

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Salivary submandibular gland (SMG) morphogenesis is regulated by the functional integration of stage-specific growth factor- , cytokine- and transcription factor-mediated signaling which mediates specific patterns of cell proliferation, cell quiescence, apoptosis, and histodifferentiation. We describe the stage-specific distribution of protein components of key signaling pathways during embryonic SMG development and correlate their protein expression patterns with cell proliferation and apoptosis. We then review the critical role of the Fibroblast Growth Factor (FGF), Hedgehog (Hh) and Ectodysplasin (Eda) signaling pathways and discuss how they may interact within the context of a nonlinear genetic network.
Introduction
Morphogenesis of complex organs such as the submandibular salivary gland (SMG) requires cooperation and coordination of multiple signaling pathways to regulate cell proliferation, quiescence, apoptosis, and histodifferentiation.14 Its development is regulated by the functional integration of stage-specific growth factor- , cytokine- and transcription factor-mediated signaling which mediates these cellular events.1,511 In this review, we will focus on those signaling pathways which have been shown to play important and essential morphoregulatory roles during SMG organogenesis and discuss their functional relationships in the context of a dynamic, nonlinear genetic network.
Mouse submandibular salivary gland organogenesis is initiated with a thickening of the oral epithelium of the mandibular arch around E11 and is best conceptualized in stages: Prebud, Initial Bud, Pseudoglandular, Canalicular and Terminal Bud Stages (Fig. 1). In the Prebud Stage, SMG development begins as a thickening of the primitive oral cavity epithelium adjacent to the tongue. During the Initial Bud Stage, this thickening epithelium grows down into the first branchial (mandibular) arch mesenchyme to form the initial SMG bud. With continued epithelial proliferation and downgrowth, the SMG primordium becomes a solid, elongated epithelial stalk terminating in a bulb. The primordium branches by repeated furcation in the distal ends of successive buds to produce a bush-like structure comprised of a network of epithelial branches and terminal epithelial buds (the Pseudoglandular Stage). These branches and buds hollow out by epithelial cell apoptosis during the Canalicular and Terminal Bud Stages to form the ductal system and presumptive acini (for details, see refs. 7, 12). Epithelial cell proliferation is found in all stages, even after well-defined lumen formation in the Terminal Bud Stage. By contrast, epithelial apoptosis begins with the onset of lumen formation in the Canalicular Stage. Moreover, our studies suggest that ductal canalization is primarily due to caspase 8-mediated apoptosis whereas p53 primarily mediates terminal bud lumina formation. 1,7,1314 In addition, although we found three pro-survival/anti-apoptotic proteins (NF-κB, bcl2, and survivin) in lumen-bounding epithelial cells during canalization, the presence of only nuclear-localized (activated) survivin in the layer of surviving cells after the completion of lumen formation suggests that survivin mediates terminal bud cell survival.15 Based on our data, we conclude that apoptosis-mediated lumen formation and cell survival are achieved through several different pathways.

To delineate the molecules and signaling pathways involved in the induction and regulation of embryonic SMG morphogenesis, we first determined the stage-specific distribution of relevant growth factors and cytokines (and their receptors) and transcription factors and correlated their protein expression patterns with cell proliferation and apoptosis (Fig. 1; see refs. 7, 12, 16–17). Our identification of the protein components of the Fibroblast Growth Factor (FGF), Epidermal Growth Factor (EGF), Hedghog (Hh), Ectodysplasin (Eda), Insulin-like Growth Factor (IGF), Transforming Growth Factor-β (TGF-β) and Tumor Necrosis Factor (TNF) mediated signaling cascades suggests their importance during embryonic SMG development. The marked overlaps in multiple ligand and receptor localization patterns suggest redundancy in their functions. In addition, the nearly exclusive epithelial localization of protein components of key signaling cascades in Pseuodoglandular Stage and older SMGs suggests that, in these stages, branching morphogenesis and histodifferentiation are primarily mediated by epithelial-epithelial interactions and not epithelial-mesenchymal interactions.
SMG organogenesis is due to the functional integration of parallel and broadly related signaling pathways (for review see refs. 1, 7, 17). To begin to understand the complex interactions within this dynamic signaling network, one must first determine the contribution of individual pathways and identify those which are important and necessary for embryonic SMG development. Analyses of knock-out, transgenic and mutant mice provide insights into which of the signaling pathways present in the developing SMG play essential morphogenetic roles810 (for review see refs. 7, 17). Of particular note are the following genotype/phenotype observations: (1) SMG aplasia in Fgfr2-IIIb-/- , Fgf10-/- , and Fgf8 conditional mutant mice; 10,1619 (2) SMG dysplasia in Edardl (downless) mutant mice;8 and (3) SMG dysplasia in Shh-/- mice.9 These results suggest that the FGF/FGFR, ShhPtc, and Eda/Edar signaling cascades are critical for SMG organogenesis.
