

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Organelles, defined as intracellular membrane-bound structures in eukaryotic cells, were described from the early days of light microscopy and the development of cell theory in the 19th century. During the 20th century, electron microscopy and subcellular fractionation enabled the discovery of additional organelles and, together with radiolabelling, allowed the first modern experiments on their biogenesis. Over the past 30 years, the development of cell-free systems and the use of yeast genetics have together established the major pathways of delivery of newly synthesised proteins to organelles and the vesicular traffic system used to transfer cargo between organelles in the secretory and endocytic pathways. Mechanisms of protein sorting, retrieval and retention have been described and give each organelle its characteristic composition. Insights have been gained into the mechanisms by which complex organelle morphology can be established. Organelle biogenesis includes the process of organelle inheritance by which organelles are divided between daughter cells during mitosis. Two inheritance strategies have been described, stochastic and ordered, which are not mutually exclusive. Among the major challenges of the future are the need to understand the role of self-organization in ensuring structural stability and the mechanisms by which a cell senses the status of its organelles and regulates their biogenesis.
Introduction
Today, cell biologists are almost overwhelmed by molecular detail about organelle composition, structure, function and biogenesis. Nevertheless during the molecular era, which has encompassed the past half century, a conceptual framework has developed to explain processes such as protein sorting, membrane traffic and organelle biogenesis. In this chapter we review this development, together with earlier work that established the existence of organelles and traffic to them. Necessarily, we cannot include specific detail about all organelles and we have concentrated, for the most part, on those found on the secretory and endocytic pathways.1 We begin with some definitions.
Definitions
Organelles are defined as intracellular membrane-bound structures in eukaryotic cells, usually specialized for a particular function.2 While many organelles are morphologically similar and perform essentially the same function in all eukaryotic cells, some are specialized and occur only in particular cell types. Among the former are the nucleus, mitochondria and or- ganelles in the secretory and endocytic pathways including the endoplasmic reticulum, Golgi complex, endosomes and lysosomes (vacuoles in yeast), whereas the latter include chloroplasts restricted to the plant kingdom. In mammalian cells there has been much study of cell type-specific specialist organelles and their relationship to common organelles. Many, if not all, of these are specialized structures in the secretory and endocytic pathways and include, for example, regulated secretory granules in neuroendocrine cells3 and melanosomes,4 which are clearly lysosome-like, in skin melanocytes.
Organelle biogenesis is the process by which new organelles are made. In a few cases, notably mitochondria and chloroplasts, some organelle proteins are encoded by the organelle's own genome. However, the amount of DNA in such organelles can encode only a very small number of the many proteins required.5 In practice, the study of organelle biogenesis includes the mechanisms by which proteins and lipids, newly synthesized elsewhere in the cell, are delivered to organelles and the process by which organelles are divided between daughter cells during mitosis. In general it is thought that new organelles are derived by proliferation of preexisting organelles.6 However, for some organelles on the secretory and endocytic pathways, e.g., the Golgi complex (see below), the extent to which they can be made de novo by a cell without a preexisting organelle or template remains a subject of controversy.7
The History of Organelle Recognition
Light Microscopy and Cell Theory
Recognition of organelles is only as feasible as the available techniques for observation. The light microscope was the essential first tool; once this existed “cells” could be and were observed, initially in plant material where substances such as cellulose made observation easier or in unicellular organisms. In 1833, Brown observed and described the nucleus, the first organelle.8 In 1838, the many and various observations were converted into a cell theory by Schleiden, who proposed that all plant tissues were composed of nucleated cells.9 The following year Schwann applied this cell theory to animal tissues.10 Schleiden and Schwann assumed that cells were formed by some kind of crystallization of intracellular substance, in spite of observations on the binary fission of nucleus and cell in plants.11 However, by 1855 Virchow proclaimed “Omnis cellula e cellula” (all cells from cells)12,13 and in 1874 Flemming began to publish detailed and correct descriptions of mitosis, culminating in a comprehensive book in 1882.14 The importance of the recognition of organelles to the development of cell theory is clear, since as Richmond15 has described, “German cell theory primarily looked to cellular structures, such as the nucleus, rather than to processes as the focal points for vital organization”.
Coincident with the emergence of Schleiden and Schwann's cell theory was the recognition that a membrane structure bounded cells (reviewed in ref. 16). The osmotic properties of plant cells led to Nageli defining the “plasmamembran” as a surface layer of protoplasm, denser and more viscous than the protoplasm as a whole. By the early 20th century, the osmotic properties of red blood cells had extended the concept of the plasma membrane to mammalian cells, but it was not until the classic experiment of Gorter and Grendel published in 192517 that the basic structure of the plasma membrane was shown to consist of a bilayer of phospholipid. In this experiment, the surface area of a compressed film of total lipid extracted from a known number of red blood cells was measured and found to be twice the total cell area. The phospholipid bilayer was incorporated as a central feature in many subsequent models of the structure of both the plasma membrane and intracellular membranes, culminating in the fluid mosaic model of Singer and Nicholson in which integral membrane proteins were distributed within the bilayer.18
The structure of the interphase nucleus was also extensively studied during the late 19th century. Brown8 had suggested the possibility of a nuclear membrane and in 1882 Flemming14 summarised the evidence for its reality. Following experiments using basic stains such as haematoxylin he also defined chromatin as “the substance in the cell nucleus which takes up color during nuclear staining” (although a stain specific for DNA was not described until 1924 by Feulgen and Rossenbeck19). The nucleolus had been observed as a feature of some nuclei many times; over 700 articles on the subject had appeared before the classic paper by Montgomery in 1898.20,21
Meanwhile, mitochondria had been seen with varying degrees of conviction by a number of scientists from Henle in 1841 onwards.22 Altmann in 1890,23 however, was the first to recognize the ubiquitous occurrence of mitochondria and to suggest that they carried out vital functions. The increasing use of chemicals, which preferentially stained some parts of the cell, led to more accurate descriptions of cell structure, although concerns over artefacts had to be addressed. In 1898, Golgi24 demonstrated the existence of the Golgi complex by staining with heavy metals such as silver nitrate or osmium tetroxide. The reality of this organelle, however, continued to be doubted until the mid 1950s when electron micrographs became available.25
Electron Microscopy and Subcellular Fractionation
Mitochondria and the Golgi complex are at the limit of resolution by the light microscope; the visualization of smaller organelles had to wait for the development of electron microscopes. However, a parallel interest in taking cells apart and studying the nature of the separated components also yielded invaluable information; the existence of lysosomes was established before they were seen. Information as to the chemical nature and function of organelles was sought as early as 1934 by Bensley and Hoerr,26 who made a crude preparation of mitochondria. Claude in 1940-1946 used similar procedures with a crucial difference.27,28 He insisted on quantitative criteria, examining the total recovery of an enzyme or chemical constituent and its relative concentrations in the fractions he prepared by differential centrifugation, rather than preparing a single fraction. He also examined the size, shape and fine structure of the particulates in the separated fractions and used an isotonic medium for homogenisation. In 1948 Hogeboom, Schneider and Palade29 improved his methodology by using a Potter-Elvehjem homogeniser to achieve quantitative gentle breakage of liver cells and sucrose in place of saline. They were then able to show that most of Claude's “large granules” had the elongated shape of mitochondria and stained with Janus Green, a specific stain for this organelle.
