

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Summary
Idiopathic Parkinson's disease (IPD) is a multisystemic synucleinopathy of the human nervous system with functional consequences and a diagnostic potential that extend beyond the nigrostriatal system. Intracerebrally, only a few predisposed types of nerve cells develop the inclusion body pathology that appears in the form of Lewy neurites, Lewy bodies, and Lewy plaques. Melanoneurons and other projection cells generating long axons that are unmyelinated or sparsely myelinated are particularly susceptible. This selective vulnerability on the part of specific neuronal populations as well as anatomically and functionally interconnected brain regions results in a distinctive topographic distribution pattern of brain lesions that is nearly consistent across autopsy cases and, as such, makes it possible to predict the intracerebral progress of IPD and stage it neuropathologically. In stage 1, the lesions are confined to predisposed induction sites: the brain stem dorsal visceromotor nucleus of the vagal nerve, intermediate reticular zone, and/or telencephalic bulbus olfactorius. In stage 2 cases, inclusion bodies begin to appear in portions of the caudal raphe nuclei (above all, the nucleus raphes magnus and obscurus), the gigantocellular reticular nucleus, and the coeruleus/subcoeruleus complex. The severity of the lesions in stage 1 typically increases in stage 2 cases, and the pathology at brain stem sites not only worsens throughout the following stages but is directed essentially upwards in the direction of the cerebral cortex. Neuronal damage begins in the mesencephalic substantia nigra, pars compacta in stage 3 and is accompanied by alterations in the tegmental pedunculopontine nucleus and in the prosencephalon (central subnucleus and basolateral complex of the amygdala, magnocellular nuclei of the basal forebrain, hypothalamic tuberomamillary nucleus). In stage 4, the disease process reaches the cerebral cortex (anteromedial temporal mesocortex) for the first time. During stage 4 and subsequent stages, IPD progresses into additional telencephalic regions, including chiefly the transentorhinal region, hippocampal formation, anterior cingulate mesocortex (all limbic loop structures), and insular and subgenual areas of the mesocortex (cortical components of the autonomic loop). The pathology that accrues in these and other nonsomatomotor system structures almost certainly leads to detectable olfactory impairment, deficits in responses to emotional stimuli, dysfunctions of visceromotor and endocrinal systems, and most probably diminished cognitive faculties, including, in some individuals, dementia. The reduced influence of input from limbic system high-order centers upon both the cerebral cortex and the brain stem reticular formation probably contributes to the affect-related deficits of the voluntary and emotional motor systems that typically become manifest in the course of IPD. In the final stages 5 and 6, the pathology advances until it occupies extensive stretches of the neocortex, beginning with the high-order sensory association and prefrontal areas, then the first order sensory and premotor fields, and eventually the primary sensory and primary motor fields of the mature neocortex.
Introduction
The term “synucleinopathy” has been used broadly to designate a spectrum of progressive degenerative disorders of the human nervous system that share, among other properties, the presence of abnormal α-synuclein-immunoreactive inclusion bodies in neurons and/or macroglial cells.1-3The term currently encompasses neurobiologically and neuropathologically diverse, but also, in part, clinically overlapping entities, such as multiple system atrophy (MSA), idiopathic Parkinson's disease (IPD), dementia with Lewy bodies (DLB), the Lewy body variant of Alzheimer's disease (LBVAD), and neurodegeneration with brain iron accumulation type 1 (NBIA I).4-15It is still unclear whether the clinicial differential diagnosis between IPD and DLB (also synonymous with diffuse Lewy body disease) is defensible on purely neuropathological grounds, but the recently proposed post-mortem staging procedure for IPD reviewed here may provide one means for helping to resolve this ongoing debate.16-24
IPD is the most frequently occurring synucleinopathy. It is a multisystemic illness of the central, peripheral, and enteric nervous systems in which specific neuronal types in functionally related peripheral sites, subcortical nuclei, and cortical areas become selectively involved alongside of others that remain intact. Based on the nonrandom neuronal deterioration and loss, a predictable topographic pattern emerges: Limbic system components and centers of the visceromotor and somatomotor systems sustain the heaviest damage.24-33The density of the brain pathology may vary somewhat from one individual to another, but the distribution of the lesions is remarkably consistent across cases. These factors make it possible not only to identify six stages of brain pathology in IPD but also to distinguish IPD from all other synucleinopathies.24,32,34,35
Involvement of the substantia nigra and obliteration of dopaminergic melanoprojection neurons in the pars compacta are universally acknowledged hallmarks of IPD.36-40Together with earlier studies, IPD-staging reveals the full extent of the multisystemic pathology that develops in nonnigral predilection sites, such as the olfactory bulb and related areas, the dorsal visceromotor nucleus of the vagal nerve and adjoining intermediate reticular zone, the gain setting nuclei, tegmental pedunculopontine nucleus, nonthalamic nuclear grays with diffuse projections to cortical and subcortical sites, intralaminar and midline thalamic nuclei, amygdala, anterior portions of the mesocortex (including the the transentorhinal periallocortex), entorhinal allocortex, and allocortical hippocampal formation (Table 1, Fig. 16).24,25,31,33,41-47 In end-stage cases, lesions consistently appear in the neocortex.24
Table 1
Select brain sites involved in stages 1-6 of idiopathic Parkinson's disease.

Assessment of LNs/LBs in distinct types of nerve cells as seen in series of 100 μm-thick sections permits recognition of the disease process both in symptomatic and nonsymptomatic persons.24,33,47Autopsy cases exhibiting the mildest pathology represent the starting point and those most heavily involved the terminus of a disease spectrum, with a tendency toward increasing severity of the overall brain pathology. The characteristic lesions appear already, to some degree, in the nervous systems of persons whose medical records did not note signs of classical IPD-associated somatomotor dysfunctions (tremor, rigor, bradykinesia, postural instability) prior to their decease.48-51
This chapter describes the predictable topographic advance and extent of the brain pathology that occurs in the preclinical stages and clinical stages of IPD. At present, it is only the end-phase of the neurodegenerative process that can be detected clinically.
To facilitate an understanding of the lesional distribution pattern together with the pathology's potential clinical implications, we address pathoanatomical issues by emphasizing the functional interrelatedness of the major systems and pathways that link involved nuclear grays and cortical structures. In so doing, we hope the current emphasis on late onset symptoms and symptomatic therapies, with its one-sided focus on deficits within the nigrostriatal system, may begin to shift towards more causally-oriented diagnostic and therapeutic strategies that take into consideration the full extent of the damage to limbic, visceromotor, and somatomotor centers.
Inclusion Bodies in IPD
A prerequisite for the post-mortem diagnosis of both the presymptomatic and symptomatic phases of IPD is the assessment of the pathognomonic lesions,52,53which are not physiological phenomena of brain aging.24,54-58Persons with clinical signs of parkinsonism but whose brain tissue at autopsy lacks these lesions, should be assigned to the heterogeneous class of nonIPD syndromes.24,34,59,60
As a result of a conformational change in the predominantly presynaptic protein α-synuclein and the subsequent failure of intracellular proteasomal recycling systems to eliminate the misfolded proteins, affected neurons develop Lewy neurites (LNs) in their cellular processes and Lewy bodies (LBs) in their somata.61-67In addition, abnormal neurofilaments, synphilin, and the stress protein ubiquitin also contribute to the formation of these nearly insoluble aggregates.1,2,62,68-74Many of the inclusion bodies can be seen in sections stained with hematoxilin-eosin for general overview. Currently, however, special silver-staining methods and/ or immunoreactions with antibodies against α-synuclein are the gold standards for visualizing fine granular and filiform LNs.75,76
Despite marked abnormalities, neurons bearing inclusion bodies may survive for years. Their mere persistence, however, is no guarantee against neuronal or axonal dysfunction, and nerve cells with LBs/LNs almost certainly forfeit some degree of functional integrity prior to cell death. After the parent cell deteriorates, the pathological material sometimes remains visible in the tissue in the form of extraneuronal LBs2,77that might be analagous to the “tombstone tangles” in Alzheimer's disease.78
Lewy Neurites
In most of the brain regions studied to date, LNs appear prior to LBs and range from club or spindle-shaped to a variety of thread-like forms.25,31Filiform and granular LNs have received attention only recently (1a-h). Dense networks of these thread-like LNs have been sighted in some of the amygdalar subnuclei, transentorhinal region7and second sector of the Ammon's horn (CA2)79-81 — LNs in the CA2 have been designated as markers for “diffuse” Lewy body disease (DLB) — but other findings indicate these lesions also occur in nearly all cases of clinically diagnosed IPD.17,24,31,49
Near the white matter, ascending cortical LNs frequently increase continually in diameter, give off side branches, and terminate in droplet-like enlargements (Fig. 1, d). Thus, the question arises whether they are pathologically altered terminal segments of axons. Retrograde accumulation of abnormal α-synuclein aggregates32 and/or a pathological sprouting process might encourage arborization. Filiform LNs often appear in the white substance beneath the cortical gray matter or in the external capsule or medial forebrain bundle. Electron microscopically, they occur in cellular processes filled with mitochondria, synaptic vesicles, and dense lamellar bodies,7,79,80reinforcing the view that most LNs are located intraxonally. Brain stem filiform LNs are observable running parallel to each other for the entire intramedullary route of the vagal nerve. They are unmyelinated and unmistakably localized within pathologically altered axon segments.82LNs probably wreak havoc with somatopetal/somatofugal transport of substances through the axon and, in so doing, seriously impair the physiological functions of the parent cells.

Lewy Bodies
LBs are spherical and weakly acidophilic structures, mostly with smoothly contoured surfaces. 31,83-85Their immunoreactivity profile resembles that of LNs, lending support to the theory that one and the same process produces both lesions. LBs only occur in the somata of a few types of neurons, usually within lipofuscin or neuromelanin deposits (Fig. 2a-d). There are no indications that LBs evolve in macroglial cells or nonneuroectodermal cells. Mature cortical LBs fill most of the cell soma (Fig. 2e-i) and, following the death of their parent cells, can be found lying free in the neuropil,86their entire surface often decorated with lipofuscin granules (Fig. 2m-o). Judging from light microscopy, they are not surrounded by a neuronal cell body that contains a cell nucleus (Fig. 2k-p).
