

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction
The Toll family of receptors comprises numerous related proteins implicated in the development and defense of plants and animals. Toll was first discovered in Drosophila melanogaster as a gene that controlled the dorsal-ventral axis of the developing embryo. Elements of its molecular structure; the extracellular leucine-rich repeat domain (LRR), short cysteine rich patches, a transmembrane portion, and an intracellular domain homologous to that of the human interleukin-1 receptor (IL-1R) are discussed in detail in other chapters. Here we are principally concerned with the role of the Toll like receptors (TLRs) and their signaling pathways in the immune system.
TLRs are found in Arabidopsis as intracellular proteins, whereas the Toll proteins of Drosophila, Tol in Caenorhabditis elegans, and the mammalian TLRs are all transmembrane proteins. The human TLRs comprise a family of ten related proteins (Fig. 1). Across mammalian species the number of TLRs differs, as does their expression in different cell types and their transcriptional regulation in activated cells. Ligands (some synthetic) for all but the tenth TLR have been identified, and their number is rapidly growing, due in no small part to the frenetic research activity in this area.

Figure 1
Human TLRs.
TLR ligands are varied, comprising bacterial cell wall components, bacterial genomic DNA, fungal, parasitic and viral products and synthetic analogs of natural products. Interestingly, TLRs can also bind autologous (self ) molecules such as heat shock proteins (HSPs), intercellular matrix products, and mammalian genomic DNA. In general terms, the ligands for mammalian TLRs are either products of microbial origin that have an unusual molecular motif (pattern) or can be derived from the host species itself. A closer look reveals that host-derived ligands are usually shielded or concealed from the immune system and their emergence, for example after tissue trauma, signals that intervention by the immune system is required.
The presence of LRR modules in plant and animal proteins suggests an evolutionarily conserved role as a molecular pattern recognition receptor. Additionally, the developmental functions of TLRs in the fruit fly and nematode point to another role for TLRs in higher vertebrates, which could be to sense tissue integrity. This role could have arisen after the developmental functions that we ordinarily attribute to TLRs. Additionally; we can envisage other functions for TLRs if we consider that their activation can give rise to ten potentially divergent signals. These signals, modulated by their intensity, the cell type (including differentiation stage) of their derivation, and cellular microenvironment, may synergize or compete with one another to generate distinct TLR signals. Thus, the action of B and T cells could depend on the type of TLR signal generated by antigen presenting cells.
TLRs help the immune system to fight the dangerous, protect the useful and neglect the vast majority of harmless microorganisms that colonize our bodies. These three functions could be generalized in a statement not dissimilar to “immunity maintains the integrity of tissues”. This statement can be taken as a start point for the “Integrity Hypothesis”, which proposes three functions for TLRs. The first is to detect unusual molecular patterns. The second is to sense the extent of tissue damage, and the third is to determine the class of immune response. Specialized cells of central immunity such as dendritic cells and T and B cells are principle players in integrating these TLR signals into a specific immune response.
Clearly a better understanding of the function of TLRs in higher vertebrates is crucial for biomedical research as it will allow us to improve health care by refining the therapeutic regimes with which we treat disease.
Developmental Functions
Drosophila
The Drosophila genome contains a total of nine Toll-like genes. Toll-1 and its signaling pathway were identified by the genetic analyses of mutants which led to the discovery of maternal signals that are important for the dorsal-ventral patterning (Fig. 2) of the embryo (for a review see ref. 1). The expression levels of the other Toll proteins also change throughout development, suggesting that they may all play a role in embryonic development.
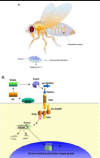
Figure 2
A) Drosophila development. B) Toll pathway in Drosophila development.
Dorsal-Ventral Polarization of the Embryo
Toll-1 mRNA is maternally transmitted and distributes evenly within the early embryo. Toll-1 is a transmembrane protein and its action begins at stage 5 of embryonic development. Prior to this stage, which is also known as the cell formation stage, the embryo is a sac containing a large number of nuclei. Cell membranes then begin to form around these nuclei, and Toll-1 inserts into the membranes to face out towards the perivitelline space (Fig. 2A). Subsequently, a proteolytic cascade is initiated by a positional signal in the ventral region of the embryo (Fig. 2B) that generates Späle, which binds Toll-1. Toll-1 then recruits the Drosophila death domain homologue of Myd88, then Pelle and Tube. Pelle, a serine/threonine kinase, phosphorylates itself as well as Tube and Toll-1. Downstream signals, possibly mediated by an unknown kinase, eventually result in the cleavage of Cactus, (equivalent to the degradation of I-κB in the I-κB/NF-κB complex) and the liberation of the transcription factor Dorsal. Dorsal translocates to the nucleus to drive the expression of genes responsible for dorsal-ventral polarization and upregulates ventral genes (such as Twist). At the same time Dorsal acts to inhibit the "dorsal" genes in a ventral patch of cells in the embryo. This action is also responsible for the formation of a dorsalventral calcium gradient at stage 5, approximately 2.5 hours after fertilization.
Muscle Development
Toll-1 is preferentially expressed in some muscle cells and it has been suggested that early development and muscle fiber growth is dependent on Toll action. The insertion of certain muscle cells into epidermal “muscle attachment cells” has also been shown to be dependent on Toll expression.
Motoneuron Growth Cone Guidance
Motoneuron growth cone guidance, also called “axonal pathfinding” is a process whereby growing neurons penetrate tissues until they form a synapse with specific muscles. This guidance is dependent on Toll expression, and specifically on Toll's modulation of the glycosylation pattern of the cells in which it is expressed. Interestingly this action provokes Toll null cells to alter their glycosylation pattern, providing a beacon with which to guide the axon to the muscle cell. It is plausible that the growth-cone receptors can recognize glycosylation patterns and use them to steer axonal growth. The role of Toll expression in other organs, including the salivary glands, pharynx, esophagus, gut, and malpighian tubules of Drosophila is unknown.
Caenorhabditis elegans
Components of the Drosophila Toll like pathway were also found in the half-millimeter long nematode called Caenorhabditis elegans and their developmental roles were examined in deletion mutants of tol-1, trf-1, pik-1, and ikb-1; the C. elegans homologues of Drosophila toll-1, dtraf, pelle, and the cactus genes respectively. These analyses showed that only tol-1 is essential for C. elegans development.
Others
Thus far, the role of TLRs in embryonic development ends with C. elegans. It is tempting to speculate that a developmental role for TLRs may exist in higher animals. However, all the murine TLR gene knockouts have been found to develop normally and their sole defects are immune related. It therefore seems unlikely that TLRs will be found to play a role in embryonic development in mammals.
It is worth noting that the somatic cells of mammalian hematopoietic lineages develop or mature upon binding TLR ligands. These include cells of myeloid and lymphoid lineage, including monocytes, precursors of dendritic cells (DCs) and B cells. Though I consider these to be developmental functions, they will be discussed later as they also dovetail with the defense functions of these cells. Similarly, osteoblasts can be induced by TLRs 4 and 9 to exhibit osteoclastogenic activity. TLRs are expressed in many somatic tissues of higher animals, but their functional significance in these tissues remains largely unknown.
Human TLR1 is ubiquitously expressed whereas TLR2 is expressed in the brain, heart, muscle and lungs. TLR3 is, in addition, expressed in the placenta and pancreas. TLR4 is abundant in the placenta, on cardiomyocytes, endothelial and smooth muscle cells and hematopoetic cells. The distribution of TLR5 is similar to TLR4, with the inclusion of its ovarian expression. TLR6 is expressed in the thymus, spleen, ovaries and lungs. However, TLRs 7 and 8 show a more restricted expression profile. TLR7 is found solely on plasmacytoid dendritic cells (pDCs) whereas TLR8 is found on monocyte derived dendritic cells. TLRs 9 and 10 are expressed on dendritic cells, and interestingly, are abundant on activated B cells.
It is possible that the primitive developmental role of TLRs has evolved in higher vertebrates into specific roles for somatic tissues, perhaps controlling their regeneration, or as physiologic mediators of homeostasis (in adults). Drawing attention to these possibilities allows us to understand how TLRs could sense tissue integrity.
The Role in Defense
Plants
The first plant protein to be discovered with any similarity to Toll was the tobacco N gene product that confers resistance to the tobacco mosaic virus. However, the similarity to Toll did not extend beyond its LRR and TIR modules. Further, in contrast to Toll proteins, the N gene product lacks a transmembrane region and is therefore intracellular. However, the N gene product was found to express another module called the nucleotide binding site, which is present in most other plant Toll-like proteins. In the Arabidopsis genome, the first plant genome to be completely sequenced, similarity searches have identified an unusually large number of proteins with modular compositions similar to the N gene product. A hallmark of most of these plant Toll-like proteins is that they are intracellular and it has been suggested that they serve in defense as pathogen sensors.
Interestingly, another possible pathogen sensor in Arabidopsis is the FLS2 protein, a transmembrane receptor, which has extracellular LRRs and a cytoplasmic serine/threonine kinase domain. Analysis of deletion mutants of FLS2 showed that it could be involved in the recognition of flagellin, the principle protein of bacterial flagella. Flagellin retards the growth of wild type plants, which may serve as a defense response as well as an infection avoidance reaction. This response may be analogous to the aversion responses seen in C. elegans (see below).
Drosophila
Humoral
Infection with the fungus Beauveria provokes Toll-1 receptor signaling which mediates the induction of the antifungal agent drosomycin. There is no direct binding of the fungus to Toll-1 receptor. Rather, the extracellular Toll-1 cascade, shown in Figure 3, is activated by the endogenous ligand Späle. Free Späle is normally unavailable, and exists as part of a larger precursor. The serine protease Persephone (whose action is tightly regulated), cleaves the Späle precursor into two unequal parts, one of which, Späle, binds to Toll-1. Persephone's action can be blocked by a protein called Necrotic. However, Persephone is rendered constitutively inactive by an unknown protease inhibitor. Blockade of this inhibitor (by Beauveria) triggers a cascade of events that leads to the anti-fungal response. Downstream of Toll-1, most of the factors involved in developmental signaling are also involved in the immune response, though with some important differences (for a review see ref. 2).
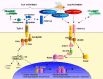
Figure 3
Humoral and cellular immunity in Drosophila.
Thus, in Drosophila adults, the Toll-1 pathway generates a quite different output. Namely, it regulates the transcription of genes that encode antimicrobial peptides, such as drosomycin and defensin, as well as regulating the genes that alter hemocyte density. Figure 3 illustrates the crucial differences between Toll-1's developmental and defense signaling pathways. In immunity the degradation of Cactus releases, instead of Dorsal, the Dorsal related immunity factor (Dif ), which then translocates to the nucleus where it acts in tandem with Relish, a nuclear factor derived from a different signaling pathway.
A Gram positive organism Micrococcus has been shown to activate the Toll-1 pathway, and drives the expression of the antifungal peptides drosomycin and defensin. Micrococcus binds extracellularly to a pattern recognition receptor called the peptidoglycan recognition protein (PGRP-SA), which is encoded by semmelweis. PGRP-SA freely circulates in the hemolymph and, using a domain that can bind to the peptidoglycan moiety of Gram positive bacteria, probably activates a serine protease that cleaves pre-Späle. This event triggers the intracellular cascade that leads to the successful defense against Micrococcus (Fig. 3) (for a review see ref. 3).