FGF/FGFR Signaling
The FGF family includes at least 23 members which have been shown to have diverse biological functions, including cell proliferation, epithelial branching and histodifferentiation (for review see refs. 20–23). FGF function is through a family of five transmembrane tyrosine-kinase receptors (FGFRs). Alternate splicing of Fgfr1, Fgfr2, and Fgfr3 generates receptor isoforms (e.g., IIIb and IIIc) with different ligand-binding specificities. Ligand binding to the appropriate receptor results in receptor dimerization, activation of the intrinsic tyrosine kinase activity and autophosphorylation which activates several intracellular cascades, including the Ras pathway, Src family tyrosine kinases, phosphoinositide 3-kinase/AKT (PI3K/AKT), the PLC-γ/PKC (phospholopase-Cγ/protein kinase C) pathway, and the STAT3 pathway (Fig. 2; for review see refs. 21–23). Although FGF receptors have the ability to activate multiple pathways simultaneously, the variable pleitrophic effects of different FGF/FGFR signaling pathways are associated with clearly distinguishable signaling which induce different downstream targets according to a cell's need. For example, the RAS pathway has been shown to be critical for cell proliferation and differentiation whereas the PI3K/AKT pathway plays a major prosurvival/anti-apoptotic role.6,2425
Figure 2
FGF/FGFR signaling pathway. Known and putative connections are based on published results (e.g., ).
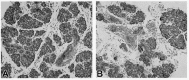
Regarding FGFR2 signaling, gene targeting has clearly demonstrated that both FGFR2 isoforms, FGFR2-IIIb and FGFR2-IIIc, play critical roles during SMG organogenesis. Although an initial SMG bud is present in Fgfr2-IIIb-/- mice (De Moerlooze, Jaskoll and Melnick, unpublished), no gland is found in E14.5 and older mutant mice.18 The observation of a similar SMG phenotype in Fgf10 null mice,19 as well as the absence of abnormalities in Fgf7 null mice,26 indicate that FGF10 is the ligand responsible for the Fgfr2-IIIb mutant phenotype. Since the absence of a SMG primordium in E14.5 and older Fgfr2-IIIb mutant mice made it impossible to determine if FGF10/FGFR2-IIIb signaling regulates branching morphogenesis, we then investigated the effect of enhanced or reduced FGF10/FGFR2-IIIb signaling on SMG development in vitro. Exogenous FGF10 peptide supplementation induced a significant 55% (P<0.01) increase in epithelial branching in embryonic SMGs compared to control. Abrogation of FGFR2-IIIb signaling with anti-Fgfr2-IIIb oligonucleotides (ODNs) resulted in a significant dose-dependent decrease in branching morphogenesis compared to sense controls (Fig. 3). Together these results indicate that FGF10/FGFR2-IIIb signaling plays a critical role for SMG branching morphogenesis and cell survival, but not initial bud formation.

Since Fgfr2-IIIc null mutations are embryolethal, we analyzed the role of FGFR2-IIIc signaling in Fgfr2-IIIc deficient mice.16 Fgfr2-IIIc+/Δ mutant SMGs are smaller, with fewer terminal buds compared to wildtype glands. We then confirmed the functional importance of the FGFR2-IIIc pathway for branching morphogenesis using our organ culture system. Anti-Fgfr2-IIIc ODN-mediated reduction in FGFR2-IIIc signaling resulted in a significant dose-dependent decrease in branching compared to sense control (Fig. 4). Based on these results, we conclude that FGFR2-IIIc signaling plays an important role during SMG branching morphogenesis. What remained unclear was whether, like the FGFR2-IIIb pathway discussed above, FGFR2-IIIc signaling is critical and essential for embryonic SMG cell survival.

To address this question, we turned our attention to which FGF ligand likely induces the FGFR2-IIIc signal during embryonic SMG development. Although FGF8 isoforms have been shown to bind with high affinity to FGFR2-IIIc, FGFR3-IIIc and FGFR4,27,28 the absence of FGFR3 and FGFR4 from Initial Bud Stage and older SMGs 16 indicates that FGF8 mediates its signal through FGFR2-IIIc. Moreover, although FGF2, FGF4, FGF6 and FGF8 and FGF9 bind with high affinity to FGFR2-IIIc (see reviews, 20–22), the normal SMG phenotype in Fgf4 null mice (A. Moon, E.J. Park, L. Francis, unpublished), the relatively normal appearance of Fgf2 and Fgf6 null mice,2930 and the absence of Fgf9 transcripts from embryonic SMGs 31 indicate that FGF8 is the important FGFR2-IIIc ligand for SMG morphogenesis. Thus, the abnormal SMG phenotype seen with interrupted FGFR2-IIIc signaling in vivo and in vitro is primarily due to diminished FGF8/FGFR2-IIIc signaling.