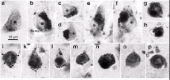
Lewy Plaques
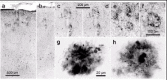
Lewy plaques (LPs) are spherical entities that consist of core-like condensations of extracellular βamyloid surrounded on their periphery by swollen, dystrophic α-synuclein immunoreactive neurites (Fig. 3g,h). These plaques attain large diameters, and groups of them meld together (Fig. 3e-h) isolated within the neuropil or surrounding small blood vessels (Fig. 3a-d). Both the fact that LPs do not exist in the absence of βamyloid precipitations and that plaque-like accumulations of α-synuclein-immunoreactive neurites are found only within βamyloid deposits suggests that the βamyloid protein itself, or its components, may be capable of inducing a pathological process that leads to formation of the LPs. LPs develop only in a subgroup of IPD cases with βamyloid deposition and are not seen in cases with lower than stage 4 pathology.
Staging in Light of Essential Anatomical Considerations
In this overview of the lesional distribution pattern that evolves during IPD, we want to emphasize the involvement not only of the more familiar sites but also of brain structures that have received little or no attention until now. As such, descriptions of the pathology at each stage are accompanied by remarks that are relevant for placing the staging schema within the larger pathoanatomical context of IPD as a multisystemic neurodegenerative disorder. Diagrams of the limbic, visceromotor, and somatomotor systems facilitate recognition of both the nonrandom pattern and multisystem aspects of the disease process.
The functional integrity of these systems depends on that of the cerebral cortex, which is divisible into two types of gray matter: an extensive neocortex (proneocortex plus mature neocortex) and a small, heterogeneously composed allocortex (allocortex proper plus periallocortex).87,88The mammalian allocortex includes (1) the olfactory bulb and related areas, and (2) limbic portions, for the most part located in the anteromedial temporal lobe: the entorhinal region, presubiculum, and hippocampal formation (Fig. 4a-c, red).89-91 The subcortical nuclear complex of the amygdala is closely interconnected with the allocortex. Whereas the olfactory bulb receives unique exteroceptive data, the hippocampal formation lacks direct input from sensory organs, depending instead on indirect input from olfactory areas and solid projections from neocortical sensory association areas for information about the world beyond the individual. Transitional zones exist between the neocortex and allocortex: a belt of periallocortical areas (including the transentorhinal region) associated with the allocortex, and a belt of proneocortical areas leading to the mature neocortex (Fig. 4a-c). Combined, both zones make up the mesocortex,87an architectonically unique entity in humans, which mediates between the allocortex and neocortex.87,92Allocortical and mesocortical areas form an unbroken ring that encircles the medial and basal components of both cerebral hemispheres (Fig. 4b).
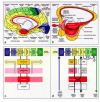
Directly adjacent to the olfactory system are the periallocortical and proneocortical portions of the insular and subgenual mesocortex (Figs. 4a,b,5a). Both represent the brain's highest organizational level for processing interoceptive data from the body's internal organs and for regulating visceromotor and endocrine functions. Bidirectional connectivities between the mesocortical insular and subgenual areas comprise the autonomic loop (Fig. 5a).
The mature neocortex of the temporal, parietal, and occipital lobes is composed respectively of (1) a highly refined primary field that receives dense input from specific thalamo-cortical projections, (2) a belt of less highly differentiated first order sensory association areas, and (3) related (but more simply organized) expansive high-order processing sensory association areas. The frontal lobe is similarly structured into (1) a primary motor field and (2) premotor areas, which in turn give way to (3) high-order motor association (prefrontal) areas (Fig. 4a,c).
The major fiber pathways that interconnect these neocortical territories are also hierarchically differentiated.87,90,93-96 Colored arrows and arrowheads (Fig. 4d) show how exteroceptive information is relayed from neocortical primary sensory fields to both the primary motor field and allocortex. Somatosensory, visual, and auditory input (deep blue arrow) flows “upstream” (terminating in layer IV) from the primary sensory field to adjoining first order association areas.97It then travels via long corticocortical projections from high-order sensory association fields to the prefrontal cortex. Short, “downstream” pathways (terminating in layer I) lead away from prefrontal to premotor areas and the primary motor field, which functions as a portal for motor programs being relayed to brain stem and spinal premotor and motor neurons (deep green arrow). The major routes for the downstream data-flow, however, are the striatal and cerebellar loops (small black semicircular arrows, Figs. 4d,5a,b), which integrate portions of the basal ganglia, lower brain stem nuclei, and the cerebellum into the regulation of cortical output.98-102
The limbic system intervenes in this data-flow via the limbic loop at the juncture where exteroceptive information is transferred from high-order sensory association areas to the prefrontal cortex (Figs. 4d,5b).103,104Some of the neocortical data is diverted from the mainstream and proceeds through the anteromedial temporal mesocortex, converging on the entorhinal allocortex and lateral subnucleus of the amygdala (Figs. 4d,5b). As such, neocortical—not olfactory—information is the dominant source of input to the human limbic loop, and the latter's components are constantly “informed” about neocortical processes.
Limbic loop components (entorhinal allocortex, hippocampal formation, and amygdala) are densely interconnected and generate efferents that terminate in the ventral striatum (accumbens nucleus and “limbic” subdivisions of the putamen, Figs. 4d,5b). This limbic input is supplemented by projections originating in the thalamic midline nuclei (Fig. 16. From there, data-transfer occurs via the ventral pallidum and magnocellular portions of the mediodorsal thalamic nucleus mainly to medial and orbitofrontal portions of the prefrontal cortex (Figs. 4d,5b,16).95
Within the larger context of the human limbic system, the limbic loop is integral to the maintenance of emotional equilibrium, learning, and memory functions.105-108At the same time, it affects somatomotor functions inasmuch as its influence on the prefrontal cortex causes an individual's motor behavior to reflect his/her emotional state.
Selective Involvement of Nuclear Grays, Cortical Areas, and Neuronal Types
IPD targets and destroys select nuclear grays and cortical areas, primarily high order motor (visceromotor, limbic, somatomotor) relay nuclei.24,31,32,109By contrast, with the exception of the olfactory bulb and olfactory-related areas, the sensory (visual, auditory, somatosensory, viscerosensory) nuclear grays maintain their structural and functional integrity or are scarcely involved.
Susceptible nerve cells display common properties: First, all of them belong to a subpopulation of projection neurons that generate disproportionately long and thin axons in relation to the size of their somata.32 Projection cells with relatively short axons withstand the disease process. Second, all of the endangered projection cells are unmyelinated or sparingly myelinated, whereas projection neurons with sturdy axons insulated by thick-caliber myelin sheaths resist forming LNs/LBs.24,31,32The fact that cholinergic projection cells of the basal forebrain nuclei with their long and sparsely myelinated axons develop the lesions (stages 3-6), whereas large short-axoned cholinergic striatal neurons are spared, not only reinforces this theory but also shows that the neurotransmitter type synthesized by a given class of neurons is not, in itself, a sufficient predictor of their predisposition to IPD.
Indirectly, the myelination process itself also may contribute to the pathogenesis of IPD. The cerebral cortex is composed hierarchically of areas that reflect phylogenetic and ontogenetic trends of cortical differentiation.87,110The average cortical myelin content steadily increases when moving from the adult frontal, insular, and temporal periallocortex through the proneocortex, high-order, and first order association areas into the neocortical primary fields (Fig. 4a,c).110,111Axonal myelination usually commences postnatally in neocortical primary fields and continues, via first order, into related high-order association areas, eventually reaching the anterior temporal mesocortex.88,112-114This means the areas of the anterior temporal mesocortex are the last to myelinate but also the sites where most of the pathology is concentrated, whereas neocortical primary fields remain impervious or show only mild pathology (Fig. 16).