Another defense pathway is the immune deficiency (Imd) pathway, named after a particular (imd) mutant. Interestingly, a related pattern recognition receptor to that involved in Toll-1 signal transduction may play an important role in this pathway. However, the two pattern recognition molecules (PGRP-LC, and -LE) involved, are transmembrane proteins, probably located in specialized cells. The Imd product is crucial to intracellular signaling, but does not interact with a pattern recognition receptor. Instead Imd signals downstream via a two-pronged pathway that involves the IKK kinase in one arm, and Dredd in the other. Both pathways cleave Relish, to release its nuclear binding portion that then translocates to the nucleus to form homodimers or heterodimers with Dif. These in turn act on various gene targets that are important in humoral defense. Figure 3 illustrates the main defense pathways of Drosophila. It appears that the Imd pathway is more potent with respect to humoral defense than the Toll-1 pathway as it controls the expression of a larger number of antimicrobials including cecropin and diptericin. These confer protection against a wide range of pathogenic microorganisms including fungi, Gram negative and Gram positive bacteria. The Gram negative diaminopimelic acid type peptidoglycan is the most potent inducer of the Imd pathway, whilst the Toll pathway is predominantly activated by the Gram positive lysine type of peptidoglycan. Thus, the ability of Drosophila to discriminate between Gram positive and Gram negative bacteria may rely on its recognition of specific forms of peptidoglycan.
Cellular
The lymph glands of adult Drosophila produce hemocytes that circulate throughout the body. These ensure host defense by encapsulating foreign bodies and phagocytosing smaller toxic objects. Encapsulation and subsequent melanization inactivates the intruder, which remains trapped in the body. Several genes that regulate cellular immunity also control the proliferation and differentiation of the hematopoetic lineage. The Polycomb group of genes regulates lymph gland cell proliferation and hemocyte numbers in the body. Mutant larvae show increased growth of the gland and excessive numbers of hemocytes that occasionally go on to invade tissue as pseudotumors. Mutants with enhanced Toll signaling show a similar phenotype, whereas those with reduced Toll signaling have fewer hemocytes (for a review see ref. 4).
In conclusion, the Toll pathway regulates cellular and some humoral aspects of immunity in Drosophila.
C. elegans
Caenorhabditis elegans expresses structural homologues of several components of the Drosophila Toll pathway. The tol-1 gene, homologous to toll, is required not only for nematode development, but also to avoid the pathogenic microbe Serratia marcescens. Analyses of deletion mutants of the C. elegans Tol pathway showed that none, save tol-1, were important for immunity. Interestingly, tol-1 deletion mutants lost their S. marcescens avoidance behavior though other chemosensory behaviors remained intact. Further analysis revealed that the Tol-1 protein is required for sensing S. marcescens by a neural system. Tol-1 is located on the sensory tip of an axon located near the oral orifice and microbial contact generates signals that then induce the evasion response (Fig. 4).5 This phenomenon is discussed in more detail by Nathalie Pujol and Jonathan Ewbank in their chapter.
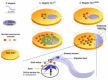
Figure 4
Defensive role of Tol-1 in Caenorhabditis elegans.
The defensive role of the neural system has not been studied sufficiently at the molecular level in higher vertebrates, due to the obvious complexity of the neural networks involved. Nevertheless, our ancestors might have expressed pattern recognition receptors in sensory neurons, similar to those of C. elegans. Perhaps taste or olfactory organelles evolved from these ancestors. It is also possible that the detection of dangerous molecular patterns on macromolecular objects using neural tissue was deselected during evolution, as the immune system developed ever more elaborate defense mechanisms.
Mammals
TLRs, 1, 2, 4 and 6, expressed at the surface of many hematopoetic cells, have been shown to bind distinct bacterial cell wall components and viral products. TLR2 associates predominantly with TLRs 1 and 6, but in some cases also with TLR4 (Fig. 5A). The ligands for TLR1, 2 and 6 includes peptidoglycan (PGN) from Gram positive bacteria (e.g., Staphylococcus aureus and Streptococcus pneumoniae), lipoproteins of spirochetes and mycobacteria, yeast and mycoplasma. The TLR1/2 heterodimer binds triacylated lipopeptides, whereas the TLR2/6 combination is specific for diacylated lipopeptides. The TLR1/2 heterodimer is also implicated in binding products derived from M. leprae and M. tuberculosis. TLR2 can bind human cytomegalovirus (HuCMV) products as well as measles virus. TLR2 has also been reported to bind bacterial lipopolysaccharides (LPS) (for example, from Leptospira interrogans and Porphyromonas gingivalis) as well as mycobacterial lipoarabinomannan (LAM). These reports implicated a second molecule, MD-2, which was found associated with TLR4. Consequently, TLR2 may associate with other TLRs in these complexes. On the other hand, TLR4 can homodimerize to bind the LPS of Gram negative bacteria (E. coli or Neisseria meningitides) in association MD-2 protein, bacterial lipoteichoic acid (LTA), and bacterial heat shock proteins such as Chlamydial HSP60, and Toxoplasma gondii HSP70. Viral products that bind TLR4 have been described in mice infected with respiratory syncytial virus and murine mammary tumor virus, a murine retrovirus.
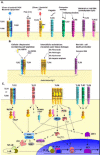
Figure 5
Exogenous TLR ligands. B) Endogenous TLR ligands. C) Intracellular signaling pathways of mammalian TLRs.
TLR3 binds double stranded RNA, a product of some viral infections, and the synthetic ligand, poly Inosine:poly Cytidine. TLR5 binds flagellin, a bacterial locomotory organelle, and TLR7 binds synthetic nucleoside analogs and, with TLR8, binds imidazoquinolines.
TLR9 binds bacterial or viral genomic DNA that contains unmethylated stretches of CpG nucleotides (or synthetic oligonucleotides that contain CpG motifs). No ligand has yet been identified for TLR10, which was identified in humans, but not in mice.
The chromosomal locations for these genes in humans and mice have been elucidated (except for TLR10), and their function in various infectious disease models extensively studied in mice deficient for various TLRs (for a review see ref. 6). Interestingly, TLRs 2 and 4 bind not only bacterial, but also host self molecules, for example the human heat shock proteins (HSP60 and HSP70), and mouse products found in the intercellular space, including fibronectin, hyaluronan, and heparan sulfate. TLR4 was implicated in binding the extra domain A of mouse fibrinogen. Similarly, in low but still notable quantities, mammalian CpG DNA can be found after necrotic cell death, which may explain the capacity for TLR9 to bind double stranded mouse genomic DNA (Fig. 5B).
TLR ligands can instigate intracellular signals similar to those of the IL-1R signaling pathway, but recently differences have begun to emerge. TLR functions have been assessed by studying murine gene deletions in vivo and ex vivo, and in vitro, using tlr null cell lines (eg HEK293) that are then transfected with the relevant tlrs. The TLR signaling pathway includes the activation of a series of adapter molecules: MyD88 and TRAF; protein kinases such as IRAK, the inhibitor of κ-B kinase (IKK), cJun N-terminal kinase (JNK), MAPK, and p38; and the activation of transcription factors such as NF-kappa;B, Rel, and AP-1 (for a review see refs. 7 and 8). Differences from the IL-1R signaling pathway include a novel signaling molecule TICAM (TRIF), the ERK-1 and ERK-2 kinase pathways and possibly Stat-1 proteins (Fig. 5C). However, a complete picture of TLR signal transduction pathways is yet to emerge. Though the expression of TLRs has been found in many somatic tissues in the human body, the research effort has (as for Drosophila) focused on cells of hematopoetic origin and the immune system.
To interpret these findings in relationship to immunity we must review the action of TLR ligands on each cellular effector of the immune system.
Divisions of Immunity
Scientific literature often mentions TLRs as a link between the innate and adaptive wings of the immune system, and discusses their pivotal role in the activation of innate immunity. Perhaps a clearer notion would be that TLRs initiate and modulate central immunity, and play an important part in the activation of autonomous immunity.
The central versus autonomous divisions of immunity will be developed in this chapter to better understand certain aspects of immunity, and especially to explain certain theories of the immune system. In order to avoid confusion by introducing a novel definition to the experienced reader, I will try to alternate the central and autonomous immune designations with the more commonly used terms, innate and adaptive immunity. Although the latter would appear inappropriate for the discussion of some aspects of immunity, it seems that none of the terminologies in present use is perfect. To describe the subtleties of the immune response is difficult, and each division is useful in its own right when addressing particular aspects of immunity (Table 1).
Table 1
Division of immunity according to various definitions.
Specific—Nonspecific (Humoral or Cellular) Immunity
A division of the immune system based on the structural features of effector entities was the first to be used. This humoral versus cellular division, each being either nonspecific or specific has helped to explain the clonal selection of lymphocytes (notably without the inclusion of inflammation, which is described as a nonspecific tissue reaction). Each B-cell receptor (BCR) or T-cell receptor (TCR) probably binds many ligands, but we usually consider them to be specific for a single antigen or peptide/MHC combination. Likewise, the cells of nonspecific immunity might be more “specific” than originally thought, as TLRs can bind specifically to certain molecular motifs. Humoral immunity consists of soluble mediators such as complement (non specific) and antibodies (specific) that can neutralize, opsonize or kill pathogens. Cellular or cell mediated immunity is represented by a number of players. First we have the _nonspecific_ phagocytes, which include the macrophages that "purge" pathogens, then specific cells such as B cells, that become antibody secreting plasma cells and T cells, including the CD4 and CD8 subsets. The CD4 T cells help B and cytotoxic T cells whilst inhibiting other T cells. The CD8 T cell subset kills virus-infected cells. The cellular and humoral divisions cannot satisfactorily describe immunoregulation. This is most evident when we discuss the roles of CD4 T cells and dendritic cells (DC). Both are effectors and, at the same time, regulators of the immune response by direct (cellular) effects as well as by cytokine production (humoral effects). Therefore, it is hard to separate humoral and cellular effects in DC and CD4 T cells.
Innate—Adaptive Immunity
The next division was suggested on the basis of development: innate or inborn immunity vs. acquired, or adaptive immunity. Here the importance was to understand how we carry, in our germline, a memory of past infection. This _memory_ is embodied by the pattern recognition receptors and their ability to bind to conserved patterns of unusual conformers present only on microbes or distantly related organisms. Such receptors would be oligo specific and could activate the cells that we had previously grouped under the umbrella of nonspecific immunity. The use of the other developmentally defined term–adaptive immunity—quickly became popular, as it cleared the confusion regarding specificity of B and T cells. Thus adaptive immunity came to describe a group of cellular and humoral mediators that each organism could acquire during its somatic development. The main components (B and T cell receptors or BCRs and TCRs) were not encoded in a mature form in the germline, but instead were assembled by rearranging variable and constant gene segments. Because these rearrangements occur randomly, BCR and TCR genes would assemble differently in the B and T cells of individuals of the same species. Therefore, acquired immunity equips every mammalian organism with a different set of randomly assembled TCRs and BCRs.
Problems with this division can be envisaged, if we wish to classify NKT cells, which have germline encoded receptors, but also rearrange their TCRs. Similarly, γδT cells rearrange the variable portions of their TCR genes, yet function at the vanguard of defense. NKT and γδT cells are generally thought to belong to the innate wing of immunity, yet both acquire their receptors in a similar fashion to αβ and B cells.
Autonomous—Central Immunity
Despite a detailed understanding of the cells and mediators of the immune system, the mechanisms that precede the induction of B and T cell effectors have remained elusive. Perhaps the problem lies in the fact that the basic function of the immune system is not only to kill pathogens, but also to tolerate commensals. To better illustrate this we could use a division based on cellular function. Autonomous immunity is the swift firstline defense whereas central immunity can be described as a nodal, regulated, or controlled type of immunity. In my opinion, it is the most suitable division to explain the role(s) of TLRs in the immune system.
Autonomous immunity comprises all but dendritic cells from previous definitions, and would include NK cells, NKT cells and T cells with γδ TCRs. The reasons for this segregation are as follows. Components of autonomous immunity are cells or factors that depend on fast local reaction (Fig. 6A). The activation of cells and components occurs at the site where effector actions are first engaged, which is usually taken to be the locale of tissue damage, inflammation, or antigen challenge (i.e., contact with the micro-organism). Thus, the cellular components require very little communication with cells that are distant from the site of pathogen intrusion.

Figure 6
A) Autonomous and central immunity. B) The central immunity: afferent and efferent loops.
