivates cyclins E and A.35 Cyclin E enters into a complex with CDK2 and collaborates with cyclin D-CDKs to complete Rb phosphorylation. Cyclin E-CDK has a broader specificity than cyclin D-CDK. For example, cyclin E-CDK phosphorylates the CDK inhibitor, p27, causing its degradation.36 The activity of cyclin E-CDK is inhibited by p21, p27, and p57.15 In contrast, these CDK inhibitors, at least at low concentrations, co-activate cyclin D-CDKs (Fig. 4). Another class of CDK inhibitors, p15, p16, and p18, specifically inhibit cyclin D-CDKs.
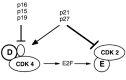
Figure 4
Effects of “CDK inhibitors”
The Restriction Point: A Knot of Mitogen and Inhibitory Signaling
Growth factors activate their receptors and other tyrosine kinases, Ras, and mitogen-activated pathways culminating in transcriptional induction of numerous genes, including protooncogenes. Similarly, genes coding many growth factors, receptors, receptor-associated proteins and kinases are all protooncogenes.37,38
One of the greatest breakthroughs in the understanding of the regulation of cell cycle is the connection of mitogenic stimulation to the cell cycle machinery. Expression of cyclins D, their nuclear location, stability, and their assembly with CDK-4 and CDK-6 into active kinase complexes are regulated by growth factors.15 Therefore, cyclins D are growth factor sensors. In turn, the ability of cyclin D-dependent kinases to trigger phosphorylation of Rb in the mid- to late-G1 phase of the cell cycle makes inactivation of the growth suppressive function of Rb a mitogen-dependent step. Rb participates in controlling the G1/S-phase transition, by binding E2F transcription factor family members (Fig. 5). Absence of functional Rb protein is sufficient for S-phase entry under growth-limiting conditions.39 E2F-1 accumulation bypasses a G1 arrest resulting from the inhibition of G1 cyclin-dependent kinase activity.40–42 GF-dependency ends with the phosphorylation of Rb, enabling the cells to pass through the restriction point at the end of mid-G1 phase and to commit to completing the remaining phases of the growth cycle.43,44 Given their ability to inactivate Rb, do cyclins D meet the criteria of R-protein? Not completely. Their ability to act as growth factor sensors depends not only on their rapid induction by mitogens but also on their protein instability, which ensures their precipitous degradation in cells deprived of growth factors. The fact that D-type cyclins are labile proteins guarantees that the subunit pool shrinks rapidly when cells are deprived of mitogens. Induction of cyclin D1 is not sufficient for the transition from quiescence through G1 into S phase.20
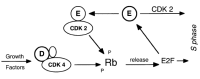
Figure 5
The restriction knot
Cyclin E in complex with CDK2 is downstream from cyclin D-CDK-4. Cyclin E/CDK2 completes Rb phosphorylation (Fig. 5). This shift from cyclin D-CDK4 to cyclin E-CDK2 accounts for the loss of dependency from growth factor. Precisely, the restriction point lies between cyclins D and E (Fig. 5). As emphasized by Kohn, the mammalian G1/S cell cycle phase transition comprises a highly nonlinear network that produces seemingly paradoxical results and makes intuitive interpretations unreliable.45 Numerous feedback loops lead to a situation that downstream events lie upstream of themselves. For example, pRb2/p130 and p27 both are involved in a negative feedback regulatory loop with cyclin E.46 Also, c-myc expression is downstream from p21, CDKs, and E2F,47,48 yet c-myc is also an upstream regulator of p21 and CDKs.49
Similarly, cyclin E is located downstream from Rb and E2F-1 because cyclin E is transactivated by E2F-1(Fig. 5). Yet, it is cyclin E that inactivates Rb and releases E2F (Fig. 5). Positive loops ensure irreversibility of commitments. Once expressed, cyclin E becomes independent of downstream GF-dependent cyclin D1. Therefore, cyclin E is a better candidate for the R-protein than cyclin D. Dependence on GF ends with the phosphorylation of Rb, enabling the cells to pass through the restriction point and to commit to completing the remaining phases of the growth cycle.43 Yet, both cyclin D and E, as well as cyclin A, phosphorylate Rb. The restriction protein is split into at least two slices (Fig. 6).
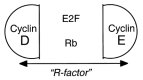
Figure 6
“R-factor”
Furthermore, E2F-1 partially expresses functional features of the R-protein. In fact, E2F is sufficient to drive a cell into S-phase.41 Under enlargement, the restriction “point” looks like a restriction “knot” (Fig. 5–6).