Six Neuropathological Stages of IPD-Pathology
The disease process begins at extranigral predilection sites: the dorsal visceromotor nucleus of the vagal nerve (dmX) and adjoining intermediate reticular zone, and the olfactory bulb and/or anterior olfactory nucleus.47 Since the earliest lesions in olfactory structures do not tend to encroach upon either related sites (olfactory tubercle, piriform cortex, periamygdalar cortex) or nonolfactory neocortical areals and subcortical nuclei, the dmX — which always is involved in stage 1 — can be viewed as the main departure point of a disease process that progresses essentially upwards through vulnerable regions of the medulla oblongata, mesencephalon, and basal forebrain until it reaches the cerebral cortex.24,32Its predictable advance within the brain is one of the pathology's major attributes and permits recognition of six neuropathological stages.24,32
Anterior Olfactory Nucleus, Olfactory Bulb, and Related Olfactory Areas
In most stage 1-2 autopsy cases, elongated and thin LNs appear within anterior olfactory structures.24,47A very dense network of these LNs, for instance, accompanied by particularly small LBs develops in the anterior olfactory nucleus and/or olfactory bulb. In microsmatic primates, such as humans, the olfactory bulb and periallocortical related areas (anterior olfactory nucleus, olfactory tubercle, and piriform cortex) are poorly developed. Also assigned to the olfactory system but well differentiated in the human brain are the periallocortical periamygdalar cortex,91the olfactory-related portion of the entorhinal region, and the medial subnucleus of the amygdala. The piriform cortex, ipsilateral amygdala, septal nuclei, and hypothalamus receive olfactory information from the lateral olfactory striae of the olfactory tracts.115 With the exception of the anterior olfactory nucleus and olfactory bulb, however, additional components of the olfactory system first begin to show mild involvement from stage 3 onwards of the disease process.24
Stages 1-2: Nonolfactory Sites Confined to the Medulla Oblongata and Tegmentum Pontis
Stage 1
The very first LNs/LBs in nonolfactory nuclei consistently occur in the dmX. (Fig. 6a)25,41 The remarkably long, thin axons (preganglionic parasympathetic fibers) generated by its projection neurons are unmyelinated, and pathological α-synuclein aggregates are detectable both in medullary and peripheral axonal portions of the vagal nerve.24,82
Other nuclei in the dorsal vagal area, including the nucleus gelatinosus, area postrema, and most of the small-celled nuclear grays clustered around the solitary tract, are barely affected or are uninvolved.32,47The neuromelanin-containing catecholaminergic projection neurons near the dmX and the melanoneurons in the inferolaterally adjoining intermediate reticular zone still are intact. These neurons do not project to the periphery but to higher levels of brain.116-118 The multipolar neurons of the ambiguus nucleus, whose myelinated fibers innervate the striated musculature of the heart, larynx, and upper esophagus do not become involved at any point in the disease process.32
Stage 2
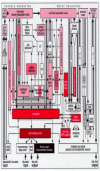
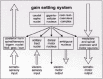
The inclusion body pathology is confined to the medulla oblongata and pontine tegmentum. The lesions in the dmX and intermediate reticular zone become exacerbated (Fig. 6b,c).24 During its ascent, the disease process additionally affects (1) portions of the caudal raphe nuclei, particularly the nucleus raphes magnus and obscurus, (2) the gigantocellular nucleus of the reticular formation, and (3) the neuromelanin-containing projection cells of the coeruleus/ subcoeruleus complex.24,47The appearance of LNs precedes the development of LBs in each of these nuclear grays, which collectively comprise the “gain” or “level setting” system of the medulla oblongata.24,45,119
Spinal and medullary centers for sensory input and motor output are illustrated schematically in (Fig. 7): Viscerosensory afferences reach the nuclei in the vicinity of the solitary tract via myelinated fiber systems, and special visceromotor efferences generated by the ambiguus nucleus also are sturdily myelinated. The preganglionic axons of the dmX are, and remain for life, unmyelinated. These spinal and medullary centers are regulated by the supervening nuclear grays of the gain setting system. Its descending fiber tracts (with long, thin, and sparely myelinated axons) function as a pain inhibitory system that delays or blocks the relay nuclei for somatosensory and viscerosensory input. At the same time, the gain setting nuclei also form a motor control system for both visceromotor and somatomotor efferences. They “set” the sensitivity and excitability levels of both medullary and spinal premotor and motor neurons; hence, the designation “gain setting” system. Taken together, the gain setting nuclei temporarily limit the conduction of incoming pain signals and place the organism's motor neurons in an intensified state of preparedness for action.45,120,121
All sensory relay nuclei, vestibular nuclei, precerebellar nuclei (e.g., inferior olive, pontine gray), the parvocellular and magnocellular portions of the red nucleus, the cerebellum, and somatomotor nuclei (III-VII, XII) develop, if at all, only slight changes (Fig. 16). Centers of the cerebellar loop commence myelination prenatally,112 and their long-axoned projection neurons develop medium- to thick-caliber myelin sheaths.
Stages 3-4: Additional Pathology in the Mesencephalic Tegmentum, Basal Forebrain, Mesocortex, and Limbic Portions of the Allocortex
Stage 3
The pathology makes inroads into the mesencephalic tegmentum and basal forebrain. The lesions within the nuclear grays affected in the preceding stages increase in severity, and inclusion bodies appear for the first time in the substantia nigra,24,38tegmental pedunculopontine nucleus, oral raphe nuclei, nonthalamic nuclear grays with diffuse projections,122 central amygdalar subnucleus, and basolateral nuclear complex of the amygdala.24,44They also begin to develop in the catecholaminergic melanoneurons of the dorsal vagal area and intermediate reticular zone.24,47
Initially, a few, in part extremely long, LNs appear in the posterolateral subnucleus of the substantia nigra followed by LBs within the somata of its dopaminergic melanoneurons.123 These nigral projection neurons have fine-caliber, thinly myelinated axons.124Thereafter, the disease process enters the posterosuperior and posteromedial cell groups, for the most part skirting the substantia nigra's magnocellular and anterior subnuclei.123 Nevertheless, because the actual loss of the melanoneurons first occurs in subsequent stages of the disease, the substantia nigra still appears macroscopically intact.24Nonmelanized projection neurons in its pars reticulata, and all of the substantia nigra's small local circuit neurons remain devoid of inclusion bodies.
Like the substantia nigra, the tegmental pedunculopontine nucleus displays remarkably long LNs at first. These can become voluminous and form a dense network. Gradually, numerous LBs also appear in the somata of the cholinergic nerve cells within the pedunculopontine nucleus' pars compacta.46,125-127Owing to its versatility, the involvement of the pedunculopontine nucleus at this stage probably impacts broadly on other systems. Its chief fiber tracts project to the gain setting nuclei, thalamic intralaminar nuclei, and nonthalamic nuclei with diffuse projections (Fig. 16). Additional bidirectional connections link the pedunculopontine nucleus to the limbic system (ventral striatum) and to components of the striatal loop (e.g., pars compacta of the substantia nigra, subthalamic nucleus, internal pallidum) (Fig. 16).128Together with the thalamic intralaminar nuclei, the tegmental pedunculopontine nucleus is a component of the ascending reticular activating system.129Along with the gain setting nuclei, it is part of a rhythmogenic complex that induces and modulates oscillatory activity patterns, including the sleep-waking cycle and states of consciousness, arousal, attention, and vigilance.130Strategically positioned between the limbic and striatal loops, the pedunculopontine nucleus influences cognitive processes and locomotion respectively.46,128,131
Striatal loop centers (dorsal striatum, external and internal pallidum, subthalamic nucleus, ventrolateral thalamus) commence myelination prenatally, so that the pallidum and subthalamic nucleus stand out in the adult human brain as remarkably well-myelinated structures. Except for the substantia nigra and pedunculopontine nucleus, whose axons are thin and poorly myelinated, the other centers of the striatal loop resist the disease process.
The nonthalamic nuclear grays that generate diffuse and extensive projections to subcortical sites and the entire cerebral cortex have long projection neurons with slender and sparsely myelinated axons.132-134 Despite different transmitter substances, all of these nuclei begin to develop the lesions: the cholinergic magnocellular nuclei of the basal forebrain, the tuberomamillary nucleus with its GABAergic neurons, the paranigral and parabrachial pigmented nuclei with their dopaminergic neurons, the coeruleus-subcoeruleus complex with its noradrenergic melanoneurons, and the oral raphe nuclei with their ascending serotonergic projections (Fig. 16). The brunt of the pathology occurs in the magnocellular basal forebrain nuclei (medial septal nucleus, interstitial nucleus of the diagonal band, basal nucleus of Meynert) (Figs. 8d-f,9a).24,25,40,122,135 LNs initially predominate over LBs, which develop afterwards. All three sites are characterized by thick-caliber serpentine and spindle-shaped LNs. The small neurons of the adjoining substantia innominata remain intact. The magnocellular nuclei receive input from olfactory areas, mesocortical insular and subgenual areas, the parabrachial pigmented nuclei, gain setting nuclei, entorhinal allocortex and amygdala, ventral striatum, ventral pallidum, and prefrontal cortex (Fig. 16). Accordingly, they serve as a relay between the autonomic and limbic loops (Figs. 5, 16).
In the nuclear complex of the amygdala, the central subnucleus begins to fill with a tight-meshed network of filiform LNs and small LBs, features that distinguish it both from the surrounding intercalated cell masses and the medial subnucleus, which evade the pathology. The central subnucleus sends dense projections to the gain setting nuclei and dmX,105,136,137 thereby bringing superordinate limbic influences to each of these modulating nuclear grays. At the same time, it receives projections from the basalolateral nuclear complex of the amygdala, which is fed by strong input from the magnocellular nuclei of the basal forebrain53 and the temporal mesocortex. The central subnucleus also regulates all of the nonthalamic nuclei mentioned above and sends major projections to visceromotor relay nuclei, thereby influencing nearly all subcortical nuclei that regulate endocrinal and autonomic functions.105,138
Lesions also are perceptible in the voluminous basolateral nuclear complex of the amygdala (Fig. 9d). The lateral subnucleus contains fewer LBs and longer LNs than the amygdalar basal and basal accessory subnuclei. The basolateral complex maintains bidirectional interconnectivities with the hippocampal formation and neocortical high-order association areas (Figs. 5b, 16). Dense efferent projections from the basal subnucleus and basal accessory subnucleus terminate chiefly in the ventral striatum, ventral pallidum, thalamic dorsomedial nucleus, insular, and prefrontal cortex (Fig. 16).136

Stage 4
The pathology in previously involved sites steadily worsens: By the end of this stage, the loss of the melanoneurons in the coeruleus-subcoeruleus complex, for example, is nearly total.24 Cases without LNs/LBs in the paranigral subnuclei and oral raphe nuclei (raphes linearis, centralis, dorsalis) already in the preceding stage, begin to develop them. Additionally, lesions appear for the first time in both the thalamic midline and intralaminar nuclei,33 as well as in the interstitial nucleus of the stria terminalis. Moreover, the disease process also invades for the first time a very vulnerable portion of the cerebral cortex, the anteromedial temporal mesocortex, particularly the transentorhinal periallocortex and adjoining ectorhinal proneocortex (Figs. 10-11). Two further cortical sites become involved at this juncture: the entorhinal allocortex and, in some instances, the CA2 (stratum oriens) of the hippocampal formation (Figs. 4,5b,16).24


The foci of the pathology in the thalamus are nuclei with poorly myelinated axons. Most thalamic relay nuclei feed the neocortex with strongly myelinated fiber tracts that form circumscribed columnar terminal arborizations within cortical layers II-V traceable to well-defined areas. LNs/LBs do not develop in these relay nuclei.33 By contrast, the intralaminar nuclei generate additional diffuse projections that terminate in layers I and VI and are poorly myelinated, less specific, and dispersed throughout multiple areas. Similarly, the axons of the midline nuclei that form both short thalamo-allocortical circuits and dense projections to the ventral striatum are sparsely myelinated. High inclusion body densities gradually accrue at all of these sites.33
The lesional pattern of the interstitial nucleus of the stria terminalis resembles that of the central subnucleus of the amygdala. The stria terminalis interconnects the amygdala and hypothalamic nuclear grays (Fig. 16).