In contrast, central immunity has afferent and efferent loops of action. As illustrated in Figure 6B, the afferent loop starts with the activation of dendritic cells in damaged tissues by micro-organisms. DCs, having sampled the antigens from their surroundings, migrate to the lymph node, which is the center of the immune response (for a review see ref. 9). There, DCs meet naive CD4 T cells, which they then stimulate. After stimulation, T cells proliferate to become effector (helper) T cells that migrate to B-cell areas of the node. Upon T-B interaction (provided that the B cell has met the same antigen as the T cell), an activatory signal is transferred and B cells go on to proliferate and develop into antibody secreting cells (called plasma cells). Alternatively, CD4 T helper cells could recruit cytotoxic CD8 T cell precursors and license them for action in the periphery.
B cells, in order to receive help from T cells, need to recognize antigen or hapten. There are numerous ways by which the stimulating antigen can be delivered to B cells. Antigen may be derived from the blood stream, usually attached to other proteins, for example an antibody/ antigen complex, or may be carried by phagocytic cells, in which case the ingested antigens could be exposed after the death of their carrier. Alternatively, circulating B cells may collect antigens along their recirculation routes to and from the lymph nodes (or spleen). B cell activation by T-dependent antigens leads to the formation of germinal centers in the lymph node cortex. B cells then proliferate and develop into effectors. During peripheral development B cells undergo a selection process by which they can increase the affinity of their BCRs. This process involves hypermutation of BCR genes and the selection of high affinity B cells on follicular dendritic cells (a stromal cell unrelated to DCs). This results in the appearance of effector germinal center (gcB) B cells with a higher affinity for antigen. These effectors can go on to develop into plasma cells.
The efferent loop of action begins with the migration of effector T and B (plasma) cells from the center to periphery, i.e., back to the tissue from whence the DCs first started their trek. Effector cytotoxic CD8 T cells (αβCR+ve) will go on to kill virally infected cells, whereas CD4 αβ cells can activate macrophages via cytokines (interferon γ) and control, or regulate (again with cytokines), other cells of the autonomous system. Plasma cells secrete antibodies and eventually migrate and lodge in a strategically superior locale from where they can ensure a long lasting supply of circulating antibodies (such as the bone marrow or spleen).
Based on these premises, immunity can be subdivided into central and autonomous. Central immunity facilitates our understanding of TLRs as pattern recognition receptors, and emphasizes their additional functions in DCs and memory B cells.
Let us now analyze the distribution of TLRs (Tables 2, 3) and the evidence for their roles in the immune system (Table 3). Later, I shall discuss these (predicted) roles in the context of various immune system models.
Table 2
TLR ligand repertoire of human cells.
Table 3
Cellular distribution and function of human TLRs.
Expression and Function of TLRs in Cells of Autonomous Immunity
Two mammalian species are considered in this section; human and mouse. TLR expression can be regulated by various substances or cellular interactions, which differ across animal species, an indication that their sensitizing or desensitizing immune responses might also differ.
Neutrophils (and Granulocytes)
Neutrophils express TLRs 1, 2, 4, 6, and 8. The lifespan of neutrophils is short, but can be prolonged by activation via TLRs 2 and 4. NF-κB, which is implicated in signaling via TLRs 2 and 4, is a known survival factor and was first implicated in the cell death mechanism delivered by TNF and related signal pathways. Only later, with the use of IKK knockout mice, did it become apparent that the survival effects of NF-kB are counter balanced by caspase driven programmed cell death.
Upon binding of TLR2 or TLR4 ligands, neutrophils upregulate the expression of chemokines, downregulate some chemokine receptors, and change their expression of adhesion molecules (integrins and selectins) and respiratory burst mediators.10, 11All these factors drive the inflammatory response in local tissue.
Cells that express small amounts of the indicated TLRs are in brackets. TLRs whose expression is regulated via another TLR ligand or signal are underlined. G = gram, Gc = germinal center.
Mast Cells
LPS and PGN (TLR2 and 4 ligands) differentially activate TNFa and IL-5, IL10, and IL13 in human mast cells. However, the release of TNFa by mast cells requires priming with IL-4 and the presence of serum components such as soluble CD14. Interestingly, regarding the possible connection with allergy and atopic diseases, PGN, but not LPS, can induce the release of histamine by mast cells.12
Monocytes
Monocytes are precursors of myeloid derived DCs and macrophages. Human monocytes express TLRs 1, 2, 4, 5, 6 and 813 and increase their expression of TLR4 when treated by IFNγ,14 whilst IL-10 has the opposite effect.15
Human monocytes can traverse the endothelium and, as for cells of the immune system, become either macrophages or dendritic cells, depending on the cytokine cocktail employed in the culture medium. GM-CSF and IL-4 induce blood monocytes to become precursors of DCs (preDCs) whilst TNFa results in CD1a+ DCs and GM-CSF and IL-15 skew development towards langerin+ DCs (similar to Langerhans cells in skin). On the other hand, the addition of fibroblasts favors the generation of CD14+ macrophages, as the fibroblasts secrete IL-6, which induce the M-CSF receptors that promote macrophage development.
Monocytes can thus be seen as precursors for both the macrophage and DC lineage, or alternatively, as tissue specific precursor cells that appear to be similar but bear different surface TLRs that are strongly expressed are in bold typeface, whereas weakly expressed TLRs are in brackets. Underlined numbers denote regulation (induction) via an exogenous factor. The brackets in the "Roles" column denote a requirement for the activation or maturation of the cells indicated. Monocytes appear twice, as some authors report them as preDC1 cells. markers. Depending on the culture conditions (or that of the tissue), precursors of one cell type might predominate over the other. Thus TLRs may contribute to monocyte differentiation by transducing differentiation specific stimuli.
Macrophages
Macrophages are generated in the tissues from monocytes, but unlike DCs, do not migrate. Human macrophages are efficient APCs and express a variety of TLRs (see Table 3). The available information about TLRs expressed on mouse macrophages is largely concordant with the human data.
Macrophages can interpret the molecular patterns expressed by intruding microorganisms, and are activated by bacterial products. Their immediate action, at the site of tissue damage, is controlled by various factors that include both the autonomous and the effector arms of central immunity. There is a direct activation of autonomous immunity via TLRs, and an indirect one, via IFNγ. The most potent activator of macrophages is IFNγ, a product of activated NK cells and effector CD4 T helper-1 cells. Activated macrophages perform a series of important functions. These include the production of extracellular oxidative radicals that can damage pathogens (and normal tissue), the production of the vasodilator, nitric oxide (NO), the secretion of proinflammatory cytokines that increase body temperature, mobilize fatty acids, and, in high enough quantities, cause septic shock. Macrophages also act to secrete chemokines that attract effector T, B, plasma and other cells to the site of inflammation, upregulate MHC class I, class II, Fc and complement receptors and kill intracellular bacteria.
Using microchip technology, a unique gene expression profile was found to be induced by Gram negative bacteria (via TLR4) in human macrophages that included the upregulation of IL-12 p70 and type I IFN. This was in stark contrast to the profile induced by Gram positive bacteria acting via TLR2.
Exposing macrophages to LPS (via TLR4) induces a hyporesponsive state to a second challenge with LPS, which is called LPS tolerance. This response is also induced by pre-exposure to TLR2 ligands. LPS signaling involves at least two pathways; a MyD88 dependent cascade that is important for the secretion of proinflammatory cytokines and a MyD88 independent pathway (via TICAM; TRIF) that controls the expression of IFNβinducible genes.16
The direct activation of macrophages via TLRs is exemplified in experiments with TLR gene knockout mice. Macrophages from mice deficient in TLR3 (that binds dsRNA) exhibited specific defects in the secretion of IL-6 and IL-12, activation of NF-κB, and the induction of type I IFN in response to poly(I:C). All of which indicate the role of macrophages in the antiviral response. Experiments with other TLR deficient macrophage cell lines have shown that macrophages also play an important role in antibacterial, antiparasitic and antifungal immunity (for a review see reference 6). Immunization preparations like bacillus Calmette-Guerin (BCG) can activate murine macrophages and induce the secretion of TNFa. This effect is mediated by both TLRs 2 and 4, as double deficient murine macrophages fail to respond to BCG by secreting TNFa, whereas single TLR2 or TLR4 deletion mutants retain some TNF production.
The expression of certain TLRs can be regulated by receptor ligand interactions that involve other TLR family members. For example, LPS (probably acting via TLR4) can induce mouse macrophages to upregulate TLR9.17 However, there is a dichotomy in antigen presentation between macrophages and DCs that is initiated by TLR ligand binding. TLR3 and TLR9 ligands are inefficient in inducing crosspresentation in macrophages and macrophages require a thousand fold higher concentration of antigen to generate the same effects as DCs.18 This would suggest that macrophages are poor stimulators of antigen specific precursor CD8 T lymphocytes (pCTL) when pulsed by TLR3 or TLR9 ligands plus antigen in somatic tissue.
NK Cells
Human NK cells express mRNA of the ubiquitous TLR1 and very low amounts of TLR7 and TLR9.19 However, TLR9 ligands can activate NK cells20 and it is possible that pattern recognition by TLRs can induce immediate action in these cells. Activated NK cells can kill somatic cells that lack particular MHC molecules as a result of their viral infection or malign transformation (as detected by killing inhibitory receptors, KIRs). NK cells can also kill cells that express unusual MHC molecules (recognized via killing activating receptors, KARs) and produce IFNγ to activate macrophages.
Expression and Function of TLRs in Cells of Central Immunity
Dendritic Cells (DCs)
Various populations and maturation stages of DCs have been described that depend on their tissue distribution and species of origin. Most DC subtypes share capabilities such as antigen presentation, the costimulation of T cells, migration to and from somatic tissues and cytokine and chemokine secretion. However, some express different sets of TLRs (Tables 2, 3). As DCs can orchestrate immune responses, the function of their TLRs is of paramount interest.
Human DCs
In human blood we can find monocyte like precursor cells (preDC or MoDCs) that develop, after 7 days in culture with GM-CSF and IL-4, into immature DCs. These can develop further, with the addition of TNFa, into mature type 1 DCs. These cells have been used extensively in cancer immunotherapy. Blood also contains immature DCs (iDCs) that are CD11c+HLA+ that can be induced to mature into mDCs. Mature DCs display a myeloid phenotype of markers, have upregulated costimulatory markers such as CD80 and CD86 and produce cytokines such as IL-12p70, IL-18 and IL-23 that help Th1 development. This DC pool, called DC1, still contains clearly divisible subpopulations and is probably derived from precursor cell preDCs. Another DC lineage found in blood is the plasmacytoid DC (pDC), with the phenotype CD11c-IL3R+CD45RA+ (for review see ref. 21). The plasmacytoid DC lineage resembles that of B plasma cells, without B cell markers but with the CD4 T cell marker. Plasmacytoid DCs secrete IFNa, and can mature into the DC2 subset, which shows a Th2 skewed cytokine secretion profile (IL-4, IL-10).
Human bone marrow cells can also yield various types of dendritic cells in vitro, if incubated with various cytokines. The mature mDC1 and mDC2 subsets probably represent the terminal stages of preDC development and it is thought that these two lineages might have originated from either lymphoid or myeloid precursors. However, it is possible that yet more DC types exist.
DCs express the widest known repertoire of TLRs (Table 3). PreDC, or monocyte like cells (MoDCs) express TLRs 1-8 excepting TLRs 3 and 7, which are expressed on mature DCs. Immature iDCs have a decreased expression of all TLRs, except TLR3. IDCs also seem to lack the expression of TLR4, though TLR4 may be present at a concentration that escapes detection. Interestingly, DCs are equipped with additional pattern recognition receptors that include the mannose receptor and DEC205, a C type lectin, both of which have roles in endocytosis or phagocytosis.
An important albeit transient capacity of immature DCs is endocytosis. It provides the means by which a microbial antigen can be taken up, processed, loaded onto MHC molecules and presented to T cells. This constitutes the first act in T cell activation (and is generally the start of the immune response). The processing and presentation of antigen will be described later. Mature DCs, on the other hand, loose this phagocytic capacity, but instead acquire an increased antigen presentation competence due to their enhanced expression of MHC II molecules. Consequently, DCs can trap their engulfed antigenic loads until they can present them to T cells in the lymph node. This makes them a unique class of cells that can capture a representative antigenic profile of each tissue that they pass.