Enzymes such as cytochrome oxidase, which appeared mostly in the large granule fraction, were clearly mitochondrial. There were also enzymes such as glucose 6-phosphatase, which appeared primarily in the smaller “microsomal” fraction. However, the work of de Duve from 1949 onwards demonstrated the existence of a group of enzymes, which were sedimented in the large granule fraction only if relatively high speeds were used in its preparation. The large granule fraction could be separated into a heavy and a light fraction. The former contained the respiratory activity characteristic of mitochondria but the light fraction contained variegated hydrolases. These were only measurable when the preparation had been subjected to hypotonic media, detergents or other insults to membrane integrity. From these results, de Duve hypothesised the existence of organelles containing primarily hydrolases and named them lysosomes. 30
Electron microscopy had meanwhile progressed to a generally available method of investigation. This necessitated the development of adequate fixing, staining, embedding and sectioning techniques as well as the development of the instruments themselves.31 In 1952, Palade published high resolution pictures of mitochondria.32 In 1954, Dalton and Felix (among others) published pictures of the Golgi complex,33 which showed that it contained cisternae and vesicles and stained with osmium tetroxide, as had the disputed structure seen by light microscopy. However, the electron microscope also revealed structures which the light microscope was completely unable to resolve. The varying forms but almost ubiquitous existence of the endoplasmic reticulum could be seen and shown to contribute largely to Claude's microsomal fraction. By a lucky chance, Porter, Claude and Fullam first saw the endoplasmic reticulum in whole tissue cells as a “lace-like” structure in 1945.34 As sectioning techniques improved over the next ten years, the endoplasmic reticulum had to be recognized in slices which were much smaller than the mesh size of the reticulum. The continuous nature of the meshes could only be demonstrated by tedious serial sectioning, although much more detailed structure could be observed and many different tissues examined to show the ubiquity of the organelle.35
Lysosomes were identified with the pericanalicular dense bodies described by Rouiller in 1954 by examination of partially purified preparations and by the development of a method for acid phosphatase localisation at both light and electron microscopic levels.36 Peroxisomes were reported by electron microscopy as microbodies in liver and kidney at about the same time, although their identity with the bodies carrying non-latent uricase and other enzymes involved with hydrogen peroxide was only established in the early sixties.37
Radiolabelling and the Dynamic Nature of Organelles
In addition to the clearly recognizable organelles, the electron microscope showed that cells contained a multiplicity of vesicles; the components of the secretory and endocytic pathways. The dynamic nature of such vesicles and of most other organelles began to be revealed when, in 1967, Jamieson and Palade used radioactive tracers and electron microscopic autoradiography. 38 They showed that newly synthesized secretory proteins during, or shortly after, synthesis, crossed the rough (ribosome-studded) endoplasmic reticulum and then moved from the endoplasmic reticulum to the Golgi region and thence to secretory granules. By 1975, Palade, if not every worker in the field, believed that movement of material through these organelles depended on vesicular traffic.39
Appreciation of the endocytic system was more diffuse. Phagocytosis was observed as early as 1887, but the fact that endocytosis was of widespread occurrence in animal cells was recognized only in the mid 1950s by electron microscopy.35 Coated pits and vesicles were observed in oocytes as early as 1964,40 but the ability of coated pits to concentrate endocytic receptors before pinching off to form coated vesicles was only recognised in 1976.41 In 1973 Heuser and Reece42 suggested that plasma membrane components inserted during exocytosis in synapses might be recycled. Quantitative electron microscopic investigations in 1976 by Steinman, Brodie and Cohn43 showed that tissue culture cells internalized plasma membrane at a rate which greatly exceeded their biosynthetic capacity. Therefore, a mechanism had to exist whereby endocytosed membrane could be recycled to the plasma membrane. Only by the 1980's was it widely accepted that endocytosed vesicles fused with an intracellular organelle called the endosome from which recycling to the plasma membrane could occur and also delivery to lysosomes and the trans-Golgi network.44 By this stage there was also widespread recognition of the various vesicle traffic pathways involved in exocytosis, endocytosis, transcytosis and biogenesis of organelles.
Protein Synthesis and Targeting
Although Palade had established that newly synthesised secretory proteins crossed into the lumen of the endoplasmic reticulum, it required the experiments of Blobel and his colleagues to establish that this sorting and targeting event was mediated by a sequence motif within the primary sequence of the secretory protein, which was named the signal sequence.45 To test the predictions of the signal hypothesis, first announced in 1971, Blobel developed a cell-free system in which protein translation and protein translocation across microsomal membrane vesicles could be measured. This cell-free system was a powerful forerunner of many others established elsewhere which faithfully recapitulated individual steps in organelle biogenesis pathways. The signal hypothesis also led directly to the concept that specific sequences within a protein could direct its targeting to a particular organelle. Thus, different consensus sequences have since been recognized as targeting motifs for import into mitochondria,46 chloroplasts,47 peroxisomes,48 nuclei49 and for the targeting of membrane proteins on secretory and endocytic pathways. Subsequent to the discovery of consensus sequences targeting proteins to particular organelles, there has been much work over the past 20 years identifying the protein machinery required for transport into such organelles, leading to an extensive understanding of transport to the mitochondrial matrix,46 inner membrane,50 outer membrane,51 into chloroplasts,52 into peroxisomes53 and through nuclear pores.54. In addition to amino acid sequence motifs, secondary modifications have also been recognized as targeting motifs, for example mannose 6-phosphate to target acid hydrolases from the Golgi complex to lysosomes55 and both glycosylation and glycosylphosphatidylinositol membrane anchors to target proteins to the apical surface of polarized epithelial cells.56
A further bequest of the signal hypothesis was that testing it provided support for the idea that in evolution the eukaryotic endoplasmic reticulum arose by invagination of the prokaryotic plasma membrane since signal sequences addressed to the eukaryotic endoplasmic reticulum function in translocation across the prokaryotic plasma membrane and signal sequences for bacterial secretory proteins function in translocation across the eukaryotic endoplasmic reticulum.57 In contrast, it had earlier been suggested that uptake of a prokaryotic progenitor cell(s) was the evolutionary origin of mitochondria and chloroplasts,58 a hypothesis largely supported by the results of subsequent genome analysis which were consistent with the origin of the mitochondrion being an endosymbiotic α-proteobacterium.59
Cell-Free Systems and Yeast Genetics
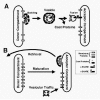
Vesicular traffic is now accepted as the central mechanism by which proteins are transported between donor and acceptor compartments on the secretory and endocytic pathways60 (Fig. 1). The discovery of clathrin by Pearse in the 1970s61 provided the first coat component of vesicles involved in membrane traffic. However, it was during the 1980s that elucidation of the molecular machinery of vesicular traffic started in earnest with the reconstitution of individual traffic steps in cell-free systems from animal cells62 and the isolation of secretory mutants in yeast.63 Probably the most informative of these early cell-free systems was one in which vesicular traffic between Golgi cisternae was reconstituted by incubating Golgi membranes with cytosol and ATP.64 In this system a population of Golgi membranes derived from cells lacking N-acetylglucosamine transferase but containing the G protein of vesicular stomatatis virus (VSV) was incubated with an population of Golgi membranes from wild type cells. Vesicular traffic resulted in addition of radioactive N-acetylglucosamine to the VSV-G as a result of the activity of the transferase in the wild type Golgi membranes. This assay led directly to the discovery of COPI (coat protein I) coated vesicles65 and the discovery of components of the general cytosolic fusion machinery required for vesicle fusion with acceptor membranes throughout the secretory and endocytic pathways.66 Subsequently, using the same principles of incubating organelle membrane fractions with cytosol and ATP, many membrane traffic steps were reconstituted in cell-free systems. Similarly, cell-free assays were established to look at the breakdown and reformation of organelles during cell division.
The isolation of secretion (sec) mutants in the budding yeast Saccharomyces cerevisiae63 provided a powerful approach to identify proteins required for traffic through the secretory pathway and to study their function. Throughout the 1980s and 1990s many proteins necessary for membrane traffic on the secretory pathway were identified almost at the same time, either by fractionating mammalian cytosol or through characterization of yeast mutants. These studies established the similarities of membrane traffic pathways at the molecular level in all eukaryotes.60 The genetic screens in yeast which allowed isolation of the original temperature-sensitive and other sec mutants were followed by many others, for example those identifying genes affected in vacuolar protein sorting (vps ) mutants67,68 and those identifying genes involved in autophagy.69 These latter screens led directly to our current understanding of the molecular mechanisms of biogenesis of the vacuole, of its mammalian equivalent the lysosome70 and of autophagosomes in both yeast and mammalian cells.69,70 In recent years, the development of cell-free systems to study homotypic yeast vacuole fusion, together with yeast genetics,71 have led to a massive expansion in our understanding of what is effectively a multi-protein machine required to achieve vacuole membrane fusion, a process essential to vacuole biogenesis in the daughter bud of a dividing yeast.
