

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The progenitors of the mitral and tricuspid valves and the membranous interventricular septum in the heart arise by an epithelial-mesenchymal cell transformation (EMT) from embryonic endothelial cells. Experiments using collagen gel cultures to mimic the three dimensional environment in the embryonic heart cushions showed that EMT was triggered by an inductive stimulus produced by the adjacent myocardium and that myocardium from a nontransforming region was insufficient to induce transformation. Studies using chick embryos have demonstrated distinct and sequential roles for two isoforms of TGFβ Bone Morphogenetic Proteins, and roles for several TGFβfamily receptors in this EMT. Additional studies have identified at least two separate transcription factors required for EMT, Slug and Mox-1. The complexity of the inductive signal is not yet fully understood, but recent studies have shown roles for several Wnt proteins and a requirement for signaling by extracellular hyaluronan. The embryonic heart provides an experimental model of relevance to congenital heart diseases that are likely to continue to provide novel insight into the regulation of EMT as a normal process of development.
Introduction
While not the first instance of phenotypic change by an epithelium in the embryo (see chapter 2: Morali et al), the formation of mesenchymal cells in the embryonic heart provides an instructive model of epithelial mesenchymal transformation (EMT). Though there are several EMT processes that take place in the heart this chapter will focus on the formation of mesenchymal cells from endothelial cells lining the atrioventricular (AV) canal. These mesenchymal cells form the earliest progenitors of the fibroblasts of the septum intermedium and the mitral and tricuspid valves. Two other observed EMT processes in the heart include a similar but not identical process in the outflow tract of the heart that forms the precursors of the aortic and pulmonary valves1 and an EMT of the epicardium that produces both the fibroblasts of the myocardium and the vascular precursors of the coronary circulation. Though some mention of complementary or conflicting information may be mentioned, there is no effort to be comprehensive concerning these other cardiac EMTs.
Study of the embryonic AV canal has several advantages for investigators attempting to understand the process of phenotypic shape change by an epithelium. EMT in the embryonic heart is an induced process that occurs with very defined timing in the embryo. Unlike several other EMTs in the embryo described in the present volume, the valvular progenitors do not appear to participate in widely divergent differentiation programs and there is no evidence to suggest that AV canal endothelial cells are divided into subsets of committed and uncommitted cell populations. The ability to identify and collect heart tissues prior to EMT enables considerable advantage in obtaining cells and tissues for examination. Studies over the last 20 years, particularly in culture, have provided significant insight into EMT and the present review will attempt to summarize the progress made in this system.
Description of EMT In Vivo
Description of embryonic heart development dates from the time of Aristotle. The obvious external location and motion of the heart in the chick embryo made it a focal point of observation. Using the light microscope, cellularity of the cardiac cushions of the AV canal was obvious although the role of these cells was not well understood.2 The modern focus on the development of the cardiac cushions and the origins of their cellular constituents dates from the investigations of Markwald and Manasek and their colleagues in the late 1970s. Using electron microscopy, light microscopy and histochemical procedures, these investigators described the formation of cushion mesenchymal cells in the AV canal and explored their interaction with the extracellular matrix that is the substance of the cardiac cushion.3-6 These investigators focused on the origin of mesenchyme from the endothelium of the atrioventricular canal and explored the interactions with the extracellular matrix. Manasek6 speculated that the formation of mesenchyme in the AV canal was due to a tissue interaction with the myocardium, but there was no experimental evidence to support this conjecture at that time. There remained some controversy as to the origin of the cardiac mesenchyme. Some argued that the myocardium of the heart was the source of mesenchyme in the AV canal. This controversy was resolved by the work of Kinsella and Fitzharris.7 These investigators utilized a microscope and a movie camera to view a cross-section of the AV canal in a culture chamber and photographed epithelial mesenchymal cell transformation in situ strictly from the endothelium. These studies combined to provide the basic picture of EMT in vivo summarized below. There has been some more recent work concerning “myocardialization” of the heart valves by cells migrating from the myocardial layer at much later stages, but it is not clear that this process represents an EMT.8
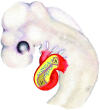
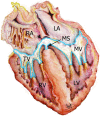