We then investigated the SMG phenotypes in Fgf8 hypomorphic and tissue-specific conditional mutant mice.10 Like the phenotype seen in Fgfr2-IIIc deficient mice, we see hypoplastic glands in Fgf8 hypomorphic mice. By contrast, SMG aplasia is seen in Fgf8 conditional mutant mice in which Fgf8 expression was completely ablated from the first pharyngeal arch ectoderm. Our observation of SMG hypoplasia and aplasia in Fgf8 hypomorphs and conditional mutant mice, respectively, indicates that FGF8/FGFR2-IIIc signaling plays an essential role for branching and survival of Pseudoglandular Stage and older SMGs, acting in a dose-dependent manner. Importantly, the functional presence of other endogenous FGF/FGFR pathways (e.g., FGF10/FGFR2-IIIb) or other signaling pathways (e.g., TGF-α/EGF/EGFR, IGRII/IGFR1) could not prevent the complete death of embryonic SMG cells in the Fgf8 conditional mutant mice. These results indicate that the FGF10/FGFR2-IIIb and FGF8/ FGFR2-IIIc signaling pathways induce unique and specific downstream cascades during embryonic SMG development that cannot be compensated by other pathways.
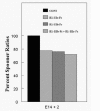
In addition, since FGFR1 is the only other FGF receptor found at all stages of embryonic SMG development (Fig. 1) and FGFR1 and FGFR2 have the ability to induce similar downstream cascades, we postulated that FGF/FGFR1 signaling also plays an important role during embryonic SMG development. However, the early embryonic death of Fgfr1 null mice 32 precluded any investigation into the role of FGFR1 signaling in later stages of development. Recently, our collaborator Mohammed Hajihosseini generated transgenic mice which express a mutant Fgfr1 gene and showed a direct relationship between mutant Fgfr1 copy number and severity of defects.33 We investigated the SMG phenotype in these Fgfr1 transgenic mice and found smaller glands composed of fewer epithelial branches in mutants compared to wildtype mice (Fig. 5). The observation of distinct lumina in terminal buds in both wildtype and mutant glands indicates that altered levels of FGFR1-mediated signaling inhibits epithelial branching but not histodifferentiation. In vitro studies are consistent with these findings. Abrogation of FGFR1 signaling in vitro with antisense ODNs34 or SU5402 peptide treatment,34 Jaskoll and Melnick, unpublished) results in a substantial decrease in branching. Taken together, these in vivo and in vitro studies indicate that the FGF/FGFR1 signaling cascade is important for SMG branching morphogenesis.
Since the IIIb and IIIc isoforms of FGFR1 bind different ligands (FGF1, 2, 4, 5, and 6 bind to FGFR1-IIIb,35 FGF1, 2 and 10 bind to FGFR1-IIIb28) and may elicit different downstream responses during embryonic SMG development, we then investigated the effect of reduced FGFR1-IIIb and FGFR1-IIIc signaling in vitro. Exogenous soluble FGFR1-IIIb-Fc or FGFR1-IIIc-Fc chimera was added to the culture medium to competitively bind endogenous FGFR1-IIIb or FGFR1-IIIc ligands. Abrogated FGFR1-IIIb or FGFR1-IIIc signaling resulted in a highly significant ˜23% decrease in branching morphogenesis compared to controls (Fig. 6). Embryonic SMGs primordia were also cultured in a combination of FGFR1-IIIb-Fc + FGFR1-IIIc-Fc chimeras. This combined treatment reduction of 28% reduction compared to control (Fig. 6) is not different from that seen with either isoform chimera alone; thus, there is no synergism. One possible explanation is that both receptor isoforms share identical ultimate downstream pathways during embryonic SMG organogenesis; thus interruption of either FGFR1-IIIb or FGFR1-IIIc signaling (or both) interrupts the same downstream cascade. What remains to be determined are the specific downstream targets of FGFR1-IIIb and FGFR1-IIIc signaling and whether they are unique and required for SMG development.
Hedgehog Signaling
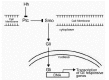
Sonic hedgehog (Shh) is a member of the hedgehog (Hh) family of signaling molecules that induces cell survival, proliferation, differentiation and pattern formation in various embryonic tissues (for review see ref. 36). The cellular response to Hh is controlled by two transmembrane proteins, Patched 1 (Ptc) and Smoothened (Smo) (Fig. 7). Ptc acts as a negative regulator of the Hh signal whereas Smo is a positive activator. Hh binding to Ptc relieves the inhibition of Smo signaling to initiate a signaling cascade that results in the activation of target genes via the Gli family of transcription factors, including genes associated with cell cycle progression.3638
Data from knockout mice clearly indicate that the Shh signaling cascade is essential for many aspects of mammalian embryogenesis, including neural tube, craniofacial, limb, and kidney development (for review see ref. 10). Given that (1) Shh null mice are characterized by cyclopia, holoprosenchepaly and the virtual absence of mandibular derivatives,39 (2) Shh is essential for neural crest cell survival,40 and (3) the SMG initial bud develops as an invagination of the oral epithelium into the underlying neural crest-derived mesenchyme of the mandibular arch,12 we postulated that the Shh null SMG would be absent. Much to our surprise, we found a severely dysplastic gland in Shh-/- mice.9 The presence of a small, severely abnormal SMG primordium, as well as our observation that enhanced Shh signaling induced, and abrogated signaling decreased, SMG branching morphogenesis in vitro,9 indicate that Shh signaling is critical for embryonic SMG branching morphogenesis, most likely by regulating epithelial cell proliferation.