Mouse DCs
Murine DCs share many markers with their human counterparts (for a review see reference 22). Certain populations have a common marker like CD11c, which is, in association with CD18, an integrin molecule. Murine DCs can also express markers common to the helper and cytotoxic T cells, CD4 and CD8a. The latter is not found on their human counterparts (thought to be pDCs). Based on these markers there could be at least 6 different murine DC populations. DCs can be further subdivided on the basis of their expression of the CD45RA and CD205 markers, a protein phosphatase and C type lectin respectively. Both, the lymphoid and myeloid lineage of DC progenitors have the potential to give rise to all known populations, however at different ratios. In addition, subpopulations differ in their production of cytokines and in their expression of additional cell surface markers, which could relate to their tissue of origin (spleen or bone marrow). The function of these subpopulations is not yet clear.
Precursors of murine homologues of the human preDC1 and preDC2 (pDC) subtypes have also been identified in blood. A CD11c-/+CD11b-CD45RA+ mouse population closely resembles human plasmacytoid cells (pDCs or preDC2) on the basis of their morphology and function, as CpG motifs can stimulate these cells to produce IFNα and develop into CD8+ DCs. A second population of preDCs has the surface phenotype CD11c+CD11b+CD45RA- and closely resembles the precursors of human preDC1 cells that go on to develop into CD8- DCs after stimulation with TNFα. In the mouse, the precursors of DC2 cells (preDC2) strongly express TLRs 7 and 9. Virus or challenge via TLR9 can trigger these cells to develop into DC2 cells that secrete type I interferon (IFNα/β. These DCs generate IL-4, which has a strong influence on the preDC1 population by enhancing the production of IL-12p70. The preDC1 population has a different TLR profile, and expresses almost all of the TLRs, except 7 and 9.
Spleen or bone marrow derived precursors of the murine preDC1 subset (myeloid precursor) and preDC2 subset (plasmacytoid precursor) can induce the development of both Th1 and Th2 effector cells depending on antigen dose. At high doses Th1 cell development is favored whilst lower antigen doses induce the development of Th2 cells.23
DC Maturation
When considering the developmental preDC/DC pathway we should clarify the term maturation when used in the context of DCs. Maturation is a series of changes that can be elicited by LPS, viral or bacterial infection, cytokines, bacterial DNA, extracellular matrix in inflamed tissue or the ligation of molecules on immature DCs (iDCs) such as CD40. Various TLR ligands can induce bone marrow derived DCs to mature. The concentration of ligand that induces maturity (as measured by the induction of costimulatory molecules CD80, CD86) varies greatly. Remarkably, peptidoglycan and flagellin require the highest concentrations, whereas LPS can induce the maturation of DCs at a thousand fold lower concentration than most other ligands. The maturation of iDCs generally leads to downregulation of most TLRs.
Thus, triggering TLRs can induce the maturation of iDCs, in terms of their secretion of cytokines (IL-6, TNFa) and upregulation of costimulatory molecules (Fig. 7A.). The intracellular pathways that lead to these events have been extensively studied (Fig. 7A) though are incomplete given that subpopulations of iDCs might differ in their developmental requirements, stimulation, and further maturation. These factors could influence the biological functions of DCs and specific immunocytes. Namely, in addition to costimulatory molecules like CD80 and CD86, there are other B7 family members whose expression on APCs, like DCs, may differentially influence the fate of stimulated T cells. The relationship of these molecules, for example ICOS and the PD-1 receptor and their ligands, ICOS-L and PDL1/PDL2, with TLR signaling is poorly understood.
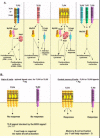
Figure 7A
A) Consequences of TLR signal transduction in DCs. B) Consequences of TLR signaling in B cells. C) Dissociation of TLR2 signaling in DCs. D) Consequences of TLR4 signal transduction. E) Consequences of TLR4/9 signal transduction.
Maturation of DCs is probably one of the main reasons for successful vaccination against infectious diseases. For example, BCG consists of a purified noninfectious material that includes a cell wall derived peptidoglycan, arabinogalactan, and mycolic acids, all of which can induce the maturation of human preDCs. This is probably mediated via TLR2 and TLR4, as murine macrophages that lack these molecules fail to secrete TNFa in response to BCG.24 Note that pathogens have also evolved counter stratagems to avoid initiating an immune response. One stratagem adopted by some bacteria is to evade DC pattern recognition receptors by "hiding" intracellularly.
Another potential function of DC TLRs is to polarize T cells towards either a Th1 or Th2 response. The migration of preDC1 cells from tissues into lymph nodes can be induced by prostaglandin PGE2 which also inhibits the Th1 polarizing cytokines in DCs. IL-4 can counteract this effect in iDCs. It would be interesting to test whether TLR signaling can influence prostaglandin secretion in somatic tissues.
B Cells
Naive human B (CD5-) cells express significant levels of TLR1 and TLRs 6-10. Activation of B cells in vivo (in the germinal center; Gc-B) results in an induction of TLRs 7, 9 and 10 but not the other TLRs.19,25 Thus, central memory B cells can upregulate certain TLRs (Tables 2, 3).
TLR ligands like unmethylated CpG DNA and LPS can also upregulate the CD80 costimulatory protein in B cells. According to some researchers, TLRs are potent, T cell-independent polyclonal activators of murine and human B cells (in terms of proliferation and differentiation). In contrast, others hold that BCR engagement is necessary for TLR mediated B cell activation.26 Simultaneous crosslinking of the BCR by BCR specific antibody enhances the TLR9 mediated intracellular response of B cells to CpG motifs. Mammalian DNA lacks stimulatory CpG motifs, and many of the remaining motifs are normally methylated. Consequently mammalian DNAs are weak stimulators. Very low affinity DNA binding or IgG binding self reactive B cells usually fall beneath the threshold required to induce anergy, deletion or editing. However, immune complexes in normal individuals (for example, anti DNA antibody bound to self DNA) may be stimulatory to naive B cells because, hypothetically, synergy between the BCR and TLR9 could trigger them. Ordinarily, this does not happen (at least not in naive B cells), because BCR signaling mediators inhibit the TLR9 signaling pathway, a situation that also applies to higher affinity DNA specific naive B cells. Analogous to the TLR9 scenario, TLR4 (or LPS) signaling could also cause the stimulation of potentially autoreactive B cells. However, the mechanism that operates in naive B cells to inhibit TLR9 signaling could also inhibit TLR4 signals and suppress autoantibody generation under normal circumstances.26
In 1974, Coutinho observed that B cell activation can be triggered by LPS.27 This prompted the idea that B cells do not necessarily need to signal via the BCR or with T cell help. The hypothesis that a single nonspecific signal could stimulate B cells was subsequently proven wrong as BCR signaling was shown to be necessary during B cell activation.28 However novel data has shown the increase of, in particular, TLRs 7, 9 and 10 in B memory cells, which leads us to an interesting possibility. Namely, we could envisage that central memory B cells can act similarly to, but not exactly as proposed by Coutinho. Naive B cells do not normally respond to CpG ligands via TLR9, because they uncouple the intracellular pathways that could lead to the development of autoreactive antibodies.26 However, in memory B cells, a different situation could arise. Renewed pathogen intrusion can cause local tissue damage, which would release double stranded genomic DNA. Memory B cells could bind genomic DNA using TLR9 and pathogenic antigen via their BCRs and become activated, but without T cell help. So, memory B cells would respond swiftly to renewed infection in the presence of recognizable antigen. Thus, TLR9 signaling might increase the speed yet retain the specificity of in vivo B cell responses (Fig. 7B).
Memory B cells are usually formed through a tight T cell controlled response, which prevents the formation of a B cell memory with a high affinity autoreactive antibody repertoire. However, if these controls fail or if B cell tolerance malfunctions, then the normal checks and controls that are exerted over memory could collapse and the potential for autoimmune pathology arise. For example, if a naive B cell clone reactive against self antigen became a memory cell, it could be activated in a T independent fashion via TLRs triggered by the products of local tissue damage. This could lead to the production of autoreactive antibodies.
An experimental autoimmune disease model in which dual engagement involving TLR9 and the BCR could induce naive B cell activation and proliferation has suggested another role for TLR9. B cells were shown to bind antibody-chromatin complexes in the serum of MRLlpr+/+ AM14 transgenic mice. Naive AM14 transgenic B cells that are specific for the Fc portion of an IgG2a antibody are normally inactive in AM14 transgenic mice. However, in animals crossed with an autoimmune prone genetic background like MRL-lpr, they become activated, and develop into cells that produce Fc specific antibodies (rheumatoid factors). Their activation was shown to be dependent on the engagement of both TLR9 and transgenic BCR. Firstly, TLR9 binds chromatin, then the transgenic BCR binds the Fc portion of the immune complex formed by chromatin and an antibody specific for chromatin. This dual binding (or crosslinking) triggers AM14 B cell activation and proliferation,29 leading to rheumatoid factor production and other autoimmune symptoms. This report seems to contradict the finding that naive B cells inhibit TLR9 signaling by contemporaneous BCR signaling.26 An explanation for this could be that the genetic background of the mice uncouples the “brake” in TLR9 signaling as the AM14 transgenic mice (with non MRL-lpr genetic background) do not normally generate rheumatoid factors. However, there is as yet, no evidence regarding the nature of the molecule(s) involved in these processes.
In mice, TLRs can affect isotype switching in B cells. The TLR9 ligand CpG (but not the TLR4 ligand, LPS) upregulates the intracellular mediator T-bet (Th1 gene regulator) in normal B cells, an effect that was abrogated in mice deficient in TLR9 and MyD88. IL-12 acts synergistically with the TLR9 ligand. In fact, upregulation of T-bet mimics, in part, the Th1 type antibody response, because of the inhibition of IgG1 and IgE switching.30 IL-4 and CD40 specific antibodies can induce these isotypes in purified B cells from normal mice. A switch to IgE is usually found when a Th2 type response is favored and is epitomized by allergy. Perhaps, TLR9 has immunomodulatory effects in suppressing some allergic responses. In conclusion, the function of TLRs in (CD5 negative) B cells might be to speed up secondary immune response to antigen, and affect isotype switching.
The Roles of TLRs in APC—T Cell Interactions
TLR function in antigen presenting cells is implicated in antigen presentation to T cells, the expression of costimulatory molecules and the capability of APCs to polarize T cell responses.
TLRs and Antigen Presentation
Professional APCs like DCs, macrophages and B cells express basal levels of the MHC class I and II proteins at their cell surface. Upon maturation or activation, APCs upregulate the expression of both classes of MHC molecule. The consequence of this is an enhanced presentation of antigens to T cells. There are two basic pathways by which an APC can process and present antigen: endogenous and exogenous.
Endogenous Pathways of Ag Presentation
The classical endogenous pathway involves MHC class I molecules. These proteins are found on all nucleated cells and usually present peptides derived from intracellular antigens. Proteins synthesized within the cell can be also degraded in the cytoplasm to generate peptide fragments that are transported across the ER membrane and loaded onto newly synthesized chains of the class I molecules in the ER. The MHC class I - peptide complex is then transported to the cell surface via the golgi apparatus and secretory pathway (Fig. 8A). Rarely, endogenously synthesized proteins are also presented on MHC class II molecules.
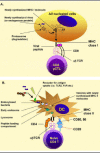
Figure 8A
A) MHC Class I (endogenous) pathway of Ag presentation. B) MHC Class II (exogenous) pathway of Ag presentation. C) Crosspresentation (crosspriming).
The Exogenous Pathway of Ag Presentation
There are two types of exogenous antigen presentation pathway. Peptides from extracellular antigens can be presented on MHC class II molecules via the classical pathway (Fig. 8B). However, peptides derived from extracellular antigens can also be presented by MHC class I molecules in a process called crosspresentation or crosspriming, in reference to the activated T cell readout employed. Crosspresentation represents the nonclassical pathway (Fig. 8C).