On the heels of the amygdala, the disease process finally succeeds in penetrating the anteromedial temporal mesocortex,24,32having taken the transentorhinal periallocortex as its point of departure (Figs. 10,11b-d). From there, the lesions make inroads into the ectorhinal proneocortex and advance in subsequent stages through the mature temporal neocortex. As if through a defile, all of the data in transit from neocortical high-order sensory association areas are channeled through the transentorhinal region to supervening centers of the limbic loop and points beyond, but chiefly to the prefrontal cortex (Fig. 5b,16). A thick network of LNs ini- tially emerges in the superficial layers II-III of the transentorhinal periallocortex, and numerous projection neurons located in the deep layers V-VI develop LBs.
In the entorhinal region, the most of the incipient pathology is concentrated in the deep layer pri-α (for terminology, see Braak and Braak).92 Later, some stage 6 cases also display a band of tightly-packed projection cells containing LBs, which is visible to the naked eye (Figs. 10,11a) and gradually encroaches upon the adjoining areas of the anterior insular mesocortex.
The entorhinal region extends over the ambient gyrus and anterior portions of the parahippocampal gyrus. None of the entorhinal region's layers corresponds to any layer of the neocortex.92Projections from olfactory areas to the entorhinal allocortex are sparse and rudimentary. Other direct entorhinal afferents come from limbic circuits via the thalamic midline nuclei and presubiculum (Fig. 16). Again, the hallmark of the human entorhinal allocortex, which serves as an interface between neocortex and hippocampus, is the enormous neocortical input that it receives by way of the intervening transentorhinal region.31
Three internal divisions of the hippocampal formation are clearly delineated in frontal sections through the posterior hippocampus at the level of the lateral geniculate body: the fascia dentata, the four sectors of the Ammon's horn, and the subiculum.139-142In the human brain, the hippocampal formation is the most important allocortical structure. Hippocampal output is generated mainly in the subiculum, which sends major projections to the entorhinal allocortex, amygdala, hypothalamus, midline and anterior thalamic nuclei, and ventral striatum.88,139-143
The data that originate in the anterior thalamic nuclei are transported to the hippocampus via two pathways: a short circuit by way of the entorhinal region and a somewhat larger one that ultimately reaches the hippocampal formation via the anterior cingulate proneocortex and amygdala (Fig. 16).
During the intermediate stages 3-4, it is very likely that the presymptomatic phase gradually yields to the clinically recognizable phase of IPD.24,32
Stages 5-6: Additional Pathology in Neocortical Areas
Stage 5
In its final stages, the neurodegenerative process reaches its greatest topographic extent, paving the way for the disease manifestation with its full spectrum of classical neurological symptoms. The uninterrupted diminishment of limbic input to the prefrontal cortex can lead to cognitive decline if patients do not die first.
In the brain stem, the vulnerable portions of the substantia nigra appear nearly denuded of melanoneurons and have an unmistakeable pallor upon macroscopic inspection.24 At this stage, the nonthalamic nuclei with diffuse projections, the central subnucleus of the amygdala, and interstitial nucleus of the stria terminalis are replete with lesions (Fig. 9b,c). The anteromedial temporal mesocortex and CA2 gradually amass some of most severe pathology of all the sites in the cerebral cortex (Figs. 12,13a).24,32 From the anteromedial temporal mesocortex, the pathology breaches and occupies portions of the adjoining mature neocortex, beginning with the expansive high-order sensory association and prefrontal areas (Figs. 4,8a). LNs no longer occur in the deep layers V-VI, and the dense networks of filiform LNs are on the decrease in layers II-III (Figs. 10,15a,b). Although in individual IPD-cases the extent of neocortical involvement can vary somewhat, the primary sensory and motor fields as well as the first order sensory association fields still are nearly fully intact. Additional lesions appear in subcortical sites in the ventral portions of the claustrum and striatum (Figs. 8c,13c), and involvement expands to include the territories of the insular and subgenual mesocortex as well as anterior cingulate mesocortex (Fig. 13b).
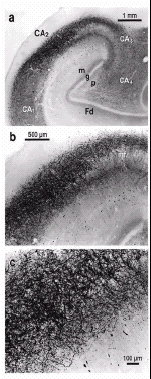
The formerly sparse lesions in CA2 that began developing in stage 4 (some isolated LNs may appear already in stage 3) evolve into a dense network of long and repeatedly branching LNs located preferentially in the stratum oriens but penetrating deeply and widely, like tendrils on a trellis, into the pyramidal layer (Fig. 12a-c). In the molecular layer of CA1 and close to the hippocampal fissure, atypically large spindle-shaped or club-shaped LNs are dispersed throughout the neuropil (Figs. 12c,13a). A further predilection site of these enigmatic lesions in CA1 is the transitional region between the amygdala and hippocampal formation. The CA2 plexus of LNs is so characteristic of the final two stages that even in the absence of sections through the substantia nigra the neuropathological diagnosis of IPD can be made based on its involvement alone.24 The hilus and CA3 exhibit a few elongated LNs and, rarely, LBs. The fascia dentata and the subiculum are, and remain for the remainder of the disease process, nearly intact (Fig. 12a).
LBs are seen in projection neurons of the ventral claustrum (Fig. 8c), and short ultrathin or elongated filiform LNs there usually lack arborizations. Located underneath the insular mesocortex and separated from it by the extreme capsule, the claustrum is interconnected with nearly all portions of the cerebral cortex. The mesocortical anterior cingulate areas, entorhinal allocortex, and subiculum send robust projections to the ventral claustrum, and dense bilateral fiber tracts link the ventral claustrum to the thalamic intralaminar nuclei, peripeduncular nucleus, and zona incerta.144 In sum, the ventral claustrum functions as a diffusely projecting relay between both limbic and autonomic system centers and the entire cerebral cortex (Figs. 5a,b,8a).

Whereas the dorsal striatum is almost intact, the ventral striatum with its somewhat sparse, but curious, pathology displays a completely different lesional pattern than the neighboring interstitial nucleus of the diagonal band or that of any other nucleus described here previously (Fig. 13c). LBs appear in clusters surrounded by milder pathology. Although, collectively speaking, striatal LBs outnumber LNs, the latter assume a wider variety of forms, and thick-caliber LNs prevail over filiform LNs. Remarkable are the exceptionally elongated, filiform LNs with scant arborizations. The ventral striatum is particularly expansive in the human brain. It receives, in addition to input from the amygdalar basal and basal accessory subnuclei, afferents from the hippocampal formation, entorhinal allocortex, thalamic midline and intralaminary nuclei (Figs. 5b,16).99,145-147

The insular and subgenual mesocortex are cortical viscerosensory and visceromotor regions that function as major components of the autonomic loop (Fig. 5a). Large, mostly spherical, LBs lend the tissue of the insular mesocortex a spotted or peppered appearance, a feature seen elsewhere in the cortex as a whole. The mesocortical insular and subgenual areas influence heart rate, blood pressure, respiration, gastrointestinal motility, and the appropriate response of skin conductance to emotional stimuli.148-155Together with the adjoining association areas, the insular agranular and dysgranular fields include gustatory regions and a topically organized representation of the innner organs and internal surface of the body. These insular fields are reciprocally connected with the subgenual and anterior cingulate mesocortex, entorhinal allocortex, amygdala, claustrum, and thalamic intralaminar nuclei. They also generate major projections to the magnocellular basal forebrain nuclei, ventral claustrum, and ventral striatum. Thus, a pathway exists that connects the insular mesocortex via the ventral pallidum and mediodorsal thalamus with the prefrontal association cortex (Figs. 5a,16). By way of the magnocellular basal forebrain nuclei, thalamic intralaminar nuclei, ventral claustrum, and ventral striatum, the insular mesocortex influences the entire cerebral cortex. Based on these connectivities, the insular mesocortex has been designated a “viscerosensory and limbic integration cortex.”156,157
The subgenual mesocortex is part of the ventromedial frontal lobe and represents a topically organized visceromotor region.158-160Bidirectionally organized projections connect the region with adjoining prefrontal areas, the insular and anterior cingulate mesocortex, entorhinal allocortex, hippocampal formation, amygdala, thalamic intralaminary and midline nuclei, lateral hypothalamus, and autonomic regions of the brain stem and spinal cord.161 Since the subgenual mesocortex also prominently projects to the ventral striatum,162 it influences the prefrontal cortex via the ventral pallidum and mediodorsal thalamus (Fig. 5a). The subgenual mesocortex can be said to fulfill the functions of a “visceromotor and limbic integration cortex.”163 Fine, elongated LNs are dispersed throughout the tissue, and thick, lengthy club-shaped LNs occur sporadically in all of its layers. Larger forms of LBs are more prevalent than smaller ones in layers V-VI.
Stage 6
The disease process in all olfactory areas (Fig. 4a,b) and brain stem sites becomes exacerbated. The periamygdalar cortex, a small portion of the adjoining entorhinal allocortex (covering the ambient gyrus) and the cortical as well as accessory cortical subnuclei of the amygdala show evidence of heavy neuronal damage. The piriform cortex is moderately involved, the olfactory tubercle to a lesser extent so. With the exception of the medial subnucleus of the amygdala, all of the components of the olfactory system eventually become affected in the course of IPD. The fact that anterior olfactory structures (in stage 1 already) and the periamygdalar region subsequently become severely involved164,165(Fig. 8a,b) is congruent with clinical evidence of hyposmia that is detectable in IPD patients, sometimes even prior to the manifestation of motor symptoms.51,166-168
The insular and subgenual mesocortex are very severely affected, whereby the proneocortical dysgranular fields (Figs. 5a,14b,d,16) are somewhat less heavily damaged than the periallocortical agranular areas (Figs. 5a,14a,c). Along with the insular and subgenual mesocortex, the limbic system anterior cingulate mesocortex169-171belongs to the cortical areas with the highest concentrations of LB-bearing projection neurons (layers V-VI).