Eda/Edar Signaling
Ectodysplasin (Eda) and its receptor (Edar) are members of the TNF superfamily shown to play critical roles during the development of ectodermal derivatives, including teeth, hair and sweat glands.4142 Eda-A1, the biologically active isoform, binds specifically to Edar activate NF-κB translocation into the nucleus to regulate the transcription of NF-κB responsive genes, including genes associated with DNA repair, cell cycle progression, cell survival, and apoptosis (Fig. 8; see refs. 43–44). (In this review, Eda is used to designate the biologically active ligand, Eda-A1).Loss of Eda or Edar function in humans results in hypohidrotic ectodermal dysplasia which is characterized by absent or hypoplastic teeth, hair, and sweat glands;4142,4546 similar phenotypes are seen in Tabby (EdaTa; Eda-less) and downless (Edardl; Edar-less) mutant mice.4142,45 Our previous demonstration that TNF/TNFR signaling is important for balancing mitogenesis and apoptosis during embryonic SMG development13 and that the NF-κB cascade (the primary downstream pathway of Eda/Edar signaling) plays an essential role for SMG epithelial cell proliferation and survival1 suggested that the Eda/Edar pathway is essential for SMG development. Thus, we analyzed Tabby (EdaTa) and downless (Edadl) mutant mouse SMGs and found mutation-specific phenotypic abnormalities.8 The Tabby SMG is hypoplastic whereas the downless gland is dysplastic. Since Eda ligand is present in downless mice, their more severe abnormal SMG phenotype indicates that no other receptor can compensate for Edar's functional absence. The decrease or absence of SMG ducts, acini and mucin protein in Tabby and downless SMGs, respectively, indicate that Eda/Edar signaling is essential for branching morphogenesis, lumina formation, and histodifferentiation. Complementary in vitro studies provide additional mechanistic data in support of our conclusion. Enhanced Eda/Edar signaling in vitro induces, and abrogated signaling decreases, branching morphogenesis as well as the activation of NF-κB.8 What remains to be determined are which signaling cascades downstream of the Eda/Edar/NF-κB signal are important for SMG organogenesis.
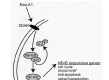
Downstream Targets Rescue SMG Phenotypes
To help delineate the morphogenetic role of any individual signaling pathway, developmental biologists have analyzed the effect of reduced/absent signaling on the expression of a select number of candidate growth factors or receptors (e.g., see refs. 47–49) and determined if supplementation with a downstream factor could rescue the abnormal phenotype in vitro (e.g., see refs. 50–52). Since FGF8 is a target downstream of the Shh signal,9,49 we determined if FGF8 peptide supplementation in vitro could rescue the abnormal SMG phenotype seen with decreased Shh/Ptc signaling.9 FGF8 supplementation restored SMG branching morphogenesis to normal. Similarly, exogenous Shh and FGF10, physiologic downstream targets of FGF8/ FGFR2-IIIc signaling,10,47,53 rescued the abnormal SMG phenotype seen with decreased FGF8/FGFR2-IIIc signaling in vitro.10 In addition, FGF7, FGF10 and BMP7, physiologic downstream targets of FGF/FGFR1 signaling, rescued the SMG phenotype seen with decreased FGF/FGFR1 in vitro.34 These rescue experiments indicate that enhancement of a downstream signaling pathway can compensate for decreased signaling in a single pathway and can restore SMG branching morphogenesis. Nevertheless, the observation of abnormal SMG phenotypes in the transgenic and mutant mice discussed above indicate that such compensatory signaling does not occur during normal in vivo SMG organogenesis.
A Nonlinear Genetic Network Regulates SMG Organogenesis
Over the last decade, it has become increasingly apparent that organogenesis is the programmed expression of regulatory genes coupled to downstream structural genes and epigenetic events.13 This process is dependent on the functional integration of diverse signal transduction pathways that transmit information between and within cells. The cellular and extracellular components may be visualized as a Connections Map which details the functional relationships within and between pathways (Fig. 9). Specific growth factor- and cytokine-mediated signal transduction pathways are parallel and largely functionally redundant; that is, some pathways differentially and combinatorially compensate for the dysfunction of a given individual pathway. However, other pathways have unique and nonredundant functions since no other pathways(s) can compensate in full for their absence/dysfunction. These nonredundant pathways are the key signaling cascades which regulate the cell dynamics of the developing SMG.
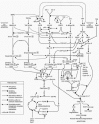
Figure 9
Connections Map. This signaling map reflects the pathways investigated in our laboratory. Known and putative connections are based on work in our laboratory and the laboratories of others.