Vesicle Budding and Delivery
When clathrin was purified and shown to be the major protein component of purified coated vesicles,61 it was not clear whether it was simply the scaffold that makes the coat, involved in vesicle budding and/or also involved in sorting cargo into the vesicles. Very soon it was realized that there were at least two classes of clathrin coated vesicles in cells, one predominantly Golgi-associated, subsequently shown to be involved in budding from the trans-Golgi network and the other at the plasma membrane responsible for a major endocytic uptake route. The two classes of clathrin-coated vesicles were distinguished by the presence of two different heterotetrameric adaptor protein complexes, AP-1 at the trans-Golgi network and AP-2 at the plasma membrane. Electron microscopy, protein-protein interaction studies and most recently structural biology72 have strongly suggested that adaptor complexes have similar structures, resembling Mickey Mouse, with a core or “head” consisting of medium (μ) and small subunits and the amino-terminal domains of two large subunits (α/γ and β), flanked by flexibly-hinged “ears” consisting of the carboxyterminal domains of the two large subunits. Work in several laboratories showed that the adaptors were involved in cargo sorting as well as recruitment of clathrin to the membrane.73 Later, further family members were discovered including heterotetrameric AP-3 and AP-4 complexes that are not associated with clathrin and the more distantly related monomeric GGAs (Golgi-localised, γ-ear-containing, ARF-binding proteins).74 All of these coat proteins function in post-Golgi membrane traffic pathways. In mammalian cells GGAs are important in trafficking mannose 6-phosphate receptors and associated newly synthesised mannose 6-phosphate-tagged acid hydrolases to the endosomes for delivery to lysosomes. AP-1 is most likely involved in traffic back to the trans-Golgi network of the empty mannose 6-phosphate receptors. AP-3 is required for efficient delivery of newly synthesised membrane proteins to lysosomes and lysosome-related organelles. Mutations in AP-3 occur naturally in animals including fruit flies (i.e., Drosophila melanogaster ) and humans, leading to alterations of eye colour in the former and a rare genetic disease in the latter as a result of defects in delivery of proteins to lysosome-related organelles such as Drosophila eye pigment granules and platelet dense core granules, respectively. AP-4 may be involved in delivery to lysosomes and/or polarized sorting in epithelial cells. The formation of clathrin coated vesicles at either the plasma membrane or at intracellular sites is now recognised to require a host of accessory and regulatory proteins, many of which interact primarily with the carboxyterminal “ear” domains of the large subunits of the heterotetrameric AP complexes. Once mechanical invagination of the donor membrane to form the vesicle is complete, pinching off occurs, mediated at least in part by the action of the GTPase dynamin.75
While clathrin and AP complexes provide the major coat components for vesicle traffic in post-Golgi pathways, different coats are required for traffic between the endoplasmic reticulum and the Golgi complex. The first coat to be identified for vesicular traffic in this part of the secretory pathway was COPI using the cell-free assays described above. In such assays it was found that non-hydrolyzable analoges of GTP, such as GTP-γS can block traffic and this was accompanied by the accumulation of 70nm coated vesicles. The COPI coat on these vesicles contains eight polypeptides, one being the small GTPase ARF (ADP-ribosylation factor) responsible for coat recruitment to the membrane and the remainder being associated in an equimolar coat protomer (coatomer) complex.60 Weak sequence similarities and information about coatomer interactions have led to the suggestion that the molecular architecture of the COPI coat is similar to that of the AP/clathrin coats.76 It is now thought that the major traffic pathway mediated by COPI coated vesicles is the retrograde pathway from the Golgi complex to the endoplasmic reticulum necessary for the retrieval of escaped resident endoplasmic reticulum proteins and for the recycling of membrane proteins required for vesicle traffic and membrane fusion.77 Whereas COPI coated vesicles were first discovered through cell-free assays (although it was rapidly realised that the mammalian coatomer γ-COP is homologous to yeast Sec21p), the COPII coat, required for vesicles to bud from the endoplasmic reticulum for traffic to the Golgi complex, was identified by analysis of yeast sec mutants. The COPII coat consists of the small GTPase Sar1p, responsible for coat recruitment to the endoplasmic reticulum membrane, and the heterodimeric protein complexes Sec23/24p and Sec13/31p. These five proteins are necessary and sufficient to produce COPII vesicles from endoplasmic reticulum microsomes or from chemically defined liposomes.78 COPII coated vesicles were the first vesicles to be reconstituted solely from purified components. Indeed they might be regarded as the first organelles to be reconstituted solely from purified components since they fulfill the essential criteria to be called organelles in being intracellular membrane-bound structures in eukaryotic cells.
Vesicular traffic between donor and acceptor organelles in the secretory and endocytic pathways requires not only vesicle formation, but subsequent loss of the vesicle coat and fusion with the acceptor organelle. In addition, it often requires interactions of the vesicle with the cytoskeleton: with microtubules via kinesin or dynein motor proteins for long distance movement and/or via unconventional myosins for efficient short distance movement through actin rich regions of the cell. Once the vesicle reaches its target acceptor organelle, membrane fusion can occur, utilizing a common cytosolic fusion machinery and cognate interacting membrane proteins specific to the particular vesicle and organelle. Discovery of the common cytosolic fusion machinery derived from the observation that low concentrations of the alkylating agent N-ethylmaleimide (NEM) inhibited many membrane traffic steps reconstituted in cell-free systems. Using essentially brute force biochemistry, Rothman's group purified the soluble cytosolic NEM sensitive protein required to reconstitute membrane fusion in their cell-free Golgi assay, calling it NSF (NEM-sensitive factor).66 This protein had ATPase activity and its sequence showed similarity to that of yeast Sec18p. The discovery of NSF led rapidly to the finding of proteins, called SNAPs (soluble NSF atachment proteins) which bind it to membranes. The next stage was discovery of SNAP receptors, or SNAREs, which are integral membrane proteins that confer specificity on individual fusion reactions.60,79 The first of these were identified in mammalian brain, a tissue highly specialized for the membrane fusion required for neurotransmission at synapses. These studies led to the proposal of the SNARE hypothesis in which each transport vesicle bears a unique address marker or v-SNARE and each target membrane a unique t-SNARE, thus allowing targeting specificity to be achieved by the v-SNARES binding to matching t-SNAREs.60,79,80 Importantly in yeast, whereas mutations in SEC18 and SEC17 (the gene encoding the yeast homologue of α-SNAP) had effects throughout the secretory and endocytic pathways, when SNARE mutants were isolated it was found that individual alleles often affected only trafficking steps related to the organelles with which a particular SNARE was associated.81 In the few cases where a SNARE complex required for membrane fusion has been fully characterized it consists of four interacting α-helices aligned in parallel. A classification of SNAREs based on sequence alignments of the helical domains and structural features observed in the crystal structure of the synaptic SNARE fusion com- plex82 has been proposed. This separates SNAREs into Q-SNAREs and R-SNAREs, with four-helix SNARE complex bundles being composed of three Q-SNAREs and one R-SNARE.83,84 Q and R represent the glutamine and arginine residues observed in the central hydrophilic layer of the helical bundle.
Although cognate SNARE proteins can be reconstituted into liposomes and themselves act as phospholipid bilayer fusion catalysts,80,85,86 membrane fusion within the cell requires the functional involvement of other proteins. Most current models of fusion suggest three steps, tethering of the vesicle to the target organelle, SNARE complex formation and phospholipid bilayer fusion. A class of small GTPases known as rab proteins was identified as generally important when it was shown that different rabs localize to different organelles on the secretory and endocytic pathways.87 Rab proteins have been proposed to play a variety of roles in membrane fusion, and current evidence suggests a major function in the recruitment of tethering and docking proteins at an early stage in membrane interaction.88 Tethering has been defined as involving links that extend over distances >25 nm from a given membrane surface, and docking as holding membranes within a bilayer's distance, <5-10 nm of one another.88 Following tether recruitment and oligomeric assembly of the tethers, SNARE complex formation occurs. Fusion may also require downstream events after SNARE complex formation. In yeast vacuole fusion, a process which has been reconstituted in cell-free assays, Ca2+ release from the vacuole lumen is required in a post-docking phase of fusion89 and there is increasing evidence that Ca2+ may have a function late in the fusion process in other membrane fusion events.90 Once fusion has taken place the SNARE complex will reside in the target organelle membrane, necessitating separation of the complex, mediated by the ATPase activity of NSF followed by retrieval of the v-SNARE for further rounds of fusion.