Growth Arrest Versus Proliferation
An isolated activation of signaling, which is downstream of the restriction point, induces S-phase entry. However, apoptosis may occur.50,51 Apoptosis is suppressed by GF-dependent events upstream of the restriction point. On the other hand, the choice between proliferation and growth arrest in response to growth factors and inhibitors is determined by the state of the restriction knot.
It is well known that exogenous stimuli such as EGF, TGF-β, CSF-1, interleukins, phorbol esters and endogenous transduction molecules such as Ras, PKC, Raf-1, and ERK can induce either growth arrest or cycle progression (Fig. 7).52–61 Yet, one consideration is so simple that it is often overlooked: growth stimulation can be detected only when an agent is applied to a resting cell, whereas to detect growth inhibition the agent must be applied to a proliferating cell.51
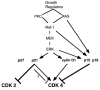
Figure 7
Proliferation vs growth arrest
There is increasing evidence that mitogenic signaling induces cyclin D and p21 simultaneously.51 Not only the ratio but the proliferative status of a cell may determine the response. In a resting cell, one can expect to find low basal levels of cyclins D1 and E but high levels of the CDK inhibitor, p27. In a proliferating cell, one would expect to find high levels of cyclin D1 and E with low levels of p27. In resting fibroblasts, low levels of Raf-1 activity induces cyclin D1 and proliferation, whereas in cycling fibroblasts, high levels of Raf-1 activity leads to growth arrest due to p21 induction.57–59 Treatment with phorbol ester increased both cyclin D1 and p21.60,62,63 While the growth of malignant melanoma cells was inhibited by TPA, the growth of normal melanocytes was stimulated.62 Similarly, stimuli that are mitogenic for mature (resting) T-cells induce arrest in proliferating tumor T-cells, inducing both p21 and cyclin D.63,60 E2F-1 and H-Ras can activate the p21 promoter, and induction of the p21 promoter by activated Ras is mediated at least in part by E2F-1.64
The same stimuli can induce different outcomes depending on the Rb status. Cytokine response gene induced p21 that blocked either G1/S or G2/M transition but did not prevent G1/S transition in Rb-negative (Rb−) cells, causing endoreduplication.65 The same stimuli caused proliferation in quiescent normal human T cells.65 Levels of p21 are also important. Thus, in HCT116 cells, low levels of endogenous p21 caused G2 but did not cause G1 arrest following DNA damage.66,67 However, much higher levels of exogenous p21 arrested these cells in G1 phase.67 In primary cells, Ras is initially mitogenic but eventually induces premature senescence. Constitutive activation of MEK induces both p53 and p16 and is required for Ras-induced senescence of normal human fibroblasts.68 Furthermore, activated MEK permanently arrests primary murine fibroblasts but forces proliferation in cells lacking either p53 or p16. This may explain the opposite response of normal and immortalized cells to constitutive activation of MEK.68
It is very important that not only inhibitors and stimulators of CDKs are induced simultaneously, but some of these molecules may act as both an inhibitor and a stimulator (Fig. 7). While inhibiting cyclin E-CDKs, p21 and p27 at least at low levels activate cyclin D1-CDKs complexes.69 For example, p21 permits growth factor-induced cell cycle progression of vascular smooth muscle.70 PKC alpha controls glioma cell cycle progression via up-regulation of p21, which facilitates active cyclin-CDK complex formation.71 In contrast, in many cell lines, PKC induces growth arrest due to p21 induction,48 but the sensitivity to p21 may be lost in very aggressive cancers.72 Increased expression of ectopic cyclin E in a mouse mammary epithelial cell line inhibits rather than stimulates growth, and this may be due to increased expression of the inhibitor p27.73
In addition to p21, p16(INK4a) and p15(INK4b), which are pure inhibitors of cyclin D-CDK-4 and -6, are induced through the MAPK pathway (Fig. 7). This pathway is involved in p21 and p15 induction by TGF-β.74 Like contact inhibition, TGF-β can arrest the cell cycle in G1 by inducing p27.75 Oncogenic Ras inhibits growth of primary cells due to induction of p16 and p19(ARF).68,76 p15 is induced by oncogenic Ras to an extent similar to that of p16, and expression of both is associated with G1 arrest and senescence.77 Ras-dependent induction of these two INK4 genes is mediated mainly by the Raf-MEK-ERK pathway.77 In addition, high expression levels of both wild-type and oncogenic Ras inhibit growth of K562 leukemia cells, which are deficient for p53, p16, p15, and p19 (ARF) genes. H-ras increases p21 and causes growth arrest in these cells.78