The MHC Class II Antigen Presentation Pathway
MHC class II molecules are normally expressed on professional APCs and, under nonphysiologic or pathological conditions, can also be expressed on other tissues.
Antigen is internalized by endocytosis into intracellular vesicles, which gradually acidify, degrading their antigen cargos. MHC class II molecules are de novo synthesized in the endoplasmic reticulum (ER) and transported from the ER via the golgi compartments to endosomal vesicles. These fuse with the vesicles in which antigen degradation occurs to form a specialist compartment in which peptide loading can take place. Subsequently, the peptide-MHC class II complexes are transported to the cell surface for presentation to T cells.
Crosspresentation: MHC Class I Antigen Presentation Pathway
Crosspriming was first described as the stimulation of class I restricted precursor cytotoxic T lymphocytes (pCTL; CD8 T cell) by exogenous, cell associated antigens. Since then, soluble forms of antigen have also been shown to induce crosspriming. Therefore, a commonly used definition for crosspriming is the capacity of exogenous antigens to stimulate class I restricted CTL responses (Fig. 8C). Crosspresentation is the presentation of peptides by class I molecules to T cells, whereas crosspriming involves, additionally, the costimulation of naive CD8 T cells. The consequence of these definitions is that peptide-MHC specific effector CTLs can kill cells by crosspresenting antigenic peptides, even if they lack costimulatory molecules. This is an important distinction: pCTLs require crosspriming for activation and proliferation, whereas effector CTLs require only crosspresentation in order to unleash (activate) their killing machinery.
Protein antigens or their peptide fragments can be taken up by the antigen presenting cell in two ways, endocytosis or direct cell-cell contact (gap junctions). Endocytosis allows APCs to engulf antigens in cellular debris, apoptotic bodies and live or dead micro-organisms. However, peptides derived from neighboring cells in a tissue can, for example, enter a dendritic cell via gap junctions. If the cell died by apoptosis (because of infection or in the course of tumor growth), the apoptotic bodies would contain pathogen specific or tumor derived antigens, and perhaps their peptides. Crosspresentation of these could induce CTL responses against pathogens or tumor cells. Particles taken up by phagocytosis have a tendency to induce crosspriming, but the molecular mechanism is still unclear. Many bacteria live in vacuoles inside phagocytic cells as a part of their life cycle and it has been shown that the antigens derived from these live intracellular bacteria can be crosspresented on MHC class I antigens to induce CTL responses that kill the infected cells. Additionally, antigens in cell debris can, after phagocytosis, be crosspresented on class I molecules. Immature DCs have several receptors for apoptotic bodies that can contribute to crosspresentation, employing the cytoplasmic pathway. These receptors include mannose binding lectin, scavenger receptor and iC3b binding molecules like CD11b, CD11c, CD21 and CD35.
TLR Ligand Linked Antigen Presentation in Immature DCs
Human immature DCs derived from bone marrow, pulsed with antagonistic TLR2 specific mAbs containing κ light chains, could stimulate a Cκ specific CD4+ T cell clone in the absence of maturation effects on iDCs (Fig. 7C). An isotype/light-chain matched control antibody produced a two to three orders of magnitude lower response, indicating enhanced antigen presentation via TLR2. Stimulation was TLR2 specific, as antibodies against other surface molecules such as CD62 and CXCR1 were not stimulatory. Inhibitors of lysosomal degradation, processing and MHC class II presentation like chloroquine, leupeptin or brefeldin A almost completely abolished T cell stimulation. Furthermore, an anti-TLR2 mAb was directly shown to reside in endosomal vesicles in pulsed iDCs.31 Thus, antigen linked to the TLR2 ligand can be endocytosed after binding TLR2, processed via the classical (exogenous) pathway of antigen presentation, and can enhance the stimulation of T cells. This same route could be exploited to generate more efficacious vaccines.
Unlinked TLR Ligand Antigen Crosspresentation in Mature DCs
TLR9 ligand (oligonucleotides containing CpG motifs; CpG) or TLR3 ligand (poly I:C) were mixed with ovalbumin, incubated with mouse bone marrow derived iDCs, then tested for their ability to crossprime ovalbumin/MHC specific precursors of cytotoxic CD8 cells (pCTL; naive or resting CD8 T cells). The antigen was not linked with the TLR ligand. Ovalbumin was endocytosed, processed, its peptides loaded onto both MHC class I and class II molecules and subsequently presented to T cells. Such pulsed mDCs were able to stimulate syngeneic ovalbumin peptide/MHC class I specific pCTL and this stimulation was MyD88 dependent, showing the importance of the TLR signals in crosspresentation. As a control, these DCs were also able to stimulate autologous CD4 T cells (bearing ovalbumin peptide/MHC class II specific TCR), showing their ability to engage in classical, exogenous antigen presentation using MHC class II molecules. A CD8+ subset of mouse DCs was shown to be able to crosspresent and present ovalbumin peptides.18
Interestingly, if DCs were first pulsed with TLR9 ligand, washed, then incubated with ovalbumin an unexpected result occurred. TLR9 binding could sensitize DCs to take up ovalbumin for crosspresentation for several hours after they had been pulsed with CpG. In contrast, TLR9 ligand pulsed, then ovalbumin pulsed DCs, were unable to present ovalbumin to autologous CD4 T cells. It seems, therefore, that TLR9 triggering engages a crosspresentation machinery (Fig. 8C), whilst at the same time uncoupling the classical, exogenous MHC class II pathway of antigen presentation in mDCs (Fig. 8B). Macrophages, on the other hand, do not possess the ability to crosspresent ovalbumin after being pulsed with CpG.18
In these experiments, antigen was not endocytosed by TLRs. DCs have plenty of molecules that facilitate antigen capture, for example, Fcγ receptors. There is however, a dichotomy between the crosspresentation pathway of FcγR and those of TLRs 3 and 9. All three use cytoplasmic pathways, but TLR mediated crosspresentation does not require acidification in an endosomal compartment. It is possible therefore that TLRs trigger an unknown mechanism that operates early in endosomes, shuttling bound antigen into the cytoplasm of DCs for further processing and crosspresentation on MHC class I molecules.
Presentation of Self Antigens that Are TLR Ligands in DCs
In mice, TLR2 and TLR4 binds exposed ends of fibronectin,32 hyaluronan,33 and heparan sulfate,34 which are present in the extracellular matrix. TLR4 has been implicated in binding mouse extravascular fibrin(ogen),35 which is found in tissues only if vascular permeability is increased, as in inflammation. Interestingly, human DCs can mature in the presence of chondroitin sulphate and hyaluronic acid (hyaluronan) when cultured together with GM-CSF36, suggesting that human TLRs bind inflamed tissue extracellular matrix. Furthermore, human HSP60 and 70 can also act as ligands for TLRs 2 and 4.37-39 Human genomic DNA may also be modified in inflamed tissues, such that CpG islands can bind TLR9 (Fig. 5B). Even without modifications, mammalian genomic DNA is weakly stimulatory.40 All these TLRs were shown (using different antigen response models) to be able to present antigens to T cells. Thus it is possible that self-antigens can be captured and processed by DCs expressing TLRs, and used to stimulate autoreactive T cells.
TLRs and T Cell Costimulation
The antigenic peptide/MHC complex binding to TCRs of naive or memory T cells is a prerequisite for the initiation of the immune response. There is ample evidence that TLRs can indirectly provide costimulatory signals to T cells via the activation or maturation of APCs.
TLR signals in immature DCs can, under certain conditions, provide only an antigen presentation facility to T cells. In other words, antigen presentation can be dissociated from costimulation (upregulation of CD80 and CD86) in immature DCs. Evidence for this comes from experiments in which authors used TLR2 as a point of antigen entry and showed that an antagonistic human TLR2 specific mAb (TL2.1) containing κ-light chains could be taken up by iDCs, processed into peptides, and subsequently loaded onto MHC class II molecules to stimulate a Cκ specific CD4+ T cell clone.31 The TL2.1 mAb was antagonistic, because it had no stimulatory effect on macrophages in comparison to agonist mAb (Fig. 7C).41 The authors showed that TL2.1 could bind to TLR2 molecules on iDCs, but neither upregulation of CD80 and CD86 nor secretion of TNF was observed. There was nothing wrong with the maturation capability of TL2.1 pulsed iDCs, because iDCs could be induced to mature and to express costimulatory molecules by the addition of TLR ligands like Pam3Cys (TLR2) or LPS (TLR4). Then, they showed that TL2.1 was endocytosed, processed into peptides, loaded onto class II molecules, and finally expressed on the cell surface for presentation, as determined by proliferation and interferon γ release of a responder T cell clone. The T cell clone did not require costimulatory molecules for these actions, because it was an effector cell. The result indicated that iDCs could dissociate antigen presentation from the costimulation of T cells. Thus, antigen internalization and processing can be initiated by binding to TLR2 for presentation on MHC class II molecules. Further, it does not necessarily hold that every time a ligand binds to TLR2, an iDC will be activated to provide both a stimulus and costimulus to naive T cells (Fig. 7C). Though the experiment with naive T cells is needed to confirm this suggestion, the result by Schjetne et al. is consistent with this hypothesis. It would be interesting to know whether the actions of other human TLRs on iDCs can be similarly dissociated. If so, perhaps the missing signal (needed for full activation of iDCs) could be called co-initiation or co-activation. Thus, TLRs can provide two kinds of signals, one that fully activates DCs, propelling them towards a more mature phenotype, and a second that only partially activate DCs, rendering them devoid of costimulatory molecules, but capable of presenting antigen.
Is there other evidence that TLRs have two modes of action? The answer is yes and comes from experiments using crosslinked TLR4 to test signaling under suboptimal ligand concentration. Crosslinking (not just binding) of TLR4 by LPS is necessary for downstream signaling from macrophages.42 Ligands such as HSPs act on TLR4 by enhancing the stimulatory effects of otherwise substimulatory concentrations of LPS (Fig. 7D).43 This could, perhaps, be understood as a dissociation of TLR4 signaling, similar to that seen with TLR2. Furthermore, evidence that intracellular TLR signaling cascades can be synergistically triggered by ligands for TLRs 2 and 9, or 4 and 9, under suboptimal conditions (Fig. 7E),44 supports the idea for twolevel TLR signaling.
TLRs and Polarization of the T Cell Response
In human DCs, a TLR4 agonist specifically promoted the production of IL-12p70, which is associated with the Th1 responses. In contrast, TLR2 stimulation resulted in the secretion of the IL-12 inhibitory p402 homodimer, producing an environment that would favor Th2 development.45 CpG DNA, as a TLR9 ligand, also has immunomodulatory effects, such as induction of the Th2 type cytokines (IL-4 and IL-10) in DCs.
Induction of costimulatory molecules on APCs is probably not the only mechanism that can control naive T cell activation. T cell responses can also be regulated by CD4+CD25+ regulatory T cells (Tr cells) (for a review see ref. 46), and TLRs might play a role in their generation. These cells are thought to be important for the maintenance of peripheral T cell tolerance, as their depletion leads to organ specific autoimmune diseases. Though the molecular mechanism of Tr cell mediated suppression is unknown, it seems to be cell contact dependent. The inhibition of IL-2 transcription in responder T cells is one of the results of such suppression.
The culture medium of DCs stimulated by TLR4 and TLR9 ligands (LPS and CpG) has been shown to inhibit the generation of regulatory T cells. This has been shown to be dependent on IL-6.47 It seems likely that the generation of regulatory T cells depends on the lack of certain TLR signals, however the precise mechanism remains unclear.
Various kinds of TLR signaling pathways may influence the expression of cytokines in APCs, which may, in turn, regulate the formation of various types of T helper (Th1, Th2 and Th3) or regulatory T cells.