Sorting, Retrieval and Retention
Vesicular traffic between organelles on the secretory pathway is the mechanism by which proteins and lipids are delivered and removed. To allow the organelles to retain their integrity as well as to ensure efficient traffic of cargo by vesicles requires mechanisms for sorting proteins into vesicles, to retrieve proteins that have been inappropriately delivered to another organelle and to retain proteins in an organelle (Fig. 1). Efficient sorting of cell surface membrane receptors into clathrin coated pits was recognized at an early stage in their biochemical characterization. By the late 1970s it was recognised that while some receptors are concentrated into clathrin-coated pits, other plasma membrane proteins are effectively excluded such that the pits act as molecular filters.91,92 An important clue about the molecular basis of such sorting came from analysis of the sequence of the low density lipoprotein receptor in a patient with familial hypocholesterolemia, patient J.D.93 In fibroblasts from this patient, receptor numbers on the cell surface were normal but they were not concentrated into coated pits. The mutation leading to this phenotype was an amino acid substitution in the cytoplasmic domain resulting in a cysteine replacing a tyrosine. Subsequent work showed that cytoplasmic tail motifs of the form NPXY(where X is any amino acid) as in the low density lipoprotein receptor, YXXØ (where Ø is a bulky hydrophobic amino acid), or dileucine motifs could act as efficient endocytosis signals as a result of their interaction with the clathrin adaptor AP-2.94 Membrane proteins without such motifs cannot be efficiently internalized. Cytoplasmic tail sequence motifs containing tyrosine and dileucine are now recognized as being important not only for internalization from the cell surface but also for targeting to organelles within the secretory and endocytic pathways. Different coated vesicle adaptor proteins show subtle differences in specificity for such sequences. The structural basis for such differences is unclear. However, the way in which a YXXØ motif binds to the μ subunit of AP-2 has been determined by X-ray crystallography.95 The recent solving of the complete structure of the core of AP-2 has shown that the μ binding site for YXXØ is blocked, implying a large structural change in the molecule to allow AP-2 to recruit receptors into clathrin-coated pits.72
Not only is there sorting into vesicles for anterograde traffic in the secretory and endocytic pathways, but also sorting into vesicles for retrieval. The concept of retrieval derived initially from studies of lumenal proteins in the endoplasmic reticulum. Munro and Pelham96 showed that a number of lumenal proteins in mammalian endoplasmic reticulum have the sequence KDEL at their carboxy-terminus (HDEL in S. cerevisiae ) and that if this is deleted the proteins escape and are secreted. Subsequently, Pelham's laboratory identified the recycling receptor, Erd2p that is responsible for the retrieval of such proteins from the Golgi complex.97,98 In this retrieval pathway, membrane proteins with di-lysine motifs in their cytoplasmic tails bind to COPI.77 The structural basis for this interaction is not yet understood.
The identity of an organelle is not maintained solely by retrieval but also by retention. Perhaps the clearest example of this is in the cisternae of the Golgi complex where a variety of glycosyl transferases must be retained to carry out their function in the biosynthesis of glycoproteins. These enzymes are type II membrane proteins with trans-membrane domains that are, on average, five amino acids shorter than the trans-membrane domains of plasma membrane proteins.99 During the 1990s it was recognised that the length of the trans-membrane domain rather than its amino acid composition is important to localization, since in the case of sialyl transferase, replacement of the trans-membrane domain by 17 leucines provides efficient retention whereas a longer stretch of leucines does not.99 However, in the case of N-acetylglucosaminyltransferase I, part of the lumenal stalk domain appeared to be sufficient and necessary for retention.100 Two hypotheses have been proposed to explain retention of glycosyl transferases in the Golgi complex, one based on phospholipid bilayer thickness,99 which differs between the Golgi complex and the plasma membrane, and the other entitled “kin recognition” based on the formation of glycosyltransferase hetero-oligomers.101 For an individual membrane protein, it is feasible that both length of trans-membrane domain and interaction with other membrane proteins may contribute to retention. In the trans-Golgi network, the localization of the protein TGN38 depends on both retention provided by the trans-membrane domain and retrieval provided by a YXXØ motif in the cytoplasmic tail.102
Organization into Complex Structures
Organelle biogenesis is not simply a question of delivering newly synthesized proteins and lipids to a specific intracellular site but may also require the establishment of a complex architecture. A dramatic example of this is seen in the case of the Golgi complex where it is clear that the observed morphology in part reflects the interaction of the structure with the cytoskeleton via appropriate motor proteins103 and in part the function of matrix proteins in the organization of the cisternae.104,105 A further complication, particularly for organelles on the secretory and endocytic pathways, is the requirement to maintain morphological form and associated functional integrity despite the large volume of through traffic of both proteins and lipids. In the case of the Golgi complex, there has long been a debate about how secreted proteins pass through it.106 The work of Rothman and colleagues described above on reconstituting traffic through the Golgi complex in a cell-free system suggested anterograde vesicular traffic between the Golgi cisternae. However, electron microscopy studies of large macromolecules, including algal scales and collagen aggregates favoured a maturation model with new cisternae forming on the cis-side and mature ones fragmenting from the trans-side. The cisternal maturation model has been refined to encompass data on retrograde vesicular traffic in COPI coated vesicles such that the present consensus is that most, if not all, anterograde movement through the Golgi complex is the result of cisternal progression with retrograde vesicular traffic ensuring that the polarized distribution of Golgi enzymes in the cisternal stack is maintained (Fig. 1).107 A recent three dimensional reconstruction of the Golgi complex from data obtained by high voltage electron microscopy has suggested that tubular and vesicular structures can bud at every level of the Golgi stack.108 Structurally, using conventional electron microscopy techniques, and functionally, the trans-Golgi network can be distinguished from the cisternal stack and is defined as the site for sorting to different post-Golgi destinations.109 Both clathrin-coated vesicles and noncoated tubular structures appear to bud from the trans-Golgi network. Experiments in which secreted proteins tagged with green fluorescent protein have been imaged as they leave the Golgi complex in living cells have shown that large tubular carriers are particularly important for constitutive traffic to the cell surface.110 In many neuroendocrine cell types, regulated secretory granules are also formed at the trans-Golgi network. Despite the biogenesis of such organelles being amongst the first to be studied by radiolabelling pulse-chase techniques (see above), the mechanisms by which proteins are sorted into these granules remain unclear, with “sorting for entry” and “sorting by retention” models still the subject of much debate.3
In the endocytic pathway, the biogenesis of individual organelles has been less well studied with the exception of lysosomes and the yeast vacuole.111113 This has partly been due to the pleiomorphic morphology of endosomes, partly to the difficulty of identifying marker proteins that, at steady state, are mainly localized in endosomes and partly because the molecular mechanisms of membrane traffic through the pathway have only started to be understood in the last few years. As in the secretory pathway, vesicular traffic between individual organelles does not explain all steps in the pathway. Clathrin-coated vesicles budding from the plasma membrane comprise a very important, but not sole, mechanism of delivery from the plasma membrane to early endosomes (defined historically as the first endosomal compartment to be entered by endocytosed ligands). Traffic from early to late endosomes, found deeper within the cell, has been studied extensively and is mediated by large endocytic carrier vesicles which some regard as matured early endosomes.114,115 Delivery from late endosomes to lysosomes involves “kiss and run” and direct fusion between the two organelles. Such fusion is SNARE-mediated and results in a hybrid organelle from which lysosomes are reformed. In addition to heterotypic fusion between late endosomes and lysosomes, the endocytic pathway is characterized by the occurrence of homotypic fusions between early endosomes and between late endosomes. These homotypic fusion events are also SNARE-mediated116,117 and allow continuous remodelling of these organelles. Organelles in the late endocytic pathway are characterised by the presence of numerous internal vesicles, leading to the alternative description of late endosomes as multivesicular bodies. Some cell surface receptors are sorted into such vesicles after internalization from the plasma membrane and prior to degradation. Recently, insights have been gained into the molecular mechanisms by which proteins are sorted into these vesicles, which have a different lipid composition from the limiting membrane of the organelle. Such mechanisms include partitioning into lipid microdomains, dependent on the composition of trans-membrane domains, and ubiquitination of cytoplamic tail domains followed by recognition of the ubiquinated domain by protein complexes involved in inward vesiculation.118,119