From Restriction- to “Check”-Points
The restriction point could be considered as a prototype of cell cycle checkpoints. The restriction point is related to mitogen deprivation. Checkpoints are mostly related to DNA damage79 and mitotic progression. Following DNA damage, a cell undergoes growth arrest to repair DNA. Damaged DNA can propagate during S-phase and mitosis. Therefore, arrest occurs in G1 (before S) and in G2 (before mitosis). Arrest in G1 prevents aberrant replication of damaged DNA, and arrest in G2 allows cells to avoid segregation of defective chromosomes. Some mechanisms of G1 and G2 checkpoints are similar, including p53-dependent p21 induction and Rb dephosphorylation.66,80 Other mechanisms are distinct because different sets of cyclins and kinases are activated in G1 and G2 phases. Massive DNA damage or deprivation of nucleotides forces a cell to be arrested in S-phase.81,82 In the G2 checkpoint, p21 and the 14-3-3 protein play distinct but complementary roles and cooperate to achieve arrest following DNA damage.83 p21 overexpression may result in senescence-like growth arrest and abnormal mitosis.84,85
In order to ensure faithful chromosome segregation, cells have evolved mechanisms that delay progress into and out of mitosis until certain events are completed.86 A spindle checkpoint in mitosis ensures equal chromosome segregation. The spindle checkpoint inhibits cell cycle progression in response to a signal generated by mitotic spindle damage or by chromosomes that have not attached to microtubules. This prevents chromosome dysfunction and exit from mitosis until all of the kinetochores (and thus chromosomes) are attached to the spindle. A canonical example is the mitotic arrest by microtubule-active drugs such as paclitaxel.87
The Restriction Point and G1 Checkpoint
As discussed, the restriction “point” lies between the accumulation of cyclins D and E and the activation of cyclin E-CDK2. What is the topological relationship of this GF-dependent restriction point and DNA damage-induced G1 checkpoint?
DNA damage targets CDKs by two mechanisms. First is rapid and transient disappearance of cyclin D1. DNA damage-induced pathways target cyclin D and then cyclin E-CDK2. Following DNA damage, cyclin D1 is rapidly degraded by the proteasome.88 Cyclin D1 degradation initiates a specific release of p21 from CDK4 complexes, leading to CDK2 inactivation. p21 may be redistributed without an increase in total amount. This is a p53-independent process. (Note: the restriction point control is also p53-independent). Second is p53-dependent induction of p21 resulting in inhibition of cyclin E-CDK2. The escape of cells with nonfunctional p53 from the initial G1 arrest probably stems from the fact that the reservoir of p21 held by cyclin D1/CDK4 complex is quickly exhausted after DNA damage.88 Simultaneously, p53 protein is accumulated. This results in p21 induction and sustained growth arrest. Importantly, pRB is a critical component of this DNA damage checkpoint. The checkpoint pathway uses the normal cell cycle regulatory machinery to induce the accumulation of the growth-suppressive form of pRb.89
Not only is the DNA damage checkpoint located nearby the restriction point, but many regulators cause growth arrest by affecting the restriction point. Progesterone, a steroid hormone, regulates proliferation and differentiation in the mammary gland and uterus and induces p27 and p18 to inhibit cyclin E-Cdk2 and Cdk4.90 Since similar models have been developed for growth inhibition by TGF-β and during adipogenesis, interaction between the Cip/Kip and INK4 families of inhibitors may be a common theme in physiological growth arrest.90 The protein kinase inhibitor staurosporine stops progression of normal nontransformed cells in the G1 phase between cyclin D and cyclin E activities.91 In contrast, the histone deacetylase inhibitor FR901228 causes nonphysiological G1 arrest with down-regulation of cyclin D1 and up-regulation of cyclin E in cancer cells lacking cycle control.92
The Restriction Point and Therapy