TLRs and Theories about the Function of the Immune System
A critical question in immunology concerns the initiation of the immune response. How do the current theories of immune system function tackle the role of TLRs? Further, how useful are the current immunological theorems and how can we put them to the test?
Ideally, theories about immunity should find fundamental and, hopefully simple rules, to explain and clarify the biological, cellular and molecular functions of immunity. The theory then becomes a “map” upon which we can plan our research. It is obvious that such a “map” will only be complete when we have obtained an all-encompassing knowledge of all the structures and molecules involved, and their interactions. Obviously, this will take some time! In the meantime we can take a short cut by making some educated guesses.
At the same time, it stands to reason that an oversimplistic theory will require multiple additional explanations or rules in order to accommodate new or controversial results. This has the effect of generating a rather complicated and unwieldy theorem of limited use in explaining, or predicting, the workings of the immune system.
Self-Nonself Discrimination and Associated Antigen Recognition
The “Self-nonself ” discrimination (S-NS) model has evolved considerably since it was first conceived by Bretscher and Cohn in 1970.48 Its basic tenet is that cells of the immune system can recognize exogenous molecules by clonally distributed receptors on immunocytes. These exogenous or “nonself ” molecules are mostly derived from pathogenic microorganisms. That TLR ligands are polyclonal activators of the immune system neither fits the original theory, nor its latest incarnation as the Associated Antigen Recognition (AAR) model (Langman and Cohn).49 This model describes the activation of the specific immune system solely by nonself antigen. This occurs by the generation of two major signals that can activate naive B or T cells. The first signal (signal-1) is derived from the antigen receptor (BCR or TCR), and causes programmed cell death (clonal deletion), which results in tolerance to a particular antigen. The second signal (signal-2) rescues the cell from death. For B cells, this signal is T cell help. For T cells, this is also T cell help, but from effector T cells. How the latter are formed is not known, but the authors predict that each naive T cell would, by default, differentiate after some period of time into an effector. Thus, in short, only novel antigen, never before encountered by the immune system can initiate specific immune responses. The model suggests that tolerance to self-antigens occurs during embryonal development or very early in the life of an individual and predicts that any novel antigen detected by the adult immune system would be seen as nonself.
The induction of antigens in the lactating breast is an example for which this model lacks a simple explanation. Why do these proteins not cause autoimmunity? The concept of regulatory or suppressor T cells is also incompatible with this model as the AAR model presupposes that simultaneous antigen driven inhibition would counteract the response.
Cytokine Burst
This model suggests that the control of the immune system rests with cytokines.50 An increase in the local concentration of certain cytokines would stimulate T cells to initiate the immune response. This hypothesis could be adapted to suggest that pattern recognition leads to the secretion of cytokine “soups” that upregulate the antigen presenting capacity of APCs, and costimulatory molecules, both of which are required (in addition to the cytokines) to stimulate T cells.
Antigen Localization (Ignorance)
This model proposes that the regulation of the immune response lies outside of the specific immune system, and is principally controlled by antigen localization.51 This model can be adapted to include TLRs as mediators of signals that mobilize adaptive immunity and implies that migrating (non effector) T cells are ignorant of antigens in somatic tissues. In other words, such T cells would not be stimulated to proliferate as they cannot see antigenic peptide/MHC complexes, at least not in the appropriate costimulatory context. The model also predicts that somatic tissues lack antigen crosspriming (for pCTLs) and the expression of costimulatory molecules in tissue residing APCs (for naive CD4 T cells). Naive and resting memory T cells would only respond to antigenic peptide/MHC ligands by proliferating in lymph nodes. In support of this model, immature DCs (mainly found in tissues) cannot crosspresent antigens, whereas mature DCs (mostly lymph node resident) can.18 The failure of macrophages to crosspresent after stimulation via TLRs 3 or 918 is also consistent with this hypothesis. Inconsistent is evidence showing that macrophages can express costimulatory molecules upon activation and are therefore capable of activating naive CD4 T cells. However, naive CD4 T cells mainly home to lymph nodes, and are not often seen in somatic tissues. Another prediction of the antigen localization model is that there would be no clonal deletion of pCTLs in somatic tissues as a mechanism of peripheral T cell tolerance (in contrast to the Danger model).
Pattern Recognition
Janeway originally proposed that infectious “nonself ” substances are initiators of the immune response.52 The TLRs, according to this model are called pattern recognition receptors (PRRs). Consequently, their ligands are pathogen associated molecular patterns (PAMPs). The model explains the function of TLRs as sensors of pathogenic “nonself ” and TLRs would raise the alertness of innate immunity, and prime adaptive immunity to discriminate self from nonself (Fig. 9A).

Figure 9
The Pattern recognition model.
The model implies that TLRs are receptors for nonself molecular patterns that are evolutionarily foreign to a species and, as such, pattern recognition receptors serve as a memory of past infection. Basically, the model suggests that antigen alone, or in conjunction with the PRR (TLRs) signal, can activate adaptive immunity. This hypothesis can be seen as an extension of the self-nonself model.
Recently, it was suggested that a lack of TLR signaling in DCs might induce regulatory T cells (Fig. 9B), whereas TLR induced maturation of DCs would cause T cell activation and thence an immune response.47
This model neglects the possible influence of somatic tissues on the activity of the adaptive immune system, lacks simple explanations for the existence of endogenous (autologous) TLR ligands, and does not explain the dissociation between antigen presentation and the expression of costimulatory molecules on iDCs brought about by antagonistic TLR2 mAb.
Danger
Matzinger's "Danger" model takes a fresh look at signal-1 and signal-2. It proposes that "danger" signals act as an alarm to kick start the specific immune response (signal-0), and that these signals include pathogen specific molecules.53,54TLR signaling is but one of the prototype danger signals (exogenous danger) (Fig. 10A). Other danger signals include tissue distress, disruption and necrotic death (endogenous danger), all of which should activate the specific (adaptive) immune response (Fig. 10B). Although the difference between “infectious nonself ” and danger seems semantic, this was the first model that allowed us to shift our understanding about the control of specific immunity away from the strict self-nonself paradigm. According to Matzinger, specific B or T cells could still react to nonself, but would only do so if danger were signaled. Her model predicted that danger signals would upregulate costimulatory molecules on APCs, and hence activate naive T cells. Without danger, even nonself antigens would be tolerated (by clonal deletion of reactive T or B cells) (Fig. 10C). Therefore, a nonspecific, "alarm" type danger signal emanating from tissues that surround APCs and lymphocytes would have ultimate control of the immune response, which is quite different from Janeway's model. However, the molecular definition of danger remains elusive. Furthermore, danger represents a conserved alarm signal, which could, in itself, be “dangerous”, as pathogens could evolve to avoid tripping the alarm. Consequently evolution could deselect nonspecific warning systems in higher vertebrates. A solution for this problem might lie in the variability of the proposed danger signal, which may be sufficient to overcome its eradication in evolutionary terms. Perhaps this is the reason for the diversified group of pattern receptors that we see in higher vertebrates. Another problem with the Danger model is the nature of “endogenous” danger signals. For example, if the binding of bacterial DNA to TLR9 represents “exogenous danger”, why does the binding of autologous genomic DNA to the same receptor not constitute “endogenous danger”? Wouldn't this predispose us to autoimmune disease? Similar problems occur in explaining the dissociation of stimulation and costimulation of T cells via TLR2 binding in iDCs (see _TLRs and T Cell Costimulation). Lastly, the danger hypothesis lacks an explanation for regulatory T cells.
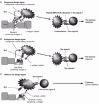
Figure 10
The Danger model. A) Exogenous danger signal. B) Endogenous danger signal. C) Induction of tolerance.
Integrity
The Integrity model suggests that three signals control the cells of central immunity (DC, T and B cells). 55-57 In general, it is well known that any cell can divide, grow, differentiate, die, or lie dormant, but we are far from knowing all of the signals that regulate these outcomes. Each tissue may have its own set of regulatory signals, perhaps overlapping with one another. Many specialized cells, like those of central immunity, have evolved another property, namely, a specific effector function that requires activation. Activation, though an ill-defined term, is used in many different contexts in cell biology. In my view, activation should be regarded as a process by which a cell acquires the capacity to unleash an effector function. Therefore, to become an effector, a cell must make a number of decisions after its initial activation. These decisions would depend on the cellular microenvironment and state of differentiation. Related intracellular signals that can perform these functions could be grouped into several clusters such that the transmission of a main activating signal would be modulated by the influence of two (in its simplest form) auxiliary ones (Fig. 11). Hence, we can distinguish three groups of stimuli. Those that descend from the main effector-function related receptor and engage various cytoplasmic intracellular mediators until the nuclear factor level. A second that assists receptors to modulate signal-1 either within the cytoplasm or nucleus, by engaging a different set of intracellular mediators and/or DNA-binding factors. These signals could compete with each other. A third set of signals could act to regulate the availability of certain nuclear factors and/or DNA accessibility (in terms of DNA binding, chromatin organization and cell-differentiation stage)(Fig. 11). These signals represent a necessarily simplified view of complex intracellular signal transduction pathways. In its most rudimentary sense, the cells of central immunity are thought to operate by receiving, modulating and transmitting incoming signals to the nucleus. These result in the expression of a set of specific genes that causes the cell to engage in a series of specific functions. The three-signal concept allows for the creation of cellular messages that could be transmitted to and thereby influence neighboring or distant cells.
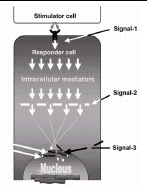
Figure 11
The cellular signaling model
In cells of central immunity, the main signal (signal-1) is provoked by antigen, peptide/MHC or TLR ligand in B, T and DCs, respectively. In naive and resting B and T cells the receipt of signal-1 alone would cause clonal deletion. Signal-2, arising from auxiliary receptors would modulate signal-1. Signal-3 would derive from the acceptor signal from supplementary inputs and facilitate, modify or prevent the acceptance of signal-1 (e.g., nuclear translocation-DNA binding effects). Examples of signal-2 can be envisaged. For B cells, signal-2 would be T cell help. For T cells it would be costimulation (if originating from CD28), or inhibition (if generated via CTLA4 or some other inhibitory B7 family receptor). Possibilities for signal-3 could be predicted and their modifying action may transform stimulatory signals into homeostatic ones for cells of central immunity. Consequently, these signals would play an important role in the regulation of the class of the immune response and the generation of regulatory T cells. Thus, for B and T cells, signal-1 and signal-2 are, in part, similar to those described in the previous models (S-NS, Danger), whereas signal-3 is a novel “integrity disruption” input that may derive, in part, from TLR signaling.
The signals for DCs would be different from those for B and T cells, in that DCs would use TLRs for pattern recognition (signal-1), whereas signal-2 would give an estimate of integrity. Thus the receipt of signal-1 and signal-2 in DCs would provide a strong stimulus for activation (maturation) together with signal-3 (Fig. 12A). Signal-3 for DCs could be provided by soluble factors such as prostaglandins and / or by a break in the cell-cell or cell-intercellular matrix interaction and homeostatic signaling.
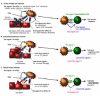
Figure 12
The Integrity model. A) Tissue damage and infection with pathogens. B) Induction of tolerance regarding commensals. C) Induction of tolerance (clonal deletion in the periphery).