Organelle Inheritance
Organelle biogenesis is closely linked to organelle inheritance in cell division. During the cell cycle, each organelle must double in size, divide and be delivered appropriately to the daughter cells. Historically, the inheritance of organelles was recognised as occurring over the same period of the late 19th and early 20th centuries as the basic mechanics of mitosis were being described.13,120122 In summarizing a large amount of earlier work, Warren and Wickner120 categorized two organelle inheritance strategies that have been described. The first is stochastic, relying on the presence of multiple copies of an organelle randomly distributed throughout the cytoplasm and the second is ordered, often, but not always, using the mitotic spindle as a means of partitioning (Fig. 2). The morphology of many organelles may differ in different cell types, which itself may be related to the use of one or other of these strategies to a greater or lesser extent. Mitochondria, for example are, in many cells, multiple copies of small bean shaped structures, but in the budding yeast S. cerevisiae form an extensive tubular reticulum beneath the plasma membrane which partitions in an ordered way into the bud. The steady-state morphology of mitochondria which continuously grow, divide and fuse throughout the cell cycle is itself largely determined by the frequency of fission events and fusion.123 It should be noted that growth and division of mitochondria also requires coordination of these processes for the inner and outer membranes. In contrast to mitochondria, the endoplasmic reticulum is always a single copy organelle, albeit a dynamic reticulum. This breaks down into tubular vesicular elements during cell division to a variable extent. It often fragments little, thus segregation of equal amounts into daughter cells during mitosis may rely mainly on the uniform and extensive distribution of the endoplasmic reticulum network throughout the cytoplasm of the mother cell. In S. cerevisiae the endoplasmic reticulum becomes anchored at the bud tip pulling the network into the bud as it enlarges.124 Whereas the bulk of the endoplasmic reticulum often does not fragment during mitosis, inheritance of the nuclear envelope, the outer membrane of which is continuous with the endoplasmic reticulum, is more complex since it has to break down during mitosis of animal cells to allow separation of the chromatids. At the end of mitosis the nuclear envelope rapidly reassembles around daughter chromosomes. During the 1980's, nuclear envelope breakdown in animal cells was shown to involve depolymerisation of the lamina underlying the membrane, fragmentation of the membrane and dissassembly of nuclear pore complexes.125 This was accompanied by reversible phosphorylation of many nuclear envelope proteins thought to lead to the formation of a discrete population of vesicles which could fuse at the end of mitosis to reform the envelope. Using Xenopus oocytes, which contain many nuclear components stored for use in early development, it was observed that injection of bacteriophage lambda DNA or its addition to cell-free extracts was sufficient to trigger nuclear assembly.126 The availability of this cell-free system enabled study, at the molecular level, of the pathway of nuclear assembly, including nuclear envelope vesicle fusion.127 Recently, it has been suggested that the nuclear envelope does not have to vesiculate completely during mitosis, but that phosphorylation may allow redistribution of nuclear envelope membrane proteins back into the endoplasmic reticulum.128 The lack of requirement for membrane vesiculation has raised the question of how the nuclear envelope ruptures, resolved by recent evidence that it is literally torn apart by motor protein attachment and movement along microtubules. 128
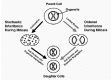
Perhaps the greatest recent controversy concerning organelle inheritance relates to how the Golgi complex is divided between daughter cells at mitosis.7 Two models have been proposed to explain this. In the first, proposed by Warren, the Golgi complex breaks down into vesicle clusters and shed vesicles which are distributed stochastically between the daughter cells where they reassemble in telophase.129 Cell-free assays have led to the identification of some of the molecular machinery for disassembly and reassembly.130 In the second model, proposed by Lippincott-Schwartz, endoplasmic reticulum is the partioning unit, with the Golgi complex merging with the endoplasmic reticulum during prometaphase and emerging from it during telophase.131 A key observation in developing this second model was that inhibition of traffic from the endoplasmic reticulum to the Golgi complex results in disintegration of the latter. Some of the discrepancies between the two models may be resolved by data from Warren's group who have shown that whilst Golgi membrane enzymes may, to a greater or lesser extent, redistribute to the endoplasmic reticulum during mitosis, matrix proteins do not, thus allowing the disassembled matrix to become the partitioning units on which the Golgi complex is reassembled after mitosis.132,133 A further twist has come from the study of the protozoan Toxoplasma gondii which has a single Golgi that divides as a result of lateral cisternal growth followed by medial fission.134 Even in mammalian cells, Golgi fragmentation-dispersion may not be obligatory for equal partitioning. Kondo and colleagues recently found that prevention of Golgi dissassembly, by microinjection of a nonphosphorylated mutant form of a soluble protein required for this process, had no effect on equal partitioning of the Golgi to daughter cells.135
Challenges
It is now clear that intracellular organelles are very dynamic structures, yet at steady state they exhibit characteristic morphology and architecture that are easily observed by microscopy. Recently Misteli133 has suggested that the generation of an overall stable configuration in such dynamic structures is consistent with organelle morphology being determined by self-organization. This is defined as “the capacity of a macromolecular complex or organelle to determine its own structure based on the functional interaction of its components”. Self-organization will ensure structural stability without loss of plasticity. Self-organization is an interesting concept, but how organelles self-organize is unclear. What is certain is that future investigations will lead us to a better understanding of the molecular machinery of organelle biogenesis and inheritance. Such investigations are likely to address a number of questions to which we have few answers at present. These include the role of lipids, in particular lipid-protein interactions in microdomains, in determining morphology and the regulation of the size, shape and number of organelles in cells.
Acknowledgements
We thank the Medical Research Council and the Wellcome Trust for supporting our experimental work on lysosome biogenesis and post-Golgi membrane traffic pathways.
References
- 1.
- Mellman I, Warren G. The road taken: Past and future foundations of membrane traffic. Cell. 2000;100:99–112. [PubMed: 10647935]
- 2.
- In: Kendrew J ed.The Encyclopedia of Molecular Biology Oxford: Blackwell Science 1994 .
- 3.
- Tooze SA. Biogenesis of secretory granules in the trans-Golgi network of neuroendocrine and endocrine cells. Biochim Biophys Acta. 1998;1404:231–244. [PMC free article: PMC7126647] [PubMed: 9714820]
- 4.
- Raposo G, Marks MS. The dark side of lysosome-related organelles: Specialization of the endocytic pathway for melanosome biogenesis. Traffic. 2002;3:237–248. [PubMed: 11929605]
- 5.
- Schatz G. What mitochondria have told me. Mol Biol Cell. 2001;12:777–778. [PMC free article: PMC32264] [PubMed: 11294884]
- 6.
- Nunnari J, Walter P. Regulation of organelle biogenesis. Cell. 1996;84:389–394. [PubMed: 8608592]
- 7.
- Check E. Will the real Golgi please stand up. Nature. 2002;416:780–781. [PubMed: 11976645]
- 8.
- Brown R. Observations on the organs and mode of fecundation in Orchidae and Asclepiadeae. Trans Linn Soc (Lond). 1833;16:685–743.
- 9.
- Schleiden MJ. Beiträge zur Phytogenesis. Müller's Arch Anat Physiol Wiss Med. 1838:136–176.
- 10.
- Schwann T. Mikroskopische untersuchungen über die uberstimmung in der Struktur und dem Wachsthum der Thiere und Pflanzen. Berlin: Verlag der Sander'schen Buchhandlung. 1839
- 11.