Selective killing of malignant cells without killing of normal cells is the ultimate goal of cancer therapy. Loss of the restriction point predisposes selective killing of cancer cells by chemotherapy.93 Autonomous growth of cancer cells lacking the restriction point can be exploited to selectively arrest growth of normal cells.94 For example, malignant transformation is associated with loss of dependency on epidermal growth factor (EGF).95 MCF-10A, EGF-dependent immortalized breast cells, underwent G0/G1 growth arrest following EGF withdrawal. In contrast, MCF-7 cancer cells were not affected by EGF withdrawal. By using inhibitors of the EGF receptor kinase, this dependence on EGF can be exploited for protection of MCF-10A cells from microtubule-active drugs.96 Microtubule-active drugs, such as paclitaxel (Taxol) and vinblastine, kill cells in a cycle-dependent manner, in mitosis. Paclitaxel causes microtubule dysfunction leading to mitotic arrest of cells that entered mitosis.97,98 Mitotic arrest is accompanied by phosphorylation of serine in multiple regulatory proteins, including Bcl-2, and this precedes cell death.87 A cell arrested in interphase by a nontoxic agent cannot enter mitosis and therefore cannot undergo mitotic arrest when exposed to paclitaxel. Hence, arresting normal cells in interphase prior to treatment with paclitaxel could increase the selectivity of microtubule-active drugs against cancer cells by minimizing undesired toxicity to normal cells. Low doses of AG1478, an inhibitor of the EGF receptor kinase, arrested the MCF-10 breast cell line but not the MCF-7 cancer cell line.96 In fact, AG1478 abrogated paclitaxel effects in MCF-10A cells but not in MCF-7 cells.96 Also, by arresting growth, CDK inhibitors, such as p21 and p16, can protect cells from apoptosis caused by cell cycle-dependent chemotherapy.67,99,100 Targeting pathways which are not essential for cancer cells that lack the restriction point can selectively protect normal cells against chemotherapy.
References
- 1.
- Pasero P, Schwob E. Think global, act local---how to regulate S phase from individual replication origins. Curr Opin Genet Dev. 2000; 10:178–186. [PubMed: 10753785]
- 2.
- Thyberg J, Moskalewski S. Role of microtubules in the organization of the Golgi complex. Exp Cell Res. 1999; 246:263–279. [PubMed: 9925741]
- 3.
- Baserga R. Oncogenes and the strategy of growth factors. Cell. 1994;79:927–930. [PubMed: 8001140]
- 4.
- Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci USA. 1974;71:1286–1290. [PMC free article: PMC388211] [PubMed: 4524638]
- 5.
- Campisi J, Medrano EE, Morro G, Pardee AB. Restriction point control of cell growth by a labile protein: Evidence for increased stability in transformed cells. Proc Natl Acad Sci USA. 1982;79:436–440. [PMC free article: PMC345758] [PubMed: 6952194]
- 6.
- Pardee AB. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. [PubMed: 2683075]
- 7.
- Croy RG, Pardee AB. Enhanced synthesis and stabilization of Mr 68,000 protein in transformed BALB/c-3T3 cells: candidate for restriction point control of cell growth. Proc NAtl Acad Sci USA. 1983;80:4699–4703. [PMC free article: PMC384111] [PubMed: 6308643]
- 8.
- Liang P, Pardee AB. Differential display of eukaryotic messenger-RNA by means of the polymerase chain-reaction. Science. 1992;257:967–971. [PubMed: 1354393]
- 9.
- Alpan RS, Sparvero S, Pardee AB. Identification of mRNAs differentially expressed in quiescence or in late G1 phase of the cell cycle in human breast cancer cells by using the differential display method. Mol Med. 1996;2:469–4. [PMC free article: PMC2230160] [PubMed: 8827717]
- 10.
- Matsushime H, Roussel MF, Ashmun RA, Sherr C. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–713. [PubMed: 1827757]
- 11.
- Sewing A, Burger C, Brusselbach S. et al. Human cyclin D1 encodes a labile nuclear protein whose synthesis is directly induced by growth factors and suppressed by cyclic AMP. J Cell Sci. 1993;104(Pt2):545–555. [PubMed: 8389378]
- 12.
- Roussel M, Theodoras A, Pagano M, Sherr C. Rescue of defective mitogenic signaling by D type cyclins. Proc Natl Acad Sci U S A. 1995;92:6837–6841. [PMC free article: PMC41424] [PubMed: 7624328]
- 13.
- Surmacz E, Reiss K, Sell C, Baserga R. Cyclin D1 messenger RNA is inducible by platelet derived growth factor in cultured fibroblasts. Cancer Res. 1992;52:4522–4525. [PubMed: 1379514]
- 14.
- Sherr CJ. Cancer cell cycles. Science. 1999;274:1672–1677. [PubMed: 8939849]
- 15.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. [PubMed: 10385618]
- 16.