In this genetic network, ligand/receptor binding initiates the activation of wide array of broadly related, not independent, transcription factor cascades. Many of these signaling cascades converge at a single factor, a so-called “hub,” which then induces a myriad of downstream cellular responses. A prime example is NF-κB (Fig. 9). A large number of pathways activate NF-κB which, in turn, induces more than 150 downstream responses.54 Regarding NF-κB's role during embryonic SMG development, we have previously demonstrated that interruption of NF-κB activation in vitro results in a small gland characterized by a highly significant decrease in cell proliferation and a significant increase in apoptosis, as well as the altered expression of a multiple genes and proteins.1 These include signal transduction, translation control (checkpoint), cell cycle, and apoptosis genes and proteins that are downstream from the Eda/Edar, TNF/TNFR, FGF/FGFR, TGF-α/EGF/EGFR, and IGFII/IGFRI signals (Fig. 9).
The identification of how these genetic pathways functionally interact during SMG organogenesis in vivo provide insight into whether an individual pathway is redundant or unique (non redundant). Such information can be obtained from knock-out, transgenic and mutant mice. The observation of normal SMGs in TGF- β1-/-, TGF- β2-/-, TGF- β3-/-, Msx2-/-, Pax9-/-, Hoxa5-/-, Gli1-/-, Gli2-/-, and Gli3-/- mice indicate that other signaling cascades completely compensate for the absence of each single pathway to restore the phenotype to normal. Thus, these pathways are not the sine qua non of embryonic SMG development; therefore they are redundant (Table 1).
Table 1
Molecular pathways: Functional relationships.
In contrast, the absence of SMGs in Fgf8 conditional mutants, Fgf10-/- and Fgfr2-IIIb-/- mice indicates that endogenous levels of other FGFR-mediated or parallel signaling pathways (TGF-α/EGF/EGFR, IGF/IGFR) could not compensate for the absence of FGF10/FGFR2-IIIb or FGF8/FGFR2-IIIc signaling. Significantly, the failure of endogenous levels of FGF10/ FGFR2-IIIb signaling in Fgf8 conditional mutants or FGF8/FGFR2-IIIc signaling in Fgf10 null mice to prevent the complete death of SMG cells indicates that the FGF8 signal transduction pathway induces specific and unique downstream responses, different from those mediated by FGF10 signaling. Thus, we conclude that the FGF8/FGFR2-IIIc and FGF10/ FGFR2-IIIb pathways can be characterized as sine qua non pathways, i.e., they are essential and non redundant (Table 1). It is interesting to note that, although EGFR is a tyrosine kinase receptor which can also induce responses identical to those downstream of FGF/FGFR signaling, 56,11,15,55 our observation of hypoplastic (and not aplastic) glands in Egfr and Tace null mice (for review see ref. 17) indicates that, in this case, other pathways compensate, although only partially, for the absence of TGF-α/EGF/EGFR signaling (Table 1). This again points to the uniqueness of FGF8 and FGF10 signaling in SMG development.
Further, our observation of a dysplastic gland in downless (Edardl; Edar-less) mutants indicates that other pathways cannot completely compensate for Edar's functional absence; thus, the Eda/Edar pathway is only partially redundant (Table 1). Since multiple pathways have been shown to activate NF-κB (Fig. 9; ref 54), it is likely that this partial compensatory affect is due to NF-κB activation through pathways other than Eda/Edar. However, the absence of abnormal SMGs in TNF or TNFR1 null mice5658 and the presence of endogenous levels of TNF/TNFR signaling in downless mutants indicate that TNF/TNFR-mediated activation of NF-κB is not responsible for the rescued phenotype. In addition, although our in vitro functional studies indicate that NF-κB is an important network “hub” during SMG organogenesis, the unavailability of appropriate Nf κb mutant mice makes it impossible to definitively ascertain if the NF-κB cascade is essential and non redundant.
Regarding other signaling cascades, the presence of a dysplastic SMG in Shh-/ - and a hypoplastic SMG in Bmp7-/-and Pax6-/- mice indicate that other parallel pathways only partially compensate for the absence of Shh/Ptc, BMP7 or Pax6 signaling (Table 1). The more severely abnormal SMG phenotype in Shh null mice indicates that other endogenous parallel pathways compensate to a lesser degree for the absence of the Shh/Ptc cascade than the absence of BMP7 or Pax6 cascade. Finally, given the absence of Fgfr1 knock-out and tissue-specific conditional null mice, the precise role of the FGFR1-mediated pathway remains unclear. However, functional underexpression or overexpression of FGFR1 signaling indicates that, at the very least, FGF/FGFR1 homeostasis is essential for normal SMG morphogenesis.
Concluding Remarks
With our present knowledge, we can begin to model the regulation of SMG branching morphogenesis (Fig. 10). The initiation of branching and the progression from the Initial Bud Stage to the Pseudoglandular Stage is dependent on FGF8/FGFR2-IIIc and FGF10/FGFR2-IIIb signaling. Subsequent epithelial branching and progression from the Pseudoglandular Stage to the Canalicular Stage also require additional signaling through the Shh/Ptc and Eda/Edar pathways. Finally, since TGF-α/EGF/EGFR, BMP7 and Pax6 signaling regulate the rate of branching morphogenesis and histodifferentiation, these additional signal transduction pathways are all essential for progression from the Canalicular Stage to the Terminal Bud Stage.