- Von Mohl H. Über die vermehrung der pflanzenzellen durch theilung (Inaugural dissertation, Tübingen). 1835 .
- 12.
- Virchow R. Cellular-Pathologie. Arch für Path Anat. 1855;VIII:3–39.
- 13.
- Wilson EB. The cell in development and heredity. 3rd edNY: MacMillan Co.,1925 . [PMC free article: PMC375141]
- 14.
- Flemming W. Zellsubstanz Kern und Zelltheilung Leipzig: FCW Vogel. 1882 .
- 15.
- Richmond M. Thomas Henry Huxley's developmental view of the cell. Nature Rev Mol Cell Biol. 2002;3:61–65. [PubMed: 11823799]
- 16.
- Robertson JD. Membrane structure. J Cell Biol. 1981;91:189s–204s. [PMC free article: PMC2112820] [PubMed: 7033238]
- 17.
- Gorter E, Grendel R. On biomolecular layers of lipids on the chromocytes of the blood. J Exp Med. 1925;41:439–443. [PMC free article: PMC2130960] [PubMed: 19868999]
- 18.
- Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. [PubMed: 4333397]
- 19.
- Feulgen RJ, Rossenbeck H. Mikroskopisch-chemischer Nachweis einer Nucleinsäure vom Typus der Thymonucleinsäure und die darauf beruhende elective Färbung von Zellkernen in mikroscopischen Präparaten. Hoppe-Seyler's Zeit physiol Chem. 1924;135:203–248.
- 20.
- Montgomery TH. Comparative cytological studies with especial reference to the morphology of the nucleolus. J Morphology. 1898;XV:204–265.
- 21.
- Miller OL Jr. The nucleolus, chromosomes and visualization of genetic activity. J Cell Biol. 1981;91:15s–27s. [PMC free article: PMC2112784] [PubMed: 6172428]
- 22.
- Ernster L, Schatz G. Mitochondria: A historical review. J Cell Biol. 1981;91:227s–255s. [PMC free article: PMC2112799] [PubMed: 7033239]
- 23.
- Altmann R. Die Elementarorganismen und ihre Beziehungen zu den Zellen. Leipzig, Veit. 1890
- 24.
- Golgi C. Sur la structure des cellules nerveuses. Arch Ital Biol. 1898;30:60–71.
- 25.
- Farquhar MG, Palade GE. Golgi apparatus (complex) - (1954-1981) - from artifact to center stage. J Cell Biol. 1981;91:77s–103s. [PMC free article: PMC2112780] [PubMed: 7033246]
- 26.
- Bensley RR, Hoerr N. Studies on cell structure by freeze-drying method; preparation and properties of mitochondria. Anat Rec. 1934;60:449–455.
- 27.
- Claude A. Fractionation of mammalian liver cells by differential centrifugation II. Experimental procedures and results. J Exp Med. 1946;84:61–89. [PubMed: 20988751]
- 28.
- De Duve C, Beaufay H. A short history of tissue fractionation. J Cell Biol. 1981;91:293s– 299s. [PMC free article: PMC2112816] [PubMed: 7033241]
- 29.
- Hogeboom GH, Schneider WC, Palade GE. Cytochemical studies of mammalian tissues 1. Isolation of intact mitochondria from rat liver; some biochemical properties of mitochondria and submicroscopic particulate material. J Biol Chem. 1948;172:619–635. [PubMed: 18901182]
- 30.
- De Duve C. Exploring cells with a centrifuge. Science. 1975;189:186–194. [PubMed: 1138375]
- 31.
- Pease DC, Porter KR. Electron microscopy and ultramicrotomy. J Cell Biol. 1981;91:287s–292s. [PMC free article: PMC2112787] [PubMed: 6798043]
- 32.
- Palade GE. Fine structure of mitochondria. Anat Rec. 1952;114:427–451. [PubMed: 12996882]
- 33.
- Dalton AJ, Felix MD. Cytologic and cytochemical characteristics of the Golgi substance of epithelial cells of the epididymis - in situ, in homogenates and after homogenisation. Am J Anat. 1954;94:171–208. [PubMed: 13148119]
- 34.
- Porter KR, Claude A, Fullam EF. A study of tissue culture cells by electron microscopy. J Exp Med. 1945;81:233–246. [PMC free article: PMC2135493] [PubMed: 19871454]
- 35.
- Palade GE. The endoplasmic reticulum. J Biophys Biochem Cytol. 1956;2:85–97. [PMC free article: PMC2229739] [PubMed: 13357527]
- 36.
- Bainton DF. The discovery of lysosomes. J Cell Biol. 1981;91:66s–76s. [PMC free article: PMC2112804] [PubMed: 7033245]
- 37.
- De Duve C, Baudhuin P. Peroxisomes (microbodies and related particles). Physiol Rev. 1966;46:323–357. [PubMed: 5325972]
- 38.
- Jamieson JD, Palade GE. Intracellular transport of secretory proteins in the pancreatic exocrine cell. I. Role of the peripheral elements of the Golgi complex. J Cell Biol. 1967;34:577–596. [PMC free article: PMC2107305] [PubMed: 6035647]
- 39.
- Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358. [PubMed: 1096303]
- 40.
- Roth TF, Porter KR. Yolk protein uptake in the oocyte of the mosquito Aedes aegypti. J Cell Biol. 1964;20:313–332. [PMC free article: PMC2106398] [PubMed: 14126875]
- 41.
- Anderson RGW, Goldstein JL, Brown MS. Localisation of low density lipoprotein receptors on plasma membrane of normal human fibroblasts and their absence in cells from a familial hypercholesterolemia homozygote. Proc Natl Acad Sci USA. 1976;73:2434–2438. [PMC free article: PMC430596] [PubMed: 181751]
- 42.
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during neurotransmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. [PMC free article: PMC2108984] [PubMed: 4348786]
- 43.
- Steinman RM, Brodie SE, Cohn ZA. Membrane flow during pinocytosis. A stereologic analysis. J Cell Biol. 1976;68:665–687. [PMC free article: PMC2109655] [PubMed: 1030706]
- 44.
- Hopkins CR. The importance of the endosome in intracellular traffic. Nature. 1983;304:684–685. [PubMed: 6310401]
- 45.
- Blobel G. Protein targeting (Nobel lecture). Chembiochem. 2000;1:86–102. [PubMed: 11828402]
- 46.
- Rassow J, Pfanner N. The protein import machinery of the mitochondrial membranes. Traffic. 2000;1:457–464. [PubMed: 11208131]
- 47.
- Cline K, Henry R. Import and routing of nucleus-encoded chloroplast proteins. Ann Rev Cell Dev Biol. 1996;12:1–26. [PubMed: 8970720]
- 48.
- Subramani S. Protein import into peroxisomes and biogenesis of the organelle. Ann Rev Cell Devel Biol. 1993;9:445–478. [PubMed: 8280468]
- 49.
- Görlich D, Kutay U. Transport between the cell nucleus and the cytoplasm. Ann Rev Cell Dev Biol. 1999;15:607–660. [PubMed: 10611974]
- 50.
- Tokatlidis K, Schatz G. Biogenesis of mitochondrial inner membrane proteins. J Biol Chem. 1999;274:35285–35288. [PubMed: 10585389]
- 51.
- Mihara K. Targeting and insertion of nuclear-encoded preproteins into the mitochondrial outer membrane. BioEssays. 2000;22:364–371. [PubMed: 10723033]
- 52.
- Schleiff E, Soll J. Travelling of proteins through membranes: Translocation into chloroplasts. Planta. 2000;211:449–456. [PubMed: 11030543]
- 53.
- Purdue PE, Lazarow PB. Peroxisome biogenesis. Ann Rev Cell Dev Biol. 2001;17:701–752. [PubMed: 11687502]
- 54.
- Bayliss R, Corbett AH, Stewart M. The molecular mechanism of transport of macromolecules through nuclear pore complexes. Traffic. 2000;1:448–456. [PubMed: 11208130]
- 55.