Ultimately, the lesions advance from the high-order sensory association and prefrontal areas into first order sensory association areas and premotor fields, sometimes even making inroads into the neocortical primary sensory and motor fields (Figs. 4,16). Only with the transition from the second to the first temporal gyrus, does a clear-cut reduction in LBs become evident (Fig. 15b,c). The same phenomenon occurs again with the transition to the primary auditory field: Like the remaining primary sensory and motor fields, it typically exhibits only scattered LNs/LBs (Fig. 15c,d). Since all of the primary fields are well myelinated, the lesional distribution pattern in the mature neocortex is similar in most cases at this advanced stage.

Concluding Remarks
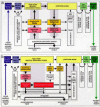
Upon surveying the overall picture of the neuropathological alterations in IPD, it is striking that particularly limbic, visceromotor, and somatomotor system centers sustain severe neuronal damage. Since components of the limbic and autonomic loops are so closely interconnected, the basic schemata of both loops can be combined (Fig. 5a,b) and selected subcortical components, together with the striatal and cerebellar circuits, added. The resulting comprehensive diagram does not presume to be definitive (Fig. 16); yet, it suffices to convey an impression of the multisystemic nature and topographic extent of the progressive brain destruction that predictably develops in IPD. Affected subcortical nuclei and cortical regions are indicated by varying degrees of shading: Black (involvement from stage 1 onwards), rust (involvement from stage 2 onwards), red (involvement from stage 3 onwards), light red (involvement from stage 4 onwards), rose (involvement from stage 5 onwards), or light pink (involvement from stage 6 onwards). White signifies noninvolvement.
Fundamental pathoanatomical studies devoted to patterns of axonal myelination, tractology, and the phenomenon of axonal transport could provide greater insight into the selective vulnerability of distinct neuronal types to IPD and, not only how the lesions progress from one brain region to another but, perhaps more importantly, whether the disease process commences within the central nervous system at all.32
Acknowledgements
This study was supported by the Deutsche Forschungsgemeinschaft (DFG) and the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie. The authors especially want to thank Helmut Bohl, M.D. (Department of Pathology, Johannes Gutenberg University, Mainz), Rob de Vos, M.D. (Laboratorium Pathologie Oost Nederland, Enschede, The Netherlands), and Prof. Hansjürgen Bratzke, M.D. (Institute for Forensic Medicine, J.W. Goethe University, Frankfurt/Main), for supplying human autopsy material in compliance with Frankfurt University Ethics Committee guidelines, and Ms. Inge Szász-Jakobi (Institute for Clinical Neuroanatomy, J.W. Goethe University, Frankfurt/Main), for her adept assistance in preparing the illustrations.
References
- 1.
- Takeda A, Mallory M, Sundsmo M. et al. Normal accumulation of NACP/α-synuclein in neurodegenerative disorders. J Pathol. 98;2:7–372. [PMC free article: PMC1857971] [PubMed: 9466562]
- 2.
- Goedert M, Spillantini MG. Nucleinopathies and α-synucleinopathiesIn: Lee VM-Y, Trojanowski, JQ, BuéL, Christen Y, eds. Fatal Attractions: Protein Aggregates in Neurodegenerative Disorders. Research and Perspectives in Alzheimer's Disease Berlin Heidelberg New York: Springer,200065–86.>.
- 3.
- Galvin JE, Lee VM-Y, Trojanowski JQ. Nucleinopathies. Clinical and pathololgical implications. Arch Neurol. 01:6–190. [PubMed: 11176955]
- 4.
- Gorell JM, Johnson CC, Rybicki BA. Parkinson's disease and its comorbid disorders: An analysis of Michigan mortality data, 1970 to 1990. Neurology. 94:65–1868.
- 5.
- Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain. 94;7:5–243. [PubMed: 8186951]
- 6.
- Jellinger K, Bancher C. Dementia with Lewy bodies: Relationships to Parkinson's and Alzheimer's diseasesIn: Perry RH, McKeith IG, Perry EK, eds. Dementia with Lewy Bodies Cambridge, New York: Cambridge University Press,1996268–286.
- 7.
- Kosaka K, Iseki E. Diffuse Lewy body disease within the spectrum of Lewy body diseaseIn: Peerry RH McKeith IG Perry EK eds. Dementia with Lewy Bodies Cambridge New York Cambridge University Press 19968–248.
- 8.
- Samuel W, Galasko D, Masliah E. et al. Cortical Lewy body counts correlate with dementia in the Lewy body variant of Alzheimer's disease. Neuropathol Exp Neurol. 96:–52. [PubMed: 8558171]
- 9.
- Arawaka S, Saito Y, Murayama S. et al. Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for α-synuclein. Neurology. 1998;51:887–889. [PubMed: 9748051]
- 10.
- Gai WP, Power JHT, Blumbergs PC. et al. Multiple-system atrophy: A new α-synuclein disease? Lancet. 1998;352:547–548. [PubMed: 9716068]
- 11.
- Bayer TA, Jäkälä P, Hartmann T. et al. α-synuclein accumulates in Lewy bodies in Parkinson's disease and dementia with Lewy bodies but not in Alzheimer's disease βamyloid plaque cores. Neurosci Lett. 1999;266:213–216. [PubMed: 10465711]
- 12.
- Dickson DW. Alzheimer-Parkinson disease overlap: NeuropathologyIn: Clark CM, Trojanowski JQ, eds. Neurodegenerative Dementias: Clinical Features and Pathological Mechanisms New York: McGraw-Hill,2000266219–216.>.
- 13.
- Dickson DW. Neuropathology of Alzheimer's disease and other dementias. Clin Geriatr Med. 2001;17:209–228. [PubMed: 11375133]
- 14.
- Dickson DW, Liu WK, Hardy J. et al. Widespread alterations of α-synuclein in multiple system atrophy. Am J Path. 1999;155:1241–1251. [PMC free article: PMC1867032] [PubMed: 10514406]
- 15.
- Dickson DW, Liu WK, Hardy J. et al. Widespread alterations of α-synuclein in multiple system atrophy. Am J Path. 1999;155:1241–1251. [PMC free article: PMC1867032] [PubMed: 10514406]
- 16.
- Wenning GK, Seppi> K, Tison F. et al. A novel grading scale for striatonigral degeneration (multiple system atrophy). J Neural Transm. 2002;109:307–320. [PubMed: 11956953]
- 17.
- Gibb WRG, Esiri MM, Lees AJ. Clinical and pathological features of diffuse cortical Lewy body disease (Lewy body dementia). Brain. 1987;110:1131–1153. [PubMed: 2823957]
- 18.
- McKeith IG, Fairbairn AF, Perry RH. et al. The clinical diagnosis and misdiagnosis of senile dementia of Lewy body type (SDLT). Br J Psychiatry. 1994;165:324–332. [PubMed: 7994501]
- 19.
- McKeith IG, Galasko D, Kosaka K. et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. [PubMed: 8909416]
- 20.
- Vos RAI, Jansen ENH, Stam FC. et al. Lewy body disease: Clinicopathological correlations in 18 consecutive cases of Parkinson's disease with and without dementia. Clin Neurol Neurosurg. 1995;97:13–22. [PubMed: 7788967]
- 21.
- Louis ED, Goldman JE, Powers JM. et al. Parkinsonian features of eight pathologically diagnosed cases of diffuse Lewy body disease. Mov Disord. 1995;10:188–194. [PubMed: 7753061]
- 22.
- Perry RH, Jaros EB, Irving D. et al. What is the neuropathological basis of dementia associated with Lewy bodies? In: Perry RH, McKeith IG, Perry EK, eds.Dementia with Lewy BodiesCambridge, New York: Cambridge University Press,1996212–23.
- 23.
- Perl DP, Olanow CW, Calne D. Alzheimer's disease and Parkinson's disease: Distinct entities or extremes of a spectrum of neurodegeneration? Ann Neurol. 1998;44:S19–S31. [PubMed: 9749570]
- 24.
- Richard ICH, Papka M, Rubio A. et al. Parkinson's disease and dementia with Lewy bodies: one disease or two? Mov Dis. 2002;17:1161–1165. [PubMed: 12465052]
- 25.
- Braak H, Del TrediciK, Rüb U. et al. Jansen Steur ENH, Braak E.† Staging of pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003a;24:197–210. [PubMed: 12498954]
- 26.
- Lewy FH. Paralysis agitans. I. Pathologische Anatomie In: Lewandowski M, ed.Handbuch der Neurologie Vol III Berlin: Springer,1912920-933.
- 27.
- den Hartog Jager WA, Bethlem J. The distribution of Lewy bodies in the central and autonomic nervous systems in idiopathic paralysis agitans. J Neurol Neurosurg Psychiatry. 1960;23:283–290. [PMC free article: PMC497426] [PubMed: 13711997]
- 28.
- Hansen LA, Galasko D. Lewy body disease. Curr Opin Neurol Neurosurg. 1992;5:889–894. [PubMed: 1467583]
- 29.
- Fearnley J, Lees A. Pathology of Parkinson's disease. In: Calne DB, ed. Neurodegenerative Diseases Philadelphia: Saunders, 1994:545-554.
- 30.
- Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. [PubMed: 8786384]
- 31.
- Braak H, de VosRAI, Jansen ENH. et al. Neuropathological hallmarks of Alzheimer's and Parkison's diseases. Progr Brain Res. 1998;117:267–285. [PubMed: 9932414]
- 32.
- Braak H, Rüb U, Gai WP. et al. Idiopathic Parkinson's disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen J Neural Transm 2003b. in press. [PubMed: 12721813]
- 33.