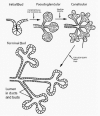
In this review, then, we have provided a glimpse into the dynamic, nonlinear network which regulates SMG organogenesis. In studying the ontogeny of the embryonic SMG, the key is to understand the functional coordination and cooperation between parallel signaling pathways to achieve the differentiated gland. The major challenge remaining is to identify the full complement of broadly related network components and to understand how they interact to regulate the cellular dynamics of SMG development. To accomplish this daunting goal requires a systems biology approach; that is, the integration of transcriptomics, proteomics, and bioinformatics.
Acknowledgements
We would like to thank Dan Witcher and Pablo Bringas, Jr. for help preparing the figures. We would also like to acknowledge the other scientists investigating salivary gland organogenesis. Due to space limitation, we were unable to cite all relevant studies. This work was supported by NIH R01 DE91142 and RO1 DE14535.
References
- 1.
- Melnick M, Chen H, Zhou Y. et al. The functional genomic response of developing embryonic ubmandibular glands to NF-κB inhibition. BMC Developmental Biology. 2001;1:15. [PMC free article: PMC59889] [PubMed: 11716784]
- 2.
- Davidson EH, Rast JP, Oliveri P. et al. A genomic regulatory network for development. Science. 2002;295:1669–1678. [PubMed: 11872831]
- 3.
- Davidson EH, McClay DR, Hood L. Regulatory gene networks and the properties of the development process. Proc Natl Acad Sci USA. 2003;100:1475–1480. [PMC free article: PMC149855] [PubMed: 12578984]
- 4.
- Gardner TS, Bernardo DD, Lorenz D. et al. Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003;301:102–105. [PubMed: 12843395]
- 5.
- Kashimata MW, Sakagami HW, Gresik EW. Intracellular signaling cascades activated by the EGF receptor and/or integrins, with potential relevance for branching morphogenesis of the fetal mouse submandibular gland. Eur J Morphol. 2000b;38:269–275. [PubMed: 10980679]
- 6.
- Koyama N, Kashimata M, Sakagami H. et al. EGF-stimulated signaling by means of P13K, PLCγ1, and PKC isozymes regulates branching morphogenesis of the fetal mouse submandibular gland. Devel Dyn. 2003;227:216–226. [PubMed: 12761849]
- 7.
- Melnick M, Jaskoll T. Mouse submandibular gland morphogenesis: A paradigm for embryonic signal processing. Crit Rev Oral Biol. 2000;11:199–215. [PubMed: 12002815]
- 8.
- Jaskoll T, Zhou Y-M, Trump G. et al. Ectodysplasin receptor-mediated signaling is essential for embryonic submandibular salivary gland development. Anat Rec. 2003;271A:322–331. [PubMed: 12629675]
- 9.
- Jaskoll T, Leo T, Witcher D. et al. Sonic Hedgehog Signaling plays an essential role during embryonic salivary gland epithelial branching morphogenesis Devel Dynam 2004. (in press). [PubMed: 15042696]
- 10.
- Jaskoll T, Witcher D, Leo T. et al. FGF8 dose-dependent regulation of embryonic submandibular salivary gland morphogenesis Devel Biol 2004. (in press). [PubMed: 15063181]
- 11.
- Larsen M, Hoffman MP, Sakai T. et al. Role of PI3-kinase and PIP3 in submandibular gland branching morphogenesis. Devel Biol. 2003;255:178–191. [PMC free article: PMC2002545] [PubMed: 12618142]
- 12.
- Jaskoll T, Melnick M. Submandibular gland morphogenesis: Stage-specific expression of TGF-alpha, EGF, IGF, TGF-beta, TNF and IL-6 signal transduction in normal mice and the phenotypic effects of TGF-beta2, TGF-beta3, and EGF-R null mutations. Anat Rec. 1999;256:252–268. [PubMed: 10521784]
- 13.
- Melnick M, Chen H, Zhou Y-M. et al. Embryonic mouse submandibular salivary gland morphogenesis and the TNF/TNF-R1 signal transduction pathway. Anat Rec. 2001;262:318–320. [PubMed: 11241200]
- 14.
- Melnick M, Chen H, Zhou Y. et al. Interleukin-6 signaling and embryonic mouse submandibular salivary gland morphogenesis. Cells Tiss Org. 2001;168:233–245. [PubMed: 11275690]
- 15.
- Jaskoll T, Chen H, Zhou Y-M. et al. Developmental expression of survivin during embryonic submandibular salivary gland development. BMC Developmental Biol. 2001;1:5. [PMC free article: PMC31339] [PubMed: 11305929]
- 16.
- Jaskoll T, Zhou Y-M, Chai Y. et al. Embryonic submandibular gland morphogenesis: Stage-specific protein localization of FGFs, BMPs, Pax 6 and Pax 9 and abnormal SMG phenotypes in Fgf/ R2-IIIc+/Δ, BMP7-/- and Pax6-/- mice. Cells Tiss Org. 2002;270:83–98. [PubMed: 11731698]
- 17.