- Kornfeld S, Mellman I. The biogenesis of lysosomes. Ann Rev Cell Dev Biol. 1989;5:483–525. [PubMed: 2557062]
- 56.
- Benting JH, Rietveld AG, Simons K. N-Glycans mediate the apical sorting of a GPI-anchored, raft-associated protein in Madin-Darby canine kidney cells. J Cell Biol. 1999;146:313–320. [PMC free article: PMC2156177] [PubMed: 10427087]
- 57.
- Blobel G. Intracellular protein topogenesis. Proc Natl Acad Sci USA. 1980;77:1496–1500. [PMC free article: PMC348522] [PubMed: 6929499]
- 58.
- Margulis L. Origin of eukaryotic cellsNew Haven: Yale University Press,1970 . [PMC free article: PMC322739]
- 59.
- Kurland CG, Andersson SGE. Origin and evolution of the mitochondrial proteome. Microbiol Mol Biol Rev. 2000;64:786–820. [PMC free article: PMC99014] [PubMed: 11104819]
- 60.
- Rothman J. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. [PubMed: 7969419]
- 61.
- Pearse BM. Clathrin: A unique protein associated with intracellular transfer of membrane by coated vesicles. Proc Natl Acad Sci USA. 1976;73:1255–1259. [PMC free article: PMC430241] [PubMed: 1063406]
- 62.
- Fries E, Rothman JE. Transport of vesicular stomatitis virus glycoprotein in a cell-free extract. Proc Natl Acad Sci USA. 1980;77:3870–3874. [PMC free article: PMC349728] [PubMed: 6253996]
- 63.
- Novick P, Field C, Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. [PubMed: 6996832]
- 64.
- Balch WE, Dunphy WG, Braell WA. et al. Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 1984;39:405–416. [PubMed: 6498939]
- 65.
- Balch W, Glick BS, Rothman JE. Sequential intermediates in the pathway of intercompartmental transport in a cell-free system. Cell. 1984;39:525–536. [PubMed: 6096009]
- 66.
- Block MR, Glick BS, Wilcox CA. et al. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci USA. 1988;85:7852–7856. [PMC free article: PMC282295] [PubMed: 3186695]
- 67.
- Robinson JS, Klionsky DJ, Banta LM. et al. Protein sorting in Saccharomyces cerevisiae: Isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol. 1988;8:4936–4948. [PMC free article: PMC365587] [PubMed: 3062374]
- 68.
- Raymond CK, Howald-Stevenson I, Vater CA. et al. Morphological classification of the yeast vacuolar protein sorting mutants: Evidence for a prevacuolar compartment in Class E vps mutants. Mol Biol Cell. 1992;3:1389–1402. [PMC free article: PMC275707] [PubMed: 1493335]
- 69.
- Noda T, Suzuki K, Ohsumi Y. Yeast autophagosomes: De novo formation of a membrane structure. Trends Cell Biol. 2002;12:231–235. [PubMed: 12062171]
- 70.
- Seaman MNJ, Luzio JP. Lysosomes and other late compartments of the endocytic pathwayIn: Endocytosis: Frontiers in Molecular BiologyOxford: Oxford University Press,2001111–148.
- 71.
- Seeley ES, Kato M, Margolis N. et al. Genomic analysis of homotypic vacuole fusion. Mol Biol Cell. 2002;13:782–794. [PMC free article: PMC99598] [PubMed: 11907261]
- 72.
- Collins BM, McCoy AJ, Kent HM. et al. Molecular architecture and fuctional model of endocytic AP-2 complex. Cell. 2002;109:523–535. [PubMed: 12086608]
- 73.
- Pearse BM, Robinson MS. Clathrin, adaptors, and sorting. Ann Rev Cell Biol. 1990;6:151–171. [PubMed: 2177341]
- 74.
- Robinson MS, Bonifacino JS. Adaptor-related proteins. Curr Opin Cell Biol. 2001;13:444–453. [PubMed: 11454451]
- 75.
- Kelly RB. New twists for dynamin. Nature Cell Biol. 1999;1:E8–E9. [PubMed: 10559872]
- 76.
- Eugster A, Frigerio G, Dale M. et al. COP I domains required for coatomer integrity, and novel interactions with ARF and ARF-GAP. EMBO J. 2000;19:3905–3917. [PMC free article: PMC306616] [PubMed: 10921873]
- 77.
- Letourneur F, Gaynor EC, Hennecke S. et al. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. [PubMed: 8001155]
- 78.
- Matsuoka K, Orci L, Amherdt M. et al. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. [PubMed: 9568718]
- 79.
- Sollner T, Whiteheart SW, Brunner M. et al. Protein SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. [PubMed: 8455717]
- 80.
- McNew JA, Parlati F, Fukuda R. et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 2000;407:153–159. [PubMed: 11001046]
- 81.
- Pelham HR. SNAREs and the secretory pathway-lessons from yeast. Exp Cell Res. 1999;247:1–8. [PubMed: 10047442]
- 82.
- Sutton RB, Fasshauer D, Jahn R. et al. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. [PubMed: 9759724]
- 83.
- Fasshauer D, Sutton RB, Brunger AT. et al. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci USA. 1998;95:15781–15786. [PMC free article: PMC28121] [PubMed: 9861047]
- 84.
- Bock JB, Matern HT, Peden AA. et al. A genomic perspective on membrane compartment organization. Nature. 2001;409:839–841. [PubMed: 11237004]
- 85.
- Weber T, Zemelman BV, McNew JA. et al. SNAREpins: Minimal machinery for membrane fusion. Cell. 1998;92:759–772. [PubMed: 9529252]
- 86.
- Fukuda R, McNew JA, Weber T. et al. Functional architecture of an intracellular membrane t-SNARE. Nature. 2000;407:198–202. [PubMed: 11001059]
- 87.
- Chavrier P, Parton RG, Hauri HP. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62:317–329. [PubMed: 2115402]
- 88.
- Pfeffer SR. Transport-vesicle targeting: Tethers before SNAREs. Nature Cell Biol. 1999;1:E17–E22. [PubMed: 10559876]
- 89.
- Peters C, Mayer A. Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature. 1998;396:575–580. [PubMed: 9859992]
- 90.
- Pryor PR, Buss F, Luzio JP. (2000) Calcium, calmodulin and the endocytic pathway ELSO Gaz 20002(http://www.the-elso-gazette.org/magazines/issue2/reviews/reviews1_pr.asp).
- 91.
- Goldstein J, Anderson RG, Brown MS. Coated pits, coated vesicles, and receptor-mediated endocytosis. Nature. 1979;279:679–685. [PubMed: 221835]
- 92.
- Bretscher MS, Thomson JN, Pearse BM. Coated pits act as molecular filters. Proc Natl Acad Sci USA. 1980;77:4156–4159. [PMC free article: PMC349789] [PubMed: 6968906]
- 93.
- Davis CG, Lehrman MA, Russell DW. et al. The J.D. mutation in familial hypercholesterolemia: Amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell. 1986;45:15–24. [PubMed: 3955657]
- 94.
- Bonifacino JS, Dell'Angelica EC. Molecular bases for the recognition of tyrosine-based sorting signals. J Cell Biol. 1999;145:923–926. [PMC free article: PMC2133128] [PubMed: 10352010]
- 95.
- Owen DJ, Evans PR. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science. 1998;282:1327–1332. [PMC free article: PMC5600252] [PubMed: 9812899]
- 96.
- Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. [PubMed: 3545499]
- 97.
- Semenza JC, Hardwick KG, Dean N. et al. ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell. 1990;61:1349–1357. [PubMed: 2194670]
- 98.
- Lewis MJ, Sweet DJ, Pelham HR. The ERD2 gene determines the specificity of the luminal ER protein retention system. Cell. 1990;61:1359–1363. [PubMed: 2194671]
- 99.
- Bretscher MS, Munro S. Cholesterol and the Golgi apparatus. Science. 1993;261:1280–1281. [PubMed: 8362242]
- 100.