- Dulic V, Lees E, Reed SI. Association of human cyclin-E with a periodic G(1)-S phase protein kinase. Science. 1992;257:1958–1961. [PubMed: 1329201]
- 17.
- Dou QP, Levin AH, Zhao SC, Pardee AB. Cyclin-E and cyclin-A as candidates for the restriction point. Cancer Res. 1993;53:1493–1497. [PubMed: 8384078]
- 18.
- Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci USA. 1993;90:1112–1116. [PMC free article: PMC45821] [PubMed: 8430082]
- 19.
- Koepp DM, Harper JW, Elledge SJ. How the cyclin became a cyclin: regulated proteolysis in the cell cycle. Cell. 1999;97:431–434. [PubMed: 10338207]
- 20.
- Won KA, Xiong Y, Beach D, Gilman MZ. Growth-regulated expression of D-type cyclin genes in human diploid fibroblasts. Proc Natl Acad Sci U S A. 1992;89:9910–9914. [PMC free article: PMC50243] [PubMed: 1409718]
- 21.
- Filmus J, Robles AI, Shi W. et al. Induction of cyclin D1 overexpression by activated ras. Oncogene. 1994;9:3627–3633. [PubMed: 7970723]
- 22.
- Aktas H, Cai H, Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol Cell Biol. 1997;17:3850–3857. [PMC free article: PMC232237] [PubMed: 9199319]
- 23.
- Winston JT, Coats SR, Wang YZ, Pledger WJ. Regulation of the cell cycle machinery by oncogenic ras. Oncogene. 1996;12:127–134. [PubMed: 8552383]
- 24.
- Tam SW, Theodoras AM, Pagano M. Kip1 degradation via the ubiquitin-proteasome pathway. Leukemia. 1997;11(Suppl 3):363–366. [PubMed: 9209391]
- 25.
- Blagosklonny MV, Wu GS, Omura S, El-Deiry WS. Proteasome-dependent regulation of p21WAF1/CIP1 expression. Biochem Biophys Res Commun. 1996;227:564–569. [PubMed: 8878553]
- 26.
- Campanero MR, Flemington EK. Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRb tumor suppressor protein. Proc Natl Acad Sci USA. 1997;94:2221–2226. [PMC free article: PMC20068] [PubMed: 9122175]
- 27.
- Winston JT, Chu C, Harper JW. Culprits in the degradation of cyclin E apprehended. Genes Dev. 1999;13:2751–2757. [PubMed: 10557204]
- 28.
- Varshavsky A, Turner G, Du F, Xie Y. The ubiquitin system and the N-end rule pathway. Biol Chem. 2000;39=81:779–789. [PubMed: 11076011]
- 29.
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. [PubMed: 9136925]
- 30.
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. [PMC free article: PMC317244] [PubMed: 9832503]
- 31.
- Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc Natl Acad Sci USA. 1998;95:1091–1096. [PMC free article: PMC18683] [PubMed: 9448290]
- 32.
- Hinds PW, Mittnacht S, Dulic V. et al. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. [PubMed: 1388095]
- 33.
- Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–2409. [PubMed: 11018009]
- 34.
- Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9:585–593. [PubMed: 9716176]
- 35.
- Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway and the restriction point. Curr Opin Cell Biol. 1996;8:805–814. [PubMed: 8939678]
- 36.
- Sheaff R, Groudine M, Gordon M. et al. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997;11:1464–1478. [PubMed: 9192873]
- 37.
- Hunter T. Oncoprotein networks. Cell. 1997;88:333–346. [PubMed: 9039260]
- 38.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. [PubMed: 10647931]
- 39.
- Almasan A, Yin Y, Kelly RE. et al. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc Natl Acad Sci USA. 1995;92:5436–5440. [PMC free article: PMC41709] [PubMed: 7777526]
- 40.
- DeGregori J, Leone G, Ohtani K. et al. E2F-1 accumulation bypasses a G1 arrest resulting from the inhibition of G1 cyclin-dependent kinase activity. Genes Dev. 1995;9:2873–2887. [PubMed: 7498785]
- 41.
- Kowalik TF, DeGregori J, Schwarz JK, Nevins JR. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol. 1995;69:2491–2500. [PMC free article: PMC188925] [PubMed: 7884898]
- 42.
- Qin XQ, Livingston DM, Kaelin WG, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA. 1994;91:10918–10922. [PMC free article: PMC45137] [PubMed: 7971984]
- 43.
- Planas-Silva MD, Weinberg RA. The restriction point and control of cell proliferation. Curr Opin Cell Biol. 1997;9:768–772. [PubMed: 9425340]
- 44.