- Rüb U, Del TrediciK, Schultz C. et al. Parkinson's disease: The thalamic components of the limbic loop are severely impaired by α-synuclein immunopositive inclusion body pathology. Neurobiol Aging. 2002;23:245–254. [PubMed: 11804710]
- 34.
- Braak H, Rüb U, Del Tredici K. Involvement of precerebellar nuclei in multiple system atrophy. Neuropathol Appl Neurobiol. 2003c;29:60–76. [PubMed: 12581341]
- 35.
- Gai WP, Power JH, Blumbergs PC. et al. Alpha-synuclein immunoisolation of glial inclusions from multiple system atrophy brain tissue reveals multiprotein components. J Neurochem. 1999;73:2093–2100. [PubMed: 10537069]
- 36.
- Mann DMA, Yates PO. Possible role of neuromelanin in the pathogenesis of Parkinson's disease. Mech Ageing Dev. 1983;21:193–203. [PubMed: 6865505]
- 37.
- Graybiel AM, Hirsch EC, Agid Y. The nigrostriatal system in Parkinson's disease. Adv Neurol. 1990;53:17–29. [PubMed: 1978514]
- 38.
- Gibb WRG, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1991;54:388–396. [PMC free article: PMC488535] [PubMed: 1865199]
- 39.
- Hirsch EC. Why are nigral catecholaminergic neurons more vulnerable than other cells in Parkinson's disease? Ann Neurol. 1992;32:88–93. [PubMed: 1510386]
- 40.
- Agid Y, Ruberg M, Javoy-Agid F. et al. Are dopaminergic neurons selectively vulnerable to Parkinson's disease. Adv Neurol. 1993;60:148–164. [PubMed: 8420132]
- 41.
- Eadie MJ. The pathology of certain medullary nuclei in parkinsonism. Brain. 1963;86:781–795. [PubMed: 14090529]
- 42.
- Goto S, Hirano A. Catecholaminergic neurons in the parabrachial nucleus of normal individuals and patients with idiopathic Parkinson's disease. Ann Neurol. 1991;30:192–196. [PubMed: 1680303]
- 43.
- Jellinger K. Pathology of Parkinson's disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol. 1991;14:153–197. [PubMed: 1958262]
- 44.
- Braak H, Braak E, Yilmazer D. et al. Amygdala pathology in Parkinson's disease. Acta Neuropathol. 1994;88:493–500. [PubMed: 7879596]
- 45.
- Braak H, Rüb U, Sandmann-Keil D. et al. Parkinson's disease: Affection of brain stem nuclei controlling premotor and motor neurons of the somatomotor system. Acta Neuropathol. 2000;99:489–495. [PubMed: 10805091]
- 46.
- Pahapill P, Lozano AM. The pedunculopontine nucleus and Parkinson's disease. Brain. 2000;123:1767–1783. [PubMed: 10960043]
- 47.
- Del TrediciK, Rüb U, de Vos RAI. et al. Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol. 2002;61:413–426. [PubMed: 12030260]
- 48.
- Calne DB, Snow BJ, Lee C. Criteria for diagnosing Parkinson's disease. Ann Neurol. 1992;32:125–127. [PubMed: 1510370]
- 49.
- de VosRAI, Jansen ENH, Yilmazer D. et al. Pathological and clinical features of Parkinson's disease with and without dementiaIn: Perry RH, McKeith IG, Perry EK, eds. Dementia with Lewy Bodies Cambridge, New York: Cambridge University1996255–267.
- 50.
- Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson's disease. Arch Neurol. 1999;56:33–39. [PubMed: 9923759]
- 51.
- Wolters EC, Francot C, Bergmans P. et al. Preclinical (premotor) Parkinson's disease. J Neurol. 2000;247(Suppl 2):103–109. [PubMed: 10991655]
- 52.
- Gibb WRG, Scott T, Lees AJ. Neuronal inclusions of Parkinson's disease. Mov Disord. 1991;6:2–11. [PubMed: 1848677]
- 53.
- Gibb WRG, Lees AJ. The significance of the Lewy body in the diagnosis of idiopathic Parkinson's disease. Neuropathol Appl Neurobiol. 1989;15:27–44. [PubMed: 2542825]
- 54.
- Forno LS. Concentric hyalin intraneuronal inclusion of Lewy type in the brain of elderly persons (50 incicental cases): Relationship to parkinsonism. J Am Geriatr Soc. 1969;17:557–575. [PubMed: 4182529]
- 55.
- Gibb WRG, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry. 1988;51:745–752. [PMC free article: PMC1033142] [PubMed: 2841426]
- 56.
- Dickson DW. Aging in the central nervous systemIn: Markesbery WR, ed. Neuropathology of Dementing Disorders London, New York: Arnold,199856–88.
- 57.
- Dickson DW, Crystal HA, Mattiace LA. et al. Identification of normal and pathological aging. Neurobiol Aging. 1992;17:365–371.
- 58.
- Braak H, Braak E, Yilmazer D. et al. Age-related changes of the human cerebral cortexIn: Cruz-Sanchez FF, Ravid R, Cuzner ML, eds. Neuropathological Diagnostic Criteria for Brain Banking. Biomedical Health Research. Vol 10 Amsterdam: IOS Press,199514–19.
- 59.
- Wenning GK, Ben ShlomoY, Magalhaes M. et al. Clinical features and natural history of multiple system atrophy: An analysis of 100 cases. Brain. 1994;117:835–845. [PubMed: 7922469]
- 60.
- Klein C, Brown R, Quinn N. The “cold hands sign.” Mov Disord. 1997;12:514–518. [PubMed: 9251069]
- 61.
- Giasson BI, Galvin JE, Lee VM-Y. et al. The cellular and molecular pathology of Parkinson's diseaseIn: Clark CM, Trojanowski JQ, eds.Neurodegenerative Dementias: Clinical Features and Pathological Mechanisms New York: McGraw Hill, 2000219–228.
- 62.
- Trojanowski JQ, Lee VM-Y. “Fatal attractions” of proteins: A comprehensive hypothetical mechanism underlying Alzheimer's disease and other neurodegenerative disorders. Ann NY Acad Sci. 2000;924:62–67. [PubMed: 11193803]
- 63.
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. [PubMed: 11121744]
- 64.
- Walker LC, LeVine H. The cerebral proteopathies. Neurodegenerative disorders of protein conformation and assembly. Mol Neurobiol. 2000;21:83–95. [PubMed: 11327151]
- 65.
- McNaught KSP, Olanow CW, Halliwell B. et al. Failure of the ubiquitin-proteasome system in Parkinson's disease. Nature Rev/Neuroscience. 2001;2:589–594. [PubMed: 11484002]
- 66.
- McNaught KStP, Shashidharan P, Perl DP. et al. Aggresome-related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16:2136–2148. [PubMed: 12473081]
- 67.
- Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in α-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. [PubMed: 11152691]
- 68.
- Gai WP, Blessing WW, Blumbergs PC. Ubiquitin-positive degenerating neurites in the brainstem in Parkinson's disease. Brain. 1995;118:1447–1459. [PubMed: 8595476]
- 69.
- Baba M, Nakajo S, Tu PH. et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article: PMC1858234] [PubMed: 9546347]
- 70.
- Spillantini MG, Crowther RA, Jakes R. et al. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. [PMC free article: PMC27806] [PubMed: 9600990]
- 71.
- Trojanowski JQ, Lee VMY. Aggregation of neurofilament and α-synuclein proteins in Lewy bodies - implications for the pathogenesis of Parkinson-disease and Lewy body dementia. Arch Neurol. 1998;55:151–152. [PubMed: 9482355]
- 72.
- Goedert M. Filamentous nerve cell inclusions in neurodegenerative diseases: Tauopathies and α-synucleinopathies. Phil Trans R Soc Lond B. 1999;354:1101–1108. [PMC free article: PMC1692614] [PubMed: 10434313]
- 73.
- Golbe LI. Alpha-synuclein and Parkinson's disease. Mov Disord. 1999146-9 [PubMed: 9918338]
- 74.
- Duda JE, Lee VMY, Trojanowski JQ. Neuropathology of synuclein aggregates: New insights into mechansim of neurodegenerative diseases. J Neurosci Res. 2000;61:121–127. [PubMed: 10878583]
- 75.
- Braak E, Braak H. Silver staining method for demonstrating Lewy bodies in Parkinson's disease and argyrophilic oligodendrocytes in multiple system atrophy. J Neurosci Methods. 1999;87:111–115 29 Idiopathic Parkinson's Disease Staging an -Synucleinopathy with a Predictable Pathoanatomy. [PubMed: 10066000]
- 76.
- Sandmann-Keil D, Braak H, Okochi M. et al. Alpha-synuclein immunoreactive Lewy bodies and Lewy neurites in Parkinson's disease are detectable by an advanced silver-staining technique. Acta Neuropathol. 1999;98:461–464. [PubMed: 10541868]
- 77.
- Ohama E, Ikuta F. Parkinson's disease: Distribution of Lewy bodies and monoamine neuron system. Acta Neuropathol. 1976;34:311–319. [PubMed: 179263]
- 78.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. [PubMed: 1759558]
- 79.
- Dickson DW, Ruan D, Crystal H. et al. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer's disease: Light and electron microscopic immunocytochemistry of CA2-3 neurites specific to DLBD. Neurology. 1991;41:1402–1409. [PubMed: 1653914]
- 80.
- Dickson DW, Schmidt ML, Lee VMY. et al. Immunoreactivity profile of hippocampal CA2/3 neurites in diffuse Lewy body disease. Acta Neuropathol. 1994;87:269–276. [PubMed: 7912027]
- 81.
- Iseki E, Li F, Odawara T. et al. Kosaka K. Hippocampal pathology in diffuse Lewy body disease using ubiquitin-immunohistochemistry. J Neurol Sci. 1997;149:165–169. [PubMed: 9171325]
- 82.