- Nilsson T, Rabouille C, Hui N. et al. The role of the membrane-spanning domain and stalk region of N-acetylglucosaminyltransferase I in retention, kin recognition and structural maintenance of the Golgi apparatus in HeLa cells. J Cell Sci. 1996;109:1975–1989. [PubMed: 8832420]
- 101.
- Nilsson T, Slusarewicz P, Hoe MH. et al. Kin recognition. A model for the retention of Golgi enzymes. FEBS Lett. 1993;330:1–4. [PubMed: 8370450]
- 102.
- Reaves BJ, Banting G, Luzio JP. Lumenal and transmembrane domains play a role in sorting type I membrane proteins on endocytic pathways. Mol Biol Cell. 1998;9:1107–1122. [PMC free article: PMC25333] [PubMed: 9571243]
- 103.
- Burkhardt JK. The role of microtubule-based motor proteins in maintaining the structure and function of the Golgi complex. Biochim Biophys Acta. 1998;1404:113–126. [PubMed: 9714769]
- 104.
- Pfeffer SR. Constructing a Golgi complex. J Cell Biol. 2001;155:873–876. [PMC free article: PMC2150916] [PubMed: 11739400]
- 105.
- Barr FA. The Golgi apparatus: Going round in circles? Trends Cell Biol. 2002;12:101–104. [PubMed: 11859015]
- 106.
- Pelham HR, Rothman JE. The debate about transport in the Golgi-two sides of the same coin? Cell. 2000;102:713–719. [PubMed: 11030615]
- 107.
- Pelham HR. Traffic through the Golgi apparatus. J Cell Biol. 2001;155:1099–1101. [PMC free article: PMC2199341] [PubMed: 11756463]
- 108.
- Marsh BJ, Mastronarde DN, Buttle KF. et al. Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT-T15, visualized by high resolution electron tomography. Proc Natl Acad Sci USA. 2001;98:2399–2406. [PMC free article: PMC30150] [PubMed: 11226251]
- 109.
- Griffiths G, Simons K. The trans Golgi network: Sorting at the exit site of the Golgi complex. Science. 1986;234:438–443. [PubMed: 2945253]
- 110.
- Lippincott-Schwartz J, Roberts TH, Hirschberg K. Secretory protein trafficking and organelle dynamics in living cells. Ann Rev Cell Dev Biol. 2000;16:557–589. [PMC free article: PMC4781643] [PubMed: 11031247]
- 111.
- Bryant NJ, Stevens TH. Vacuole biogenesis in Saccharomyces cerevisiae: Protein transport pathways to the yeast vacuole. Microbiol Mol Biol Rev. 1998;62:230–247. [PMC free article: PMC98912] [PubMed: 9529893]
- 112.
- Luzio JP, Rous BA, Bright NA. et al. Lysosome-endosome fusion and lysosome biogenesis. J Cell Sci. 2000;113:1515–1524. [PubMed: 10751143]
- 113.
- Mullins C, Bonifacino JS. The molecular machinery for lysosome biogenesis. Bioessays. 2001;23:333–343. [PubMed: 11268039]
- 114.
- Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. [PubMed: 7495576]
- 115.
- Gu F, Gruenberg J. Biogenesis of transport intermediates in the endocytic pathway. FEBS Lett. 1999;452:61–66. [PubMed: 10376679]
- 116.
- Antonin W, Holroyd C, Fasshauer D. et al. A SNARE complex mediating fusion of late endosomes defines conserved properties of SNARE structure and function. EMBO J. 2000;19:6453–6464. [PMC free article: PMC305878] [PubMed: 11101518]
- 117.
- Antonin W, Fasshauer D, Becker S. et al. Crystal structure of the endosomal SNARE complex reveals common structural principles of all SNAREs. Nat Struct Biol. 2002;9:107–111. [PubMed: 11786915]
- 118.
- Piper RC, Luzio JP. Late endosomes: Sorting and partitioning in multivesicular bodies. Traffic. 2001;2:612–621. [PubMed: 11555415]
- 119.
- Raiborg C, Bache KG, Gillooly DJ. et al. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol. 2002;4:394–398. [PubMed: 11988743]
- 120.
- Warren G, Wickner W. Organelle inheritance. Cell. 1996;84:395–400. [PubMed: 8608593]
- 121.
- Mitchison TJ, Salmon ED. Mitosis: A history of division. Nat Cell Biol. 2001;3:E17–21. [PubMed: 11146645]
- 122.
- Paweletz N. Walther Flemming: Pioneer of mitosis research. Nature Reviews Molecular Cell Biology. 2001;2:72–75. [PubMed: 11413469]
- 123.
- Shaw JM, Nunnari J. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 2002;12:178–184. [PMC free article: PMC3785940] [PubMed: 11978537]
- 124.
- Fehrenbacher KL, Davis D, Wu M. et al. Endoplasmic reticulum dynamics, inheritance, and cytoskeletal interactions in budding yeast. Mol Biol Cell. 2002;13:854–865. [PMC free article: PMC99604] [PubMed: 11907267]
- 125.
- Gerace L, Burke B. Functional organization of the nuclear envelope. Ann Rev Cell Biol. 1988;4:335–374. [PubMed: 2461721]
- 126.
- Newport J. Nuclear reconstitution in vitro: Stages of assembly around protein-free DNA. Cell. 1987;48:205–217. [PubMed: 3026635]
- 127.
- Grant TM, Wilson KL. Nuclear Assembly. Ann Rev Cell Dev Biol. 1997;13:669–695. [PubMed: 9442884]
- 128.
- Aitchison JD, Rout MP. A tense time for the nuclear envelope. Cell. 2002;108:301–304. [PubMed: 11853664]
- 129.
- Shima DT, Haldar K, Pepperkok R. et al. Partitioning of the Golgi apparatus during mitosis in living HeLa cells. J Cell Biol. 1997;137:1211–1228. [PMC free article: PMC2132532] [PubMed: 9182657]
- 130.
- Rabouille C, Kondo H, Newman R. et al. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell. 1998;92:603–610. [PubMed: 9506515]
- 131.
- Zaal KJ, Smith CL, Polishchuk RS. et al. Golgi membranes are absorbed into and reemerge from the ER during mitosis. Cell. 1999;99:589–601. [PubMed: 10612395]
- 132.
- Seemann J, Pypaert M, Taguchi T. et al. Partitioning of the matrix fraction of the Golgi apparatus during mitosis in animal cells. Science. 2002;295:848–851. [PubMed: 11823640]
- 133.
- Horter J, Warren G. Golgi architecture and inheritance. Annu Rev Cell Dev Biol. 2002;18:379–420. [PubMed: 12142281]
- 134.
- Pelletier L, Stern CA, Pypaert M. et al. Golgi biogenesis in Toxoplasma gondii. Nature. 2002;418:548–552. [PubMed: 12152082]
- 135.
- Uchiyama K, Jokitalo E, Lindman M. et al. The localization and phosphorylation of p47 are important for Golgi disassembly-assembly during the cell cycle. J Cell Biol. 2003;161:1067–1079. [PMC free article: PMC2173005] [PubMed: 12810701]
- 136.
- Misteli T. The concept of self-organization in cellular architecture. J Cell Biol. 2001;155:181–185. [PMC free article: PMC2198832] [PubMed: 11604416]
- 137.
- Warren G. Membrane traffic and organelle division. Trends Biochem Sci. 1985;10:439–443.
- Theory of Organelle Biogenesis: A Historical Perspective - Madame Curie Bioscien...Theory of Organelle Biogenesis: A Historical Perspective - Madame Curie Bioscience Database
- Pseudouridine Formation, the Most Common Transglycosylation in RNA - Madame Curi...Pseudouridine Formation, the Most Common Transglycosylation in RNA - Madame Curie Bioscience Database
- Modeling Metastasis In Vivo - Madame Curie Bioscience DatabaseModeling Metastasis In Vivo - Madame Curie Bioscience Database
- The Immune-Suppressive Effects of Pain - Madame Curie Bioscience DatabaseThe Immune-Suppressive Effects of Pain - Madame Curie Bioscience Database
- β-Secretase: Progress and Open Questions - Madame Curie Bioscience Databaseβ-Secretase: Progress and Open Questions - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...