- Jaskoll T, Melnick M. Embryonic SMG Development< http://www. usc.edu/hsc/ dental/odg/ index.htm>). [PMC free article: PMC135602]
- 18.
- De Moerlooze L, Spencer-Dene B, Revest J-M. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signaling during mouse organogenesis. Development. 2000;127:483–492. [PubMed: 10631169]
- 19.
- Ohuchi H, Hori Y, Yamasaki M. et al. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Bioch Biophys Res Com. 2000;277:643–649. [PubMed: 11062007]
- 20.
- McKeehan WL, Wang F, Kan M. The heparin-sulfate fibroblast growth factor family-diversity of structure and function. Prog Nucl Acid Res Mol Biol. 1999;59:135–176. [PubMed: 9427842]
- 21.
- Ornitz DM. 2000. FGFs, heparan sulfate and FGFRs. Complex interactions essential for development. Bioessays. 2000;22:108–112. [PubMed: 10655030]
- 22.
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2002;5:1–12. [PMC free article: PMC138918] [PubMed: 11276432]
- 23.
- Szebenyi G, Fallon J. Fibroblast growth factors as multifunctional signaling factors. International Review of Cytology. 1999;185:45–106. [PubMed: 9750265]
- 24.
- Brunet A, Bonni A, Zigmond MJ. et al. Akt promotes cell survival by phosphorylating and inihibiting a forkhead transcription factor. Cell. 1999;96:857–868. [PubMed: 10102273]
- 25.
- Chen Y, Li X, Eswarakumar VP. et al. Fibroblast growth factor (FGF) signaling through PI 3 kinase and AKT/PKB is required for embryoid body differentiation. Oncogene. 2000;19:3750–3756. [PubMed: 10949929]
- 26.
- Guo L, Degenstein L, Fuchs E. Keratinocyte growth factor is required for hair development but not for wound healing. Genes Dev. 1996;10:165–175. [PubMed: 8566750]
- 27.
- MacArthur CA, Lawshe A, Xu J. et al. FGF-8 isoforms activate receptor splice forms that are expressed in mesenchymal regions of mouse development. Development. 1995;121:3603–3613. [PubMed: 8582274]
- 28.
- Ornitz DM, Xu J, Colvin JS. 1996. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. [PubMed: 8663044]
- 29.
- Miller DL, Ortega S, Bashayan O. et al. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol. 2000;20:2260–2268. [PMC free article: PMC110842] [PubMed: 10688672]
- 30.
- Fiore F, Planche J, Gibier P. et al. Apparent normal phenotype in Fgf6-/- mice. Int J Dev Biol. 1997;41:639–642. [PubMed: 9303352]
- 31.
- Colvin JS, Feldman B, Nadeau JH. et al. Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev Dynam. 1999;216:72–88. [PubMed: 10474167]
- 32.
- Deng CX, Wynshaw-Boris A, Shen MM. et al. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. [PubMed: 8001823]
- 33.
- Hajihosseini MK, Lalioti MD, Arthaud S. et al. Skeletal development is regulated by fibroblast growth factor receptor 1 signaling. Development. 2004;131:325–335. [PubMed: 14668415]
- 34.
- Hoffman MP, Kidder BL, Steinberg ZL. et al. Gene expression profiles of mouse submandibular gland development:FGFR1 regulates branching morphogenesis in vitro through BMP- and FGFdependent mechanisms. Development. 2002;129:5767–5778. [PubMed: 12421715]
- 35.
- Beer H-D, Vindevoghel L, Gait MJ. et al. Fibroblast growth factor (FGF) receptor 1-IIIb is a naturally occurring functional receptor for FGFs that is preferentially expressed in the skin and the brain. J Biol Chem. 2000;275:16091–16097. [PubMed: 10821861]
- 36.
- McMahon AP, Ingham PW, Tabin C. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Bio. 2003;53:1–114. [PubMed: 12509125]
- 37.
- Kenney AM, Cole MD, Rowitch DH. N-myc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar neuron precursors. Development. 2003;130:15–26. [PubMed: 12441288]
- 38.
- Lowry JA, Stewart GA, Lindey S. et al. Sonic hedgehog promotes cell cycle progression in activated CD4(+) T lymphocytes. J Immunol. 2002;169:1869–1875. [PubMed: 12165511]
- 39.
- Chiang C, Litingtung Y, Lee E. et al. Cyclopia and defective axial patterning in mice lacking Sonic Hedgehog gene function. Nature. 1996;383:407–413. [PubMed: 8837770]
- 40.
- Ahlgren SC, Thackur V, Bronner-Fraser M. et al. Sonic Hedgehog rescues cranial neural crest cells from cell death induced by ethanol exposure. Proc Natl Acad Scienc. 2002;99:10476–10481. [PMC free article: PMC124946] [PubMed: 12140368]
- 41.
- Monreal AW, Zonana J, Ferguson B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am J Hum Genet. 1998;63:380–389. [PMC free article: PMC1377324] [PubMed: 9683615]
- 42.