- Braak H, Sandmann-Keil D, Gai WP. et al. Extensive axonal Lewy neurites in Parkinson's disease: A novel pathological feature revealed by α-synuclein immunocytochemistry. Neurosci Lett. 1999;265:67–69. [PubMed: 10327208]
- 83.
- Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52:183–191. [PubMed: 7684074]
- 84.
- Lowe J. Lewy bodiesIn: Calne DP, ed. Neurodegenerative Diseases Philadelphia: Saunders,199451–69.
- 85.
- Bergeron C, Pollanen MS. Pathogenesis of the Lewy bodyIn: Perry RH, McKeith IG, Perry EK, eds.Dementia with Lewy BodiesCambridge, New York: Cambridge University Press,1996302–307.
- 86.
- Rezaie P, Cairns NJ, Chadwick A. et al. Lewy bodies are located preferentially in limbic areas in diffuse Lewy body disease. Neurosci Lett. 1996;212:111–114. [PubMed: 8832651]
- 87.
- Braak H. Architectonics of the Human Telencephalic Cortex. Berlin: Springer, 1980.
- 88.
- Zilles K. Cortex. In: Paxinos G, ed. The Human Nervous System. Academic New York: Press, 1990:757-802.
- 89.
- Dart RA. The dual structure of the neopallium: Its history and significance. J Anat. 1934;69:3–19. [PMC free article: PMC1249252] [PubMed: 17104513]
- 90.
- Sanides F. Comparative architectonics of the neocortex of mammals and their evolutionary interpretation. Ann NY Acad Sci. 1969;167:404–423.
- 91.
- Price JL. Olfactory system. In: Paxinos G, ed. The Human Nervous System. San Diego: Academic Press, 1990:979-1000.
- 92.
- Braak H, Braak E. The human entorhinal cortex: Normal morphology and lamina-specific pathology in various diseases. Neurosci Res. 1992;15:6–31. [PubMed: 1336586]
- 93.
- Sanides F. The cyto-myeloarchitecture of the human frontal lobe and its relation to phylogenetic differentiation of the cerebral cortex. J Hirnforsch. 1964;6:267–282. [PubMed: 14227452]
- 94.
- Pandya DN, Yeterian EH. Architecture and connections of cortical association areasIn: Jones EG, Peters A, eds. Cerebral Cortex. Association and Auditory Cortices. Vol 4 New York, London: Plenum Press, 19853–61.
- 95.
- Pandya DN, Yeterian EH. Architecture and connections of cerebral cortex: Implications for brain evolution and function In: Scheibel AB, Wechsler AF, eds. Neurobiology of Higher Function New York: Guilford Press,199053–84.
- 96.
- Mesulam MM. From sensation to cognition. Brain. 1998;121:1013–1052. [PubMed: 9648540]
- 97.
- Rockland KS, Pandya DN. Laminar origins and terminations of cortical connections of the occipital lobe in the rhesus monkey. Brain Res. 1979;179:3–20. [PubMed: 116716]
- 98.
- Alexander GE, Crutcher MD, DeLong MR. Basal ganglia-thalamocortical circuits: Parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Progr Brain Res. 1990;85:119–146. [PubMed: 2094891]
- 99.
- Alheid GF, Heimer L, Switzer RC. Basal ganglia. In: Paxinos G, ed. The Human Nervous System. San Diego: Academic Press, 1990:483-582.
- 100.
- Braak H, Braak E. Anatomy of the human basal gangliaIn: Szelenyi I, ed. Inhibitors of Monoamine Oxidase Basel: Birkhäer,19933–23.
- 101.
- Albin RL, Young AB, Peney JB. The functional anatomy of disorders of the basal ganglia. Trends Neurosci. 1995;18:63–64. [PubMed: 7537410]
- 102.
- Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The corticobasal ganglia-thalamo-cortical loop. Brain Res Rev. 1995;20:91–127. [PubMed: 7711769]
- 103.
- Nauta HJW. Circuitous connections linking cerebral cortex, limbic system, and corpus striatumIn: Doane BK, Livingstone KE, eds. The Limbic System New York: Raven Press,198643–54.
- 104.
- Heimer L, De OlmosJ, Alheid GF. et al. “Perestroika” in the basal forebrain: Opening the border between neurology and psychiatry. Progr Brain Res. 1991;87:109–130. [PubMed: 1866444]
- 105.
- Amaral DG, Price JL, Pitkän A. et al. Anatomical organization of the primate amygdaloid complex. In: Aggleton JP, ed. The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction. New York: Wiley-Liss, 1987:1-66.
- 106.
- Hyman BT, van HoesenGW, Damasio AR. Memory-related neural systems in Alzheimer's disease: An anatomic study. Neurology. 1990;40:1721–1730. [PubMed: 2234428]
- 107.
- Damasio AR, Damasio H. Disorders of higher brain function. In: Rosenberg RN, ed. Comprehensive Neurology. New York: Raven Press, 1991:639-657.
- 108.
- Zola-Morgan S, Squire LR. Neuroanatomy of memory. Annu Rev Neurosci. 1993;16:547–563. [PubMed: 8460903]
- 109.
- Braak H, Braak E. Pathoanatomy of Parkinson's disease. J Neurol. 2000;247(Suppl 2):3–20. [PubMed: 10991663]
- 110.
- Braak H. Architectonics as seen by lipofuscin stainsIn: Jones EG, Peters A, eds. Cerebral Cortex. Cellular Components of the Cerebral Cortex. Vol I New York, London: Plenum Press,198459–104.
- 111.
- Yakovlev PI, Lecours AR. The myelogenetic cycles of regional maturation of the brain. In: Minkowksi A, ed. Regional Development of the Brain in Early Life. Oxford: Blackwell Sci Publ, 1967:3-70.
- 112.
- Kinney HC, Brody BA, Kloman AS. et al. Sequence of central nervous system myelination in human infancy. II. Patterns of myelination in autopsied infants. J Neuropathol Exp Neurol. 1988;47:217–234. [PubMed: 3367155]
- 113.
- van der Knapp MS, Valk J, Bakker CJ. et al. Myelination as an expression of the functional maturity of the brain. Dev Med Child Neurol. 1991;33:849–857. [PubMed: 1743407]
- 114.
- Hasegawa M, Houdou S, Mito T. et al. Development of myelination in the human fetal and infant cerebrum: A myelin basic protein immunohistochemical study. Brain Dev. 1992;141-146 [PubMed: 1375444]
- 115.
- Wszolek ZK, Markopoulou K. Olfactory dysfunction in Parkinson's disease. Clinical Neurosci. 1998;5:94–101. [PubMed: 10785834]
- 116.
- Saper CB, Sorrentino DM, German DC. et al. Medullary catecholaminergic neurons in the normal human brain and in Parkinson's disease. Ann Neurol. 1991;29:577–584. [PubMed: 1892359]
- 117.
- Huang XF, Törk I, Paxinos G. Dorsal motor nucleus of the vagus nerve: A cyto- and chemoarchitectonic study in the human. J Comp Neurol. 1993;330:158–182. [PubMed: 7684048]
- 118.
- Hopkins DA, Bieger D, de Vente J. et al. Vagal efferent projections: Viscerotopy, neurochemistry and effects of vagotomy. Progr Brain Res. 1996;107:79–96. [PubMed: 8782514]
- 119.
- Martin GF, Hostege G, Mehler WM. Reticular formation of the pons and medulla. In: Paxinos G, ed. The Human Nervous System. San Diego: Academic Press, 1990:203-220.
- 120.
- Holstege G. The emotional motor system. Eur J Morphol. 19923067-79 [PubMed: 1642954]
- 121.
- Randich A, Gebhart GF. Vagal afferent modulation of nociception. Brain Res Rev. 1992;17:77–99. [PubMed: 1327371]
- 122.
- Whitehouse PJ, Hedreen JC, White CL. et al. Basal forebrain neurons in the dementia of Parkinson's disease. Ann Neurol. 1983;13:243–248. [PubMed: 6847136]
- 123.
- Braak H, Braak E. Nuclear configuration and neuronal types of the nucleus niger in the brain of the human adult. Human Neurobiol. 1986;5:71–82. [PubMed: 2426228]
- 124.
- Nieuwenhuys R. The greater limbic system, the emotional motor system and the brain. Progr Brain Res. 1996;107:551–580. [PubMed: 8782542]
- 125.
- Hirsch EC, Graybill AM, Duyckaerts C. et al. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci. 1987;84:5976–5980. [PMC free article: PMC298986] [PubMed: 3475716]
- 126.
- Lee MS, Rinne JO, Marsden CD. The pedunculopontine nucleus: Its role in the genesis of movement disorders. Yonsei Med J. 2000;41:167–184. [PubMed: 10817016]
- 127.
- Zweig RM, Jankel WR, Hedreen JC. et al. The pedunculopontine nucleus in Parkinson's disease. Ann Neurol. 1989;26:41–46. [PubMed: 2549845]
- 128.
- Inglis WL, Winn P. The pedunculopontine tegmental nucleus: Where the striatum meets the reticular formation. Progr Neurobiol. 1995;47:1–29. [PubMed: 8570851]
- 129.
- Rye D, Saper CB, Lee HJ. et al. Pedunculopontine tegmental nucleus of the rat: Cytoarchitecture, cytochemistry, and some extrapyramidal connections of the mesopontine tegmentum. J Comp Neurol. 1988;259:483–528. [PubMed: 2885347]
- 130.
- Gottesmann C. The neurophysiology of sleep and waking, intracerebral connections, functioning and ascending influences of the medulla oblongata. Progr Neurobiol. 1999;59:1–54. [PubMed: 10416960]
- 131.
- Garcia-Rill E. The pedunculopontine nucleus. Progr Neurobiol. 1991;36:363–389. [PubMed: 1887068]
- 132.
- Saper CB. Diffuse cortical projection systems: Anatomical organization and role in cortical function. In: Plum F, ed. Handbook of Physiology. The Nervous System. Bethesda: Am Physiol Soc, 1987:5:169-210.
- 133.