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. [PubMed: 7736585]
- 45.
- Kohn KW. Functional capabilities of molecular network components controlling the mammalian G1/S cell cycle phase transition. Oncogene. 1998;16:1065–1075. [PubMed: 9519880]
- 46.
- Howard CM, Claudio PP, De Luca A. et al. Inducible pRb2/p130 expression and growth suppressive mechanisms: evidence of a pRb2/p130, p27Kip1, and cyclin E negative feedback regulatory loop. Cancer Res. 2000;60:2737–2744. [PubMed: 10825149]
- 47.
- Zou X, Rudchenko S, Wong K, Calame K. Induction of c-myc transcription by the v-Abl tyrosine kinase requires Ras, Raf1, and cyclin-dependent kinases. Genes Dev. 1997;11:654–662. [PubMed: 9119229]
- 48.
- Blagosklonny MV, Prabhu NS, El-Deiry WS. Defects in p21WAF1/CIP1, Rb, c-myc signaling in phorbol ester-resistant cancer cells. Cancer Res. 1997;57:320–325. [PubMed: 9000576]
- 49.
- Mitchell , El-Deiry WS. Overexpression of c-myc inhibits p21WAF1/CIP1 expression and induces S-phase entry in 12-O-tetradecanoylphorbol-13-acetate (TPA)-sensitive human cancer cells. Cell Growth Differ. 1999;10:223–230. [PubMed: 10319992]
- 50.
- Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. [PubMed: 9721090]
- 51.
- Blagosklonny MV. A node between proliferation, apoptosis, and growth arrest. Bioessays. 1999;21:704–709. [PubMed: 10440867]
- 52.
- MacLeod CL, Luk A, Castagnola J. et al. EGF induces cell cycle arrest of A431 human epidermoid carcinoma cells. J Cell Physiol. 1986;127:175–182. [PubMed: 3007537]
- 53.
- Cowley S, Peterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. [PubMed: 7911739]
- 54.
- Liu Y, Martindale JL, Gorospe M, Holbrook NJ. Regulation of p21 WAF1/CIP1 expression through mitogen-activated protein kinase signaling pathway. Cancer Res. 1996;56:31–35. [PubMed: 8548769]
- 55.
- Marshall CJ. Specificity of receptor tyrosine kinase signaling - transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. [PubMed: 7834738]
- 56.
- Pumiglia KM, Decker SJ. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 1997;94:448–452. [PMC free article: PMC19532] [PubMed: 9012803]
- 57.
- Lloyd AC, Obermuller F, Staddon S. et al. Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes Dev. 1997;11:663–677. [PubMed: 9119230]
- 58.
- Sewing A, Wiseman B, Lloyd AC, Land H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5588–5597. [PMC free article: PMC232407] [PubMed: 9271434]
- 59.
- Woods D, Parry D, Cherwinski H. et al. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–5611. [PMC free article: PMC232408] [PubMed: 9271435]
- 60.
- Blagosklonny MV. The mitogen-activated protein kinase pathway mediates growth arrest or E1A-dependent apoptosis in SKBr3 human breast cancer cells. Int J Cancer. 1998;78:511–517. [PubMed: 9797142]
- 61.
- Lee A W M, Nambirajan S, Moffat JG. CSF-1 activates MAPK-dependent and p53 independent pathways to induce growth arrest of hormone-dependent human breast cancer cells. Oncogene. 1999;18:7477–7494. [PubMed: 10602507]
- 62.
- Coppock DL, Buffolino P, Kopman C, Nathanson L. Inhibition of the melanoma cell cycle and regulation at the G1/S transition by 12-O-tetradecanoylphorbol-13-acetate (TPA) by modulation of CDK2 activity. Exp Cell Res. 1995;221:92–102. [PubMed: 7589260]
- 63.
- Desrivieres S, Volarevic S, Mercep L, Ferrari S. Evidence for different mechanisms of growth inhibition of T-cell lymphoma by phorbol esters and concanavalin A. J Biol Chem. 1997;272:2470–2476. [PubMed: 8999961]
- 64.
- Gartel AL, Najmabadi F, Goufman E, Tyner AL. A role for E2F1 in Ras activation of p21(WAF1/CIP1) transcription. Oncogene. 2000;19:961–964. [PubMed: 10702805]
- 65.
- Fan W, Richter G, Cereseto A. et al. Cytokine response gene 6 induces p21 and regulates both cell growth and arrest. Oncogene. 1999;18:6573–6582. [PubMed: 10597261]
- 66.