- Srivastava AK, Pispa J, Hartung AJ. et al. The Tabby phenotype is caused by mutation in mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains. Proc Natl Acad Sci. 1997;94:13069–13074. [PMC free article: PMC24264] [PubMed: 9371801]
- 43.
- Yan M, Wang L-C, Hymowitz SG. et al. Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science. 2000;290:523–526. [PubMed: 11039935]
- 44.
- Kumar A, Eby MT, Sinha S. et al. The ectodermal dysplasia receptor activates the nuclear factor ?B, JNK, and cell death pathways and binds to ectodysplasin A. J Biol Chem. 2001;276:2668–2677. [PubMed: 11035039]
- 45.
- Pinheiro M, FreireMaia N. Ectodermal dysplasias: A clinical classification and a causal review. Am J Med Genet. 1994;53:153–162. [PubMed: 7856640]
- 46.
- Sundberg J. 1994. The Downless (dl) and Sleek (Dl sleek) mutations, Chromosome 10In: Maibach H, ed.Handbook of Mouse Mutations with Skin and Hair AbnormalitiesBoca Raton, FL: CRC Press.
- 47.
- Moon AM, Capecchi MR. Fgf8 is required for outgrowth and patterning of the limbs. Nat Genet. 2000;26:455–459. [PMC free article: PMC2001274] [PubMed: 11101845]
- 48.
- Frank DF, Fotheringham LK, Brewer JA. et al. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4601. [PMC free article: PMC1876665] [PubMed: 12223415]
- 49.
- Aoto K, Nishimura TEK, Motoyama J. Mouse GLI3 regulates Fgf8 expression and apoptosis in the developing neural tube, face and limb bud. Dev Biol. 2002;251:320–332. [PubMed: 12435361]
- 50.
- Taya Y, O'Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. 1999. pp. 3869–3879. [PubMed: 10433915]
- 51.
- Zhao J, Chen H, Wang YL. et al. Abrogation of tumor necrosis factor-alpha converting enzyme inhibits embryonic lung morphogenesis in culture. Int J Dev Biol. 2001;4:623–631. [PubMed: 11460998]
- 52.
- Sarkar L, Cobourne M, Naylor S. et al. Wnt/Shh interactions regulate ectodermal boundary formation during mammalian tooth development. Proc Natl Acad Sci USA. 2000;9:4520–4524. [PMC free article: PMC18267] [PubMed: 10781055]
- 53.
- Macatee TL, Hammond BP, Arenkial BR. et al. Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development. 2003;130
- 54.
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–686652. [PubMed: 10602461]
- 55.
- Kashimata M, Sayeed S, Ka A. et al. The ERK-1/2 signaling pathway is involved in the stimulation of branching morphogenesis of fetal mouse submandibular glands by EGF. Develop Biol. 2000;220:183–196. [PubMed: 10753509]
- 56.
- Marin MW, Dunn A, Grail D. et al. Characterization of tumor necrosis factor-deficient mice. Pro Natl Acad Sci USA. 1997;94:8093–8098. [PMC free article: PMC21562] [PubMed: 9223320]
- 57.
- Peschon JJ, Torrance DS, Stocking KL. et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed: 9551933]
- 58.
- Rothe J, Mackay F, Bluethmann H. et al. Phenotypic analysis of TNFR1-deficient mice and characterization of TNFR1-deficient fibroblasts in vitro. Cir Shock. 1994;44:51–56. [PubMed: 7743600]
- 59.
- Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: Signaling pathways, biological responses and therapeutic inhibition. Trends Pharm Sci. 2001;22:201–201. [PubMed: 11282421]
- 60.
- Kodaki T, Woscholski R, Hallberg B. et al. The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol. 1994;9:798–806. [PubMed: 7820549]
- 61.
- Lu H-C, Swindell EC, Sierralta WD. et al. Evidence for a role of protein kinase C in FGF signal transduction in the developing chick limb bud. Development. 2002;128:2451–2460. [PubMed: 11493562]
- 62.
- Klint P, Kanda S, Kloog Y. et al. Contribution of Src and Ras pathways in FGF-2 induced endothelial cell differentiation. Oncogene. 1999;18:3354–3364. [PubMed: 10362356]
- Embryonic Salivary Gland Branching Morphogenesis - Madame Curie Bioscience Datab...Embryonic Salivary Gland Branching Morphogenesis - Madame Curie Bioscience Database
- Earthworm Immunity - Madame Curie Bioscience DatabaseEarthworm Immunity - Madame Curie Bioscience Database
- Overview of Intracellular Compartments and Trafficking Pathways - Madame Curie B...Overview of Intracellular Compartments and Trafficking Pathways - Madame Curie Bioscience Database
- Subcellular Compartmentalization of Insulin Signaling Processes and GLUT4 Traffi...Subcellular Compartmentalization of Insulin Signaling Processes and GLUT4 Trafficking Events - Madame Curie Bioscience Database
- Immunity and Autoimmunity Induced by Polyomaviruses: Clinical, Experimental and ...Immunity and Autoimmunity Induced by Polyomaviruses: Clinical, Experimental and Theoretical Aspects - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...