- Saper CB. Cholinergic systemIn: Paxinos G, ed.The Human Nervous System San Diego: Academic Press,19901095–1113.
- 134.
- Mesulam MM, Hersh LB, Mash DC. et al. Differential cholinergic innervation within functional subdivisions of the human cerebral cortex - a choline acetyltransferase study. J Comp Neurol. 1992;318:316–328. [PubMed: 1374768]
- 135.
- Candy JM, Perry RH, Perry EK. et al. Pathological changes in the nucleus of Meynert in Alzheimer's and Parkinson's diseases. J Neurol Sci. 1983;59:277–289. [PubMed: 6854353]
- 136.
- De OlmosJS. Amygdala. In: Paxinos G, ed. The Human Nervous System. San Diego: Academic Press, 1990:583-710.
- 137.
- Sims KS, Williams RS. The human amygdaloid complex: A cytologic and histochemical atlas using Nissl, myelin, acetylcholinesterase and nicotinamide adenine dinucleotide phosphate diaphorase staining. Neuroscience. 1990;36:449–472. [PubMed: 1699167]
- 138.
- Bohus B, Koolhaas JM, Luiten PGM. et al. The neurobiology of the central nucleus of the amygdala in relation to neuroendocrine and autonomic outflow. Prog Brain Res. 1996;107:447–460. [PubMed: 8782536]
- 139.
- Rosene DL, van HoesenGW. The hippocampal formation of the primate brain. A review of some comparative aspects of cytoarchitecture and connections In: Jones EG, Peters A, eds. Cerebral Cortex. Further Aspects of Cortical Function, Including Hippocampus New York, London: Plenum Press, 19876345–456.
- 140.
- Amaral DG, Insausti R. Hippocampal formation. In: Paxinos G, ed. The Human Nervous System San Diego: Academic Press, 1990:711-755.
- 141.
- van HoesenGW, Hyman BT. Hippocampal formation: Anatomy and the patterns of pathology in Alzheimer's disease. Progr Brain Res. 1990;83:445–457. [PubMed: 2392569]
- 142.
- Braak H, Braak E, Yilmazer D. et al. Functional anatomy of human hippocampal formation and related structures. J Child Neurol. 1996;11:265–275. [PubMed: 8807415]
- 143.
- Witter MP. Organization of the entorhinal-hippocampal system: A review of current anatomical data. Hippocampus. 1993;3(Suppl):33–44. [PubMed: 8287110]
- 144.
- Sherk H. The claustrum and the cerebral cortexIn: Jones EG, Peters A, eds. Cerebral Cortex. Sensory-motor Areas and Aspects of Cortical Connectivity. Vol 5 New York, London: Plenum Press, 1986467–499.
- 145.
- Heimer L, Switzer RD, van Hoesen GW. Ventral striatum and ventral pallidum. Components of the motor system? Trends Neurosci. 1982;5:83–87.
- 146.
- Haber SN, Kunishio K, Mizobuchi M. et al. The orbital and medial prefrontal circuit through the primate basal ganglia. J Neurosci. 1995;15:4851–4867. [PMC free article: PMC6577885] [PubMed: 7623116]
- 147.
- Saper CB. Role of the cerebral cortex and striatum in emotional motor response. Progr Brain Res. 1996;107:537–550. [PubMed: 8782541]
- 148.
- Damasio AR, Tranel D, Damasio H. Individuals with sociopathic behavior caused by frontal damage fail to respond automatically to social stimuli. Behav Brain Res. 1990;41:81–94. [PubMed: 2288668]
- 149.
- Neafsy EJ. Prefrontal cortical control of the autonomic nervous system: anatomical and physiological observations. Progr Brain Res. 1990;85:147–166. [PubMed: 2094892]
- 150.
- Loewy AD. Forebrain nuclei involved in autonomic control. Progr Brain Res. 1991;87:253–268. [PubMed: 1866449]
- 151.
- Oppenheimer SM, Gelb A, Girvin JP. et al. Cardiovascular effects of human insular cortex stimulation. Neurology. 1992;42:1727–32. [PubMed: 1513461]
- 152.
- Drevets WC, Price JL, Simpson JR. et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. [PubMed: 9126739]
- 153.
- Lane RD, Reiman EM, Ahern GL. et al. Neuroanatomical correlates of happiness, sadness, and disgust. Am J Psychiatr. 1997;154:926–933. [PubMed: 9210742]
- 154.
- Reiman EM, Lane RD, Ahern GL. et al. Neuroanatomical correlates of externally and internally generated human emotion. Am J Psychiatr. 1997;154:918–925. [PubMed: 9210741]
- 155.
- Damasio AR. Emotion in the perspective of an integrated nervous system. Brain Res Rev. 1998;26:83–86. [PubMed: 9651488]
- 156.
- Mesulam MM, Mufson EJ. The insula of Reil in man and monkeyIn: Jones EG, Peters A, eds.Cerebral Cortex. Association and Auditory Cortices New York, London: Plenum Press,19934179–225.
- 157.
- Augustine JR. Circuitry and functional aspects of the insular lobe in primates including humans. Brain Res Rev. 1996;22:229–244. [PubMed: 8957561]
- 158.
- Cechetto DF, Saper CB. Role of the cerebral cortex in autonomic functionIn: Loewy AD, Spyer KM, eds.Central Regulation of Autonomic Function New York: Oxford University Press, 1990208–223.
- 159.
- Morecraft RJ, Geula C Mesulam MM. Architecture of connectivity within a cingulo-fronto-parietal neurocognitive network for directed attention. Arch Neurol. 1993;50:279–284. [PubMed: 8442707]
- 160.
- Price JL, Carmichael ST, Drevets WC. Networks related to the orbital and medial prefrontal cortex: a substrate for emotional behavior? Progr Brain Res. 1996;107:528–536. [PubMed: 8782540]
- 161.
- Ögür D, An X, Price JL. Prefrontal cortical projections to the hypothalamus in macaque monkeys. J Comp Neurol. 1998;401:480–505. [PubMed: 9826274]
- 162.
- Meredith GE, Pattiselanno A, Groenewegen HJ. et al. Shell and core in monkey and human nucleus accumbens identified with antibodies to calbindin-D28k. J Comp Neurol. 1996;365:628–639 Peptide Nucleic Acids Morpholinos and Related Antisense Biomolecules 32. [PubMed: 8742307]
- 163.
- Chu CC, Tranel D, Damasio AR. et al. The autonomic-related cortex: pathology in Alzheimer's disease. Cerebral Cortex. 1997;7:86–95. [PubMed: 9023436]
- 164.
- Pearce RK, Hawkes CH, Daniel SE. The anterior olfactory nucleus in Parkinson's disease. Mov Disord. 1995;10:283–287. [PubMed: 7651444]
- 165.
- Hawkes CH, Shephard BC, Daniel SE. Olfactory dysfunction in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1997;62:436–446. [PMC free article: PMC486843] [PubMed: 9153598]
- 166.
- Doty RL, Deems DA, Stellar S. Olfactory dysfunction in parkinsonism: A general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology. 1988;38:1237–1244. [PubMed: 3399075]
- 167.
- Mesholam RL, Moberg PJ, Mahr RN. et al. Olfaction in neurodegenerative disease. A meta-analysis of olfactory functioning in Alzheimer's and Parkinson's diseases. Arch Neurol. 1998;55:84–90. [PubMed: 9443714]
- 168.
- Liberini P, Parola S, Spano PF. et al. Olfaction in Parkinson's disease: Methods of assessment and clinical relevance. J Neurol. 2000;247:88–96. [PubMed: 10751109]
- 169.
- Vogt BA, Finch DM, Olson CR. Functional heterogeneity in cingulate cortex: The anterior executive and posterior evaluative regionsIn: Jones EG, Peters A, eds. Cerebral Cortex. Functional Properties of Cortical Cells New York, London: Plenum Press,19922435–443. [PubMed: 1477524]
- 170.
- Vogt BA, Sikes RW, Vogt LJ. Anterior cingulate cortex and the medial pain systemIn: Vogt BA, Gabriel M, eds.Neurobiology of Cingulate Cortex and Limbic Thalamus Boston, Birkhäer:1993313–344.
- 171.
- van HoesenGW, Morecraft RJ, Vogt BA. Connections of the monkey cingulate cortexIn: Vogt BA, Gabriel M, eds.Neurobiology of Cingulate Cortex and Limbic Thalamus Boston, Birkhäer:1993249–284.
- 172.
- Smithson KG, MacVicar BA, Hatton GI. Polyethylene glycol embedding: A technique compatible with immunocytochemistry, enzyme histochemistry, histofluorescence and intracellular staining J Neurosci Methods 1983. >727–41. [PubMed: 6188002]
- 173.
- Lopes da Silva FH, Witter MP, Boeijinga PH. et al. Anatomic organization and physiology of the limbic cortex. Physiol Rev. 1990;70:453–511. [PubMed: 2181500]
- Idiopathic Parkinson's Disease: Staging an α-Synucleinopathy with a Predictable ...Idiopathic Parkinson's Disease: Staging an α-Synucleinopathy with a Predictable Pathoanatomy - Madame Curie Bioscience Database
- The Role of Heat Shock Proteins during Neurodegeneration in Alzheimer's, Parkins...The Role of Heat Shock Proteins during Neurodegeneration in Alzheimer's, Parkinson's and Huntington's Disease - Madame Curie Bioscience Database
- Abl Family Kinases in Mammalian Development - Madame Curie Bioscience DatabaseAbl Family Kinases in Mammalian Development - Madame Curie Bioscience Database
- Cell-Cell Channels and Their Implications for Cell Theory - Madame Curie Bioscie...Cell-Cell Channels and Their Implications for Cell Theory - Madame Curie Bioscience Database
- Antibiotic Resistance in Bacteria Caused by Modified Nucleosides in 23S Ribosoma...Antibiotic Resistance in Bacteria Caused by Modified Nucleosides in 23S Ribosomal RNA - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...