- Bunz F, Dutriaux A, Lengauer C. et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. [PubMed: 9822382]
- 67.
- Blagosklonny MV, Robey R, Bates S, Fojo T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J Clin Invest. 2000;105:533–539. [PMC free article: PMC289166] [PubMed: 10683383]
- 68.
- Lin AW, Barradas M, Stone JC. et al. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. [PMC free article: PMC317198] [PubMed: 9765203]
- 69.
- Cheng M, Olivier P, Diehl JA. et al. The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. [PMC free article: PMC1171245] [PubMed: 10075928]
- 70.
- Weiss RH, Joo A, Randour C. 21(Waf1/Cip1) is an assembly factor required for platelet derived growth factor-induced vascular smooth muscle cell proliferation. J Biol Chem. 2000;275:10285–10290. [PubMed: 10744715]
- 71.
- Besson A, Yong VW. Involvement of p21(Waf1/Cip1) in protein kinase C alpha-induced cell cycle progression. Mol Cell Biol. 2000;20:4580–4590. [PMC free article: PMC85854] [PubMed: 10848585]
- 72.
- Salnikow K, Costa M, Figg WD, Blagosklonny MV. Hyperinducibility of hypoxia responsive genes without p53/p21-dependent checkpoint in aggressive prostate cancer. Cancer Res. 2000;60:5630–5634. [PubMed: 11059752]
- 73.
- Sgambato A, Han EK, Zhou P. et al. Overexpression of cyclin E in the HC11 mouse mammary epithelial cell line is associated with growth inhibition and increased expression of p27(Kip1). Cancer Res. 1996;56:1389–1399. [PubMed: 8640830]
- 74.
- Hu P P C, Shen X, Huang D. et al. The MEK pathway is required for stimulation of p21(WAF1/CIP1) by transforming growth factor-beta. J Biol Chem. 1999;274:35381–35387. [PubMed: 10585406]
- 75.
- Polyak K, Kato JY, Solomon MJ. et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. [PubMed: 8288131]
- 76.
- Sherr CJ. The Pezcoller lecture: Cancer Cell Cycle Revisited. Cancer Res. 2000;60:3689–3695. [PubMed: 10919634]
- 77.
- Malumbres M, De Castro IP, Hernandez MI. et al. Cellular response to oncogenic Ras involves induction of the Cdk4 and Cdk6 inhibitor p15(INK4b). Mol Cell Biol. 2000;20:2915–2925. [PMC free article: PMC85529] [PubMed: 10733595]
- 78.
- Delgado MD, Vaque JP, Arozarena I. et al. H-, K- and N-Ras inhibit myeloid leukemia cell proliferation by a p21(WAF1)-dependent mechanism. Oncogene. 2000;19:783–790. [PubMed: 10698496]
- 79.
- Zhou B -B S, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. [PubMed: 11100718]
- 80.
- Flatt PM, Tang LJ, Scatena CD. et al. p53 regulation of G2 checkpoint is retinoblastoma protein dependent. Mol Cell Biol. 2000;20:4210–4223. [PMC free article: PMC85790] [PubMed: 10825186]
- 81.
- Wyllie FS, Haughton MF, Bond JA. et al. S phase cell-cycle arrest following DNA damage is independent of the p53/p21(WAF1) signalling pathway. Oncogene. 1996;12:1077–1082. [PubMed: 8649799]
- 82.
- Agarwal ML, Agarwal A, Taylor WR. et al. A p53-dependent S-phase checkpoint helps to protect cells from DNA damage in response to starvation for pyrimidine nucleotides. Proc Natl Acad Sci U S A. 1998;95:14775–14780. [PMC free article: PMC24525] [PubMed: 9843965]
- 83.
- Chan TA, Hwang PM, Hermeking H. et al. Cooperative effects of genes controlling the G2/M checkpoint. Genes Dev. 2000;14:1584–1588. [PMC free article: PMC316737] [PubMed: 10887152]
- 84.
- Chang B -D, Broude EV, Dokmanovic M. et al. A senescence-like phenotype distinquishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer drugs. Cancer Res. 1999;59:3761–3767. [PubMed: 10446993]
- 85.
- Chang BD, Broude EV, Fang J. et al. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000;19:2165–2170. [PubMed: 10815808]
- 86.
- Clarke DJ, Gimenez-Abian JF. Checkpoints controlling mitosis. Bioessays. 2000;22:351–363. [PubMed: 10723032]
- 87.