The Integrity model proposes that TLRs take part in sensing tissue integrity as well as in pattern recognition. DCs perform these functions more efficiently than any other antigen presenting cell, and would sense aberrant molecular patterns (signal-1) together with the extent of damage (signal-2). Evidence suggests that TLR4 can engage endogenous ligands that appear after tissue damage, for example, fragments of fibronectin, hyaluronan, heparan sulphate in intercellular matrix, and extravascular fibrin(ogen).32-35 Based on these findings, some authors have proposed that TLRs are surveillance receptors for tissue damage.58 Furthermore, additional endogenous ligands have been reported for the TLR2/4 heterodimer and TLR9 such as the stress induced heat shock proteins HSP60 and 70, and mammalian genomic DNA, respectively. 37-40 In programmed cell death, apoptotic bodies would shield the release of such products and prevent their binding to TLRs. Apoptotic bodies could also inhibit DC maturation via the C3d fragments with which they are coated. During necrotic cell death these same endogenous TLR ligands can stimulate DC maturation. However, the Integrity model predicts that this would happen only when endogenous ligands were present at optimal concentrations, as the level of tissue damage would be “measured” by the strength of TLR signaling and the synergy between signals. When signal-1 is limiting (i.e., monomeric TLR ligands, insufficient crosslinking of TLRs), a second (coactivating) signal for iDCs would be required for the expression of costimulatory molecules. Maturation would require, additionally, signal-3. On the other hand, bacterial DNA would provide a sufficiently strong signal to iDCs for maturation (via TLR9, thus linking signal-1 with signal-2, provided that the putative signal-3 is also present). Supporting evidence includes data showing that crosslinking (not just binding) of TLR4 by LPS is necessary for macrophages to signal downstream.42 Further, ligands such as HSPs can act on TLR4 by enhancing the effects of otherwise substimulatory concentrations of LPS (Fig. 7D).43 The intracellular TLR signaling cascade can also be synergistically triggered by the ligands of TLRs 2 and 9, or TLRs 4 and 9, under suboptimal conditions (Fig. 7E).44 In all of these experiments, the in vitro handling of cells would provide signal-3. Differences between this and the Danger model are best illustrated in the explanation of the experiment described in the _TLRs and T Cell Costimulation_ section.31 This experiment showed that iDCs, pulsed with a TLR2 specific mAb (TL2.1), could present a TL2.1 derived antigenic peptide but failed to upregulate their costimulatory molecules (Fig. 7C). The Danger model cannot, in simple terms, explain why iDCs did not upregulate costimulatory molecules when they received a “danger signal” via TLR2. The Integrity model explains it by proposing that DCs need more than one signal for maturation. Hence, TL2.1 mAb binding to TLR2 would transmit signal-1 to iDCs, which would be insufficient to upregulate costimulatory molecules. The prediction is that the missing “coactivating” signal (signal-2) could be provided by other TLR ligands under suboptimal concentrations.
In the afferent loop of central immunity, a weak TLR signal like danger without tissue damage (or vice versa) would cause DCs to migrate to the lymph node, where they would tolerize rather than activate the immune response (Fig. 12B). Such tolerogenic DCs have been found to exist (reviewed in ref. 59). Using this mechanism, TLRs (on DCs) could detect commensals, but regulatory T cells would alleviate any deleterious responses to them. This would allow the individual to benefit from, for example, vitamin K producing bacteria in the intestine. The DCs that would initiate such a reaction would be stimulated by TLRs under suboptimal conditions and, as such, would not elicit T cell activation, but would instead generate regulatory T cells. The latter would be able to protect commensals more assiduously than the rather passive intervention offered by clonal deletion. Clonal deletion would depend on an alternative signal-3, or the lack of it, (Fig. 12C). Evolutionarily selected to preserve commensal microorganisms, regulatory T cells might also have been selected for their protection of vital organs, as a last barrier against autoimmunity. The mechanism by which TLRs might regulate the generation of regulatory T cells would, perhaps, depend on clonal competition between various TLR signals during the initiation of the immune response. DCs would generate two kinds of responses, one that activates T cells specific for a particular antigen, and another that would generate regulatory T cells (with different antigen specificity). The immune response might thus depend on the predominant TLR signal (i.e., how much tissue damage there is).
In summary, the Integrity model conceives the immune system as a delicate web of cells that senses the integrity of tissues and reacts to these as well as to external threats. The immune system actively responds to information about the internal milieu and distinguishes between three possible actions: destruction of the harmful, protection of the useful and neglect of the rest (non dangerous microorganisms). Destruction would involve the activation of autonomous and central immunity effector cells and soluble mediators. Protection possibly includes the selection of regulatory immunocytes in central immunity and neglect could operate by the deletion of potentially autoreactive clones in the thymus, bone marrow or peripheral lymphoid organs.
TLRs in Health and Disease
Targeting TLRs could be an efficient way to prepare vaccines against infectious diseases and for cancer immunotherapy. The use of agonistic ligands coupled (linked) to antigen would greatly enhance the potency of vaccine preparations. In addition, the vaccine might be designed to target various subpopulations of DCs in order to produce the desired effect; presentation or crosspresentation that would in turn yield regulatory cells or a strong cytotoxic T cell response. In the case of the former, using monomeric TLR ligands or antagonistic mAbs like anti-TLR2 (see _TLRs and T Cell Costimulation_ ) linked to autologous antigens might help in the treatment of autoimmune diseases. For the latter, antigens mixed with ligands for TLR3 or TLR9 could cause crosspriming in pDCs and be useful in cancer immunotherapy. The predicted therapeutic advantage could be enormous.
Likewise, the animal models of infectious diseases using TLR deficient mouse strains will undoubtedly be useful in studying the role of TLRs in infection and immunity.6 The detection of polymorphisms in patients with various diseases (including cancer) might also yield valuable information concerning risk profiles for the susceptibility to bacterial infections, autoimmunity and perhaps other diseases not necessarily linked to immunity.
In humans, many studies have attempted to identify allelic variants of TLRs. Thus far, several allelic variants of TLRs 2, 4 and 9 have been identified, and used in case control studies to identify any predisposition to infectious diseases, atherosclerosis or autoimmunity.60 The detection of allelic variants of other TLRs in human populations is also rapidly expanding. It is interesting that hypomorphic (underactive) variants of TLR4 genes are found in increased frequencies in individuals prone to develop toxic septicemia in the course of infectious disease with meningococcus.61
Two polymorphisms of the TLR-2 gene have been described: Arg753Gln, which correlates with sepsis in a Caucasian population and Arg677Trp, which correlates with lepromatous leprosy in an Asian population. Leprosy is caused by M. leprae, an intracellular bacterium that lives in the Schwann cells around the axons of peripheral nerves. There are two types of leprosy. A localized tuberculoid form of the disease that causes disfigurement. Comparatively few bacteria populate the lesions and a pronounced cell mediated host response is characteristic. The second type is a lepromatous form, which can be distinguished from the former by disseminated (non tuberculoid) lesions with a large bacillary load and a relatively weak host immune response. TLRs 1 and 2 are strongly expressed in lesions from the localized tuberculoid form compared with the lepromatous type of disease.62 TLR1/2 heterodimers can bind M. leprae products. However, the TLR2 Arg677Trp mutation abolishes the recognition of mycobacterial products when transfected into HEK293 cells.63
Several allelic variants have been described for TLR4 in humans, however two seems to have functional significance, TLR4 Asp299Gly and TLR4 Thr399Ile. Both are less efficient in signaling than the wild type allele. Tested in vitro, the effects produced by Asp299Gly and Thr399Ile were as low as 5-10% and 20-30% that of the wildtype. These variants lead to a shortened immunologic response to inhaled LPS and to lower levels of proinflammatory cytokines, acute phase reactants, and soluble adhesion molecules. Interestingly, they are also associated with a reduced extent and progression of carotid atherosclerosis.64 Atherosclerosis is associated with chronic infection or inflammation, and may have an infectious origin. The TLR4 Asp299Gly polymorphism is associated with a risk of coronary artery disease. Strikingly, patients that carry this allele (in particular those with elevated CRP levels) benefit more from prevastatin treatment (to prevent cardiovascular events), than patients that express the wild type variant. On the other hand, the TLR4 Asp299Gly genotype is not associated with disease progression.65 One explanation for these clinical findings might be that the oxidized low density lipoprotein (LDL), which is elevated in such patients, is a potent upregulator of TLR4. TLR4 proinflammatory activity might therefore be reduced in patients with the Asp299Gly allele, which diminishes the extent of vascular tissue damage. The TLR4 Asp299Gly allele has also been suggested to have a beneficial role in the systemic inflammatory response syndrome.
Premature birth can be a consequence of urogenital infection, which is often caused by Gram negative bacteria. The Asp299Gly allele was associated with an increased risk of premature birth. The same allelic variant may also predispose to septic shock with Gram negative bacteria though does not influence the susceptibility to, or severity of, meningococcal disease.
Data showing that several otherwise healthy children, with an inherited deficiency in the IRAK4 molecule, developed infections with pyogenic bacteria show that deficiencies in the downstream mediators of TLR signaling can also predispose to infectious diseases.
TLRs have also been implicated in the pathogenesis and severity of some autoimmune diseases. For example, the overexpression of TLR2 has been reported in the synovial fluid cells of patients with rheumatoid arthritis.66 DNA specific IgG can trigger low affinity autoreactive antibodies like rheumatoid factor when complexed with autologous CpG DNA in the circulation of autoimmune prone mice.29 Moreover, one of the defects that is associated with susceptibility to systemic autoimmune diseases is the defective clearance of dying cells and self DNA. This would exacerbate the potential for an autoimmune response.
In conclusion, hypomorphic TLR allelic variants may predispose to many infectious diseases and perhaps autoimmunity. The genetic analysis of allelic frequencies in patients with various diseases is a research field in its infancy. Future studies will undoubtedly show the extent of TLR functions in health and disease. The knowledge that will be accumulated will allow us to improve health care by the genetic tailoring of therapies and the development of novel pharmacological and biological agents.
Acknowledgements
I thank Bjarne Bogen and Karoline Schjetne for sharing information about their work and comments, Hanne Quarsten for the inspiration for Figure 6A, and Trond S. Halstensen for comments on the text.
References
- 1.
- Belvin MP, Anderson KV. A conserved signaling pathway: the Drosophila toll-dorsal pathway. Annu Rev Cell Dev Biol. 1996;12:393–416. [PubMed: 8970732]
- 2.
- Hultmark D. Drosophila immunity: paths and patterns. Curr Opin Immunol. 2003;15:12–9. [PubMed: 12495727]
- 3.
- Hoffmann JA, Reichhart JM. Drosophila innate immunity: an evolutionary perspective. Nat Immunol. 2002;3:121–126. [PubMed: 11812988]
- 4.
- Remillieux-Leschelle N, Santamaria P, Randsholt NB. Regulation of Larval Hematopoiesis in Drosophila melanogaster. A role for the multi sex combs gene. Genetics. 2002;162:1259–1274. [PMC free article: PMC1462314] [PubMed: 12454071]
- 5.
- Pujol N, Link EM, Liu LX. et al. A reverse genetic analysis of components of the Toll signaling pathway in Caenorhabditis elegans. Curr Biol. 2001;11:809–821. [PubMed: 11516642]
- 6.
- Qureshi ST, Medzhitov R. Toll-like receptors and their role in experimental models of microbial infection. Genes Immun. 2003;4:87–94. [PubMed: 12618855]
- 7.
- Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. [PubMed: 12527213]
- 8.
- Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. [PubMed: 12791976]
- 9.
- Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol. 2002;2:982–987. [PubMed: 12461571]
- 10.
- Kurt-Jones EA, Mandell L, Whitney C. et al. Role of toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood. 2002;100:1860–1868. [PubMed: 12176910]
- 11.
- Sabroe I, Prince LR, Jones EC. et al. Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J Immunol. 2003;170:5268–5275. [PubMed: 12734376]
- 12.
- Varadaradjalou S, Feger F, Thieblemont N. et al. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human mast cells. Eur J Immunol. 2003;33:899–906. [PubMed: 12672055]
- 13.
- Kadowaki N, Ho S, Antonenko S. et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–869. [PMC free article: PMC2195968] [PubMed: 11561001]
- 14.
- Bosisio D, Polentarutti N, Sironi M. et al. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood. 2002;99:3427–3431. [PubMed: 11964313]
- 15.
- Muzio M, Bosisio D, Polentarutti N. et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol. 2000;164:5998–6004. [PubMed: 10820283]
- 16.
- Sato S, Takeuchi O, Fujita T. et al. A variety of microbial components induce tolerance to lipopolysaccharide by differentially affecting MyD88-dependent and -independent pathways. Int Immunol. 2002;14:783–791. [PubMed: 12096038]
- 17.