- Blagosklonny MV, Fojo T. Molecular effects of paclitaxel: myths and reality. Int J Cancer. 1999; 83:151–156. [PubMed: 10471519]
- 88.
- Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell. 2000;102:55–66. [PubMed: 10929713]
- 89.
- Brugarolas J, Moberg K, Boyd SD. et al. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc Natl Acad Sci U S A. 1999;96:1002–1007. [PMC free article: PMC15340] [PubMed: 9927683]
- 90.
- Swarbrick A, Lee CS, Sutherland RL, Musgrove EA. Cooperation of p27(Kip1) and p18(INK4c) in progestin-mediated cell cycle arrest in T-47D breast cancer cells. Mol Cell Biol. 2000;20:2581–2591. [PMC free article: PMC85474] [PubMed: 10713180]
- 91.
- Gong J, Traganos F, Darzynkiewicz Z. Staurosporine blocks cell progression through G1 between the cyclin D and cyclin E restriction points Cancer Res 199454(12):3136–9.1994; 54:3136-3139 . [PubMed: 8205531]
- 92.
- Sandor V, Senderowicz A, Mertins S. et al. P21-dependent G1 arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer. 2000;83:817–825. [PMC free article: PMC2363539] [PubMed: 10952788]
- 93.
- Pardee AB, James LJ. Selective killing of transformed baby hamster kidney (BHK) cells. Proc Natl Acad Sci USA. 1975;72:4994–4998. [PMC free article: PMC388861] [PubMed: 174089]
- 94.
- Darzynkiewicz Z. Apoptosis in antitumor strategies: modulation of cell-cycle or differentiation. J Cell Biochem. 1995;58:151–159. [PubMed: 7673323]
- 95.
- Cherington PV, Smith BL, Pardee AB. Loss of epidermal growth factor requirement and malignant transformation. Proc Natl Acad Sci U S A. 1979;76:3937–3941. [PMC free article: PMC383951] [PubMed: 226990]
- 96.
- Blagosklonny MV, Bishop PC, Robey R. et al. Loss of cell cycle control allows selective microtubule-active drug-induced Bcl-2 phosphorylation and cytotoxicity in autonomous cancer cells. Cancer Res. 2000;60:3425–2428. [PubMed: 10910051]
- 97.
- Horwitz SB. Mechanism of action of taxol. Trends Pharmacol Sci. 1992;13:134–136. [PubMed: 1350385]
- 98.
- Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol. 1998;10:123–130. [PubMed: 9484604]
- 99.
- Li W, Fan J, Banerjee D, Bertino JR. Overexpression of p21 (waf1) decreases G2-M arrest and apoptosis induced by paclitaxel in human sarcoma cells lacking both p53 and functional Rb protein. Mol Pharmacol. 1999;55:1988–1093. [PubMed: 10347252]
- 100.
- Stone S, Dayananth P, Kamb A. Reversible, p16-mediated cell cycle arrest as protection from chemotherapy. Cancer Res. 1996;56:3199–3202. [PubMed: 8764106]
- Mitogen-Dependent and -Independent Phases of the Cell Cycle
- The Restriction Point
- In Search of Mediators of the Restriction Point
- Cyclins: From Mitogen Signaling to the Restriction Point
- The Restriction Point: A Knot of Mitogen and Inhibitory Signaling
- Growth Arrest Versus Proliferation
- From Restriction- to “Check”-Points
- The Restriction Point and G1 Checkpoint
- The Restriction Point and Therapy
- References
- The Restriction Point of the Cell Cycle - Madame Curie Bioscience DatabaseThe Restriction Point of the Cell Cycle - Madame Curie Bioscience Database
- RNA Modification Subsystems in the SEED Database - Madame Curie Bioscience Datab...RNA Modification Subsystems in the SEED Database - Madame Curie Bioscience Database
- Origin, Recognition, Signaling and Repair of DNA Double-Strand Breaks in Mammali...Origin, Recognition, Signaling and Repair of DNA Double-Strand Breaks in Mammalian Cells - Madame Curie Bioscience Database
- Protein Misassembly: Macromolecular Crowding and Molecular Chaperones - Madame C...Protein Misassembly: Macromolecular Crowding and Molecular Chaperones - Madame Curie Bioscience Database
- p53's Dilemma in Transcription: Analysis by Microarrays - Madame Curie Bioscienc...p53's Dilemma in Transcription: Analysis by Microarrays - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...