- An H, Xu H, Yu Y. et al. Up-regulation of TLR9 gene expression by LPS in mouse macrophages via activation of NF-kappaB, ERK and p38 MAPK signal pathways. Immunol Lett. 2002;81:165–169. [PubMed: 11947920]
- 18.
- Datta SK, Redecke V, Prilliman KR. et al. A subset of Toll-like receptor ligands induces crosspresentation by bone marrow-derived dendritic cells. J Immunol. 2003;170:4102–4110. [PubMed: 12682240]
- 19.
- Bourke E, Bosisio D, Golay J. et al. The Toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood. 2003;10:10. [PubMed: 12689944]
- 20.
- Stacey KJ, Sweet MJ, Hume DA. Macrophages ingest and are activated by bacterial DNA. J Immunol. 1996;157:2116–2122. [PubMed: 8757335]
- 21.
- Banchereau J, Paczesny S, Blanco P. et al. Dendritic cells: controllers of the immune system and a new promise for immunotherapy. Ann N Y Acad Sci. 2003;987:180–7. [PubMed: 12727638]
- 22.
- Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. [PubMed: 11913066]
- 23.
- Boonstra A, Asselin-Paturel C, Gilliet M. et al. Flexibility of mouse classical and plasmacytoidderived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J Exp Med. 2003;197:101–109. [PMC free article: PMC2193804] [PubMed: 12515817]
- 24.
- Tsuji S, Matsumoto M, Takeuchi O. et al. Maturation of human dendritic cells by cell wall skeleton of Mycobacterium bovis bacillus Calmette-Guerin: involvement of toll-like receptors. Infect Immun. 2000;68:6883–6890. [PMC free article: PMC97794] [PubMed: 11083809]
- 25.
- Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: upregulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. 2003;30:30. [PubMed: 12560217]
- 26.
- Rui L, Vinuesa CG, Blasioli J. et al. Resistance to CpG DNA-induced autoimmunity through tolerogenic B cell antigen receptor ERK signaling. Nat Immunol. 2003;4:594–600. [PubMed: 12740574]
- 27.
- Coutinho A, Moller G. Editorial: Immune activation of B cells: evidence for _one nonspecific triggering signal_ not delivered by the Ig receptors. Scand J Immunol. 1974;3:133–146. [PubMed: 4132599]
- 28.
- Mamchak AA, Hodgkin PD. Absence of lipopolysaccharide high-dose paralysis in B-cell responses: implications for the one-signal theory. Immunol Cell Biol. 2000;78:133–141. [PubMed: 10762413]
- 29.
- Leadbetter EA, Rifkin IR, Hohlbaum AM. et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. [PubMed: 11948342]
- 30.
- Liu N, Ohnishi N, Ni L. et al. CpG directly induces T-bet expression and inhibits Ig(gamma1) and Ig(epsilon) switching in B cells. Nat Immunol. 2003;25:25. [PubMed: 12766768]
- 31.
- Schjetne KW, Thompson KM, Nilsen N. et al. Link between innate and adaptive immunity: Toll-like receptor 2 internalizes antigen for presentation to CD4+ T cells and could be an efficient vaccine target. J Immunol. 2003;171:32–36. [PubMed: 12816980]
- 32.
- Okamura Y, Watari M, Jerud ES. et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–10233. [PubMed: 11150311]
- 33.
- Termeer C, Benedix F, Sleeman J. et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. [PMC free article: PMC2196009] [PubMed: 11781369]
- 34.
- Johnson GB, Brunn GJ, Kodaira Y. et al. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. [PubMed: 11994480]
- 35.
- Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–2894. [PubMed: 11509636]
- 36.
- Yang R, Yan Z, Chen F. et al. Hyaluronic acid and chondroitin sulphate A rapidly promote differentiation of immature DC with upregulation of costimulatory and antigen-presenting molecules, and enhancement of NF-kappaB and protein kinase activity. Scand J Immunol. 2002;55:2–13. [PubMed: 11841687]
- 37.
- Vabulas RM, Ahmad-Nejad P, da Costa C. et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–31339. [PubMed: 11402040]
- 38.
- Asea A, Rehli M, Kabingu E. et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. [PubMed: 11836257]
- 39.
- Vabulas RM, Ahmad-Nejad P, Ghose S. et al. HSP70 as endogenous stimulus of the Toll/interleukin- 1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. [PubMed: 11842086]
- 40.
- Ishii KJ, Suzuki K, Coban C. et al. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol. 2001;167:2602–2607. [PubMed: 11509601]
- 41.
- Flo TH, Halaas O, Torp S. et al. Differential expression of Toll-like receptor 2 in human cells. J Leukoc Biol. 2001;69:474–481. [PubMed: 11261796]
- 42.
- Latz E, Visintin A, Lien E. et al. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J Biol Chem. 2002;277:47834–47843. [PubMed: 12324469]
- 43.
- Wallin RP, Lundqvist A, More SH. et al. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002;23:130–135. [PubMed: 11864840]
- 44.
- Equils O, Schito ML, Karahashi H. et al. Toll-like receptor 2 (TLR2) and TLR9 signaling results in HIV-long terminal repeat trans-activation and HIV replication in HIV-1 transgenic mouse spleen cells: implications of simultaneous activation of TLRs on HIV replication. J Immunol. 2003;170:5159–5164. [PubMed: 12734363]
- 45.
- Re F, Strominger JL. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J Biol Chem. 2001;276:37692–37699. [PubMed: 11477091]
- 46.
- McGuirk P, Mills KH. Pathogen-specific regulatory T cells provoke a shift in the Th1/Th2 paradigm in immunity to infectious diseases. Trends Immunol. 2002;23:450–455. [PubMed: 12200067]
- 47.
- Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. [PubMed: 12532024]
- 48.
- Bretscher P, Cohn M. A theory of self-nonself discrimination: paralysis and induction involve the recognition of one and two determinants on an antigen, respectively. Science. 1970;169:1042–1049. [PubMed: 4194660]
- 49.
- Langman RE, Cohn M. Self-nonself discrimination revisited. Semin Immunol. 2000;12:159–162. [PubMed: 10910734]
- 50.
- Weigle WO. Immunologic tolerance: development and disruption. Hosp Pract (Off Ed). 1995;30:81–4 89-92. [PubMed: 7531711]
- 51.
- Zinkernagel RM. Immunology taught by viruses. Science. 1996;271:173–8. [PubMed: 8539616]
- 52.
- Janeway CAJr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. [PubMed: 2700931]
- 53.
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. [PubMed: 8011301]
- 54.
- Matzinger P. An innate sense of danger. Semin Immunol. 1998;10:399–415. [PubMed: 9840976]
- 55.
- Dembic Z. Do we need integrity? Scand J Immunol. 1996;44:549–550. [PubMed: 8972734]
- 56.
- Dembic Z. Immune system protects integrity of tissues. Mol Immunol. 2000;37:563–569. [PubMed: 11163392]
- 57.
- Dembic Z. About theories and the integrative function of the immune system. The Immunologist. 2000;8:141–146.
- 58.
- Johnson GB, Brunn GJ, Tang AH. et al. Evolutionary clues to the functions of the Toll-like family as surveillance receptors. Trends Immunol. 2003;24:19–24. [PubMed: 12495720]
- 59.
- Steinman RM, Hawiger D, Liu K. et al. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann N Y Acad Sci. 2003;987:15–25. [PubMed: 12727620]
- 60.
- Lorenz E, Mira JP, Cornish KL. et al. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun. 2000;68:6398–6401. [PMC free article: PMC97725] [PubMed: 11035751]
- 61.
- Smirnova I, Mann N, Dols A. et al. Assay of locus-specific genetic load implicates rare Toll-like receptor 4 mutations in meningococcal susceptibility. Proc Natl Acad Sci USA. 2003;100:6075–6080. [PMC free article: PMC156328] [PubMed: 12730365]
- 62.
- Krutzik SR, Ochoa MT, Sieling PA. et al. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat Med. 2003;9:525–532. [PubMed: 12692544]
- 63.
- Bochud PY, Hawn TR, Aderem A. Cutting edge: a toll-like receptor 2 polymorphism that is associated with lepromatous leprosy is unable to mediate mycobacterial signaling. J Immunol. 2003;170:3451–3454. [PubMed: 12646604]
- 64.
- Kiechl S, Lorenz E, Reindl M. et al. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med. 2002;347:185–192. [PubMed: 12124407]
- 65.
- Boekholdt SM, Agema WR, Peters RJ. et al. Variants of toll-like receptor 4 modify the efficacy of statin therapy and the risk of cardiovascular events. Circulation. 2003;107:2416–2421. [PubMed: 12742999]
- 66.
- Seibl R, Birchler T, Loeliger S. et al. Expression and regulation of toll-like receptor 2 in rheumatoid arthritis synovium. Am J Pathol. 2003;162:1221–1227. [PMC free article: PMC1851232] [PubMed: 12651614]
- 67.
- Kadowaki N, Antonenko S, Liu YJ. Distinct CpG DNA and polyinosinic-polycytidylic acid doublestranded RNA, respectively, stimulate CD11c- type 2 dendritic cell precursors and CD11c+ dendritic cells to produce type I IFN. J Immunol. 2001;166:2291–2295. [PubMed: 11160284]
- 68.
- Visintin A, Mazzoni A, Spitzer JH. et al. Regulation of Toll-like receptors in human monocytes and dendritic cells. J Immunol. 2001;166:249–255. [PubMed: 11123299]
- 69.
- Takeuchi O, Sato S, Horiuchi T. et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–14. [PubMed: 12077222]
- Introduction
- Developmental Functions
- Dorsal-Ventral Polarization of the Embryo
- Muscle Development
- Motoneuron Growth Cone Guidance
- Others
- The Role in Defense
- Humoral
- Cellular
- Mammals
- Divisions of Immunity
- Specific—Nonspecific (Humoral or Cellular) Immunity
- Innate—Adaptive Immunity
- Autonomous—Central Immunity
- Expression and Function of TLRs in Cells of Autonomous Immunity
- Neutrophils (and Granulocytes)
- Mast Cells
- Monocytes
- Macrophages
- NK Cells
- Expression and Function of TLRs in Cells of Central Immunity
- The Roles of TLRs in APC—T Cell Interactions
- TLRs and Antigen Presentation
- Endogenous Pathways of Ag Presentation
- The Exogenous Pathway of Ag Presentation
- The MHC Class II Antigen Presentation Pathway
- Crosspresentation: MHC Class I Antigen Presentation Pathway
- TLR Ligand Linked Antigen Presentation in Immature DCs
- Unlinked TLR Ligand Antigen Crosspresentation in Mature DCs
- Presentation of Self Antigens that Are TLR Ligands in DCs
- TLRs and T Cell Costimulation
- TLRs and Polarization of the T Cell Response
- TLRs and Theories about the Function of the Immune System
- Self-Nonself Discrimination and Associated Antigen Recognition
- Cytokine Burst
- Antigen Localization (Ignorance)
- Pattern Recognition
- Danger
- Integrity
- TLRs in Health and Disease
- Acknowledgements
- References
- The Function of Toll-Like Receptors - Madame Curie Bioscience DatabaseThe Function of Toll-Like Receptors - Madame Curie Bioscience Database
- Gastroenterologic and Hepatic Diseases - Madame Curie Bioscience DatabaseGastroenterologic and Hepatic Diseases - Madame Curie Bioscience Database
- Human, Mouse and Yeast Telomerase - Madame Curie Bioscience DatabaseHuman, Mouse and Yeast Telomerase - Madame Curie Bioscience Database
- In Situ Activation of Caspases Revealed by Affinity Labeling Their Enzymatic Sit...In Situ Activation of Caspases Revealed by Affinity Labeling Their Enzymatic Sites - Madame Curie Bioscience Database
- The Origins of Species - Madame Curie Bioscience DatabaseThe Origins of Species - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...