As shown in (Fig. 1), the looped heart of the stage 17 chick9 embryo shows an expanded extracellular matrix in the atrioventricular canal that forms two opposing cushions. These cushions consist of an acellular extracellular matrix covered by an endothelium. Radioactive studies with labeled glucosamine suggest that the vast majority of the extracellular matrix is produced by the myocardium.10 By this stage, the endothelium shows morphological signs of EMT. Endothelial cells are hypertrophied,3 have polarized golgi apparatus,10 show loss of cell-cell adhesion11 and have extended filopodia.3,7 Mesenchymal cells have invaded the underlying extracellular matrix.3,12 Movies by Kinsella and Fitzharris enabled the appreciation of the remarkable length of these filopodia as they appeared to probe more than 50 µm into the ECM.7 Scanning electron microscope pictures11,13 showed that the hypertrophy of the endothelia was accompanied by a loss of cell-cell adhesion as openings became visible in the endothelial sheet and their appeared to be lateral migration in the endothelial layer. After probing the matrix, a subset of endothelial cells left the endothelial sheet and entered the ECM of the cushion tissue. The ECM of the cushion was shown to by a hyaluronan-rich ECM while the passage of mesenchymal cells through the cushion produced a matrix that was rich in chondroitin sulfate proteoglycans.14 EMT by the AV canal endothelium continued at least through stage 19 and produced highly cellular cushions that would be remodeled into valvular structures with subsequent development. Figure 2 shows that the mitral and tricuspid valves and the membranous septum of the ventricle are developed by remodeling of the progenitor cells that invaded the extracellular matrix of the cardiac cushions. It is worth noting that although there are micrographs that suggest a broad outline of cushions remodeling into valves,15,16 there is very little knowledge concerning these mechanisms and the development of connections to the papillary muscles. For example, during mouse heart development immunolocalization results indicate expression of type VI collagen during the period when the AV endocardial cushions differentiate into valve leaflets and the membranous septa. These findings support the hypothesis of a role of collagen VI in normal AV endocardial cushion differentiation. In the more mature AV valves of the mouse, high expression of collagen VI is confined to a thin layer on the ventricular side of the AV valve leaflets.15
Development of Collagen Gel Culture
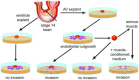
Our understanding of the mechanisms underlying EMT in the heart arose out of the development of a three-dimensional collagen gel culture system. Though the utilization of collagen gels as substrates for three-dimensional migration arose out of the work of Elsdale and Bard,17 it is not clear that cells were actually invasive into their dense substrates. However, using gelling solutions of collagen enabled several investigators of the early 1980s to explore the formation and migration of mesenchyme or fibroblasts in three dimensions.18-21 Differences in behavior by epithelial cells and mesenchyme were observed in natural ECM at approximately the same time by Overton.22 Collagen gels were first used in the heart by Bernanke and Markwald21 to explore the interactions between cushion mesenchyme and extracellular glycosaminoglycans. In 1983, collagen gel cultures were used to demonstrate that, as predicted by Manasek,6 that EMT in the heart was the product of a tissue interaction between the endothelium and the myocardium.23 These experiments consisted of the timed culture and removal of the myocardium with staged AV canal explants. These data showed that the myocardium produced an inductive stimulus that was required to initiate the EMT and that ventricular explants had little potential for EMT. Subsequent experiments24-26 showed that the stimulus for EMT could be provided by either an extract of the ECM or by myocardium-conditioned medium. Comparison of ventricular and AV canal explants supported the regional specificity of EMT in vivo. Ventricular myocardium was incapable of inducing EMT in either ventricular endothelium or AV canal endothelium.26,27 The timing and appearance of the cultures suggested a faithful replication of the EMT process in vivo. These cultures have been subsequently used by a number of investigators to explore the molecular and cellular aspects of EMT in this system. Figure 3 depicts the EMT process in vitro and summarizes the regional nature of the inductive stimulus.
Identification of Components of the EMT Process
The initial exploration of the EMT process focused upon the role of the extracellular matrix. As EMT was shown to be a response to a myocardial stimulus, the first approach was to collect and test the extracellular matrix intervening between the cell layers. Krug et al10 showed that a hyaluronidase-treated fraction of heart ECM produced an apparent activation by endothelial cells but it was insufficient to induce invasion of collagen cultures by mesenchymal cells. Subsequently it was shown that either an ECM extract of the heart collected without hyaluronidase or myocardium-conditioned medium was sufficient to induce EMT in a competent monolayer of stage 14 endothelial cells.24,25 Though, at the time, experiments combining hyaluronidase-treated ECM with exogenous hyaluronan did not produce EMT,10 it should be noted that hyaluronidase disrupts AV endocardial cushion EMT in vitro, and an active role for hyaluronan in EMT was recently demonstrated in heart explant tissues from genetically engineered mouse models (Fig. 4).28 A similar phenotype to the Has2-/- mouse embryos occurs in Zebrafish containing a mutated UDP-glucose dehydrogenase gene, encoding an upstream enzyme of Has2 (jekyll mutant).29

Because of its ubiquitous distribution in the cardiac jelly and the embryonic ECM, it is unlikely that hyaluronan alone stimulates AV canal EMT. Based on their role as upstream activators of Ras, restricted patterns of expression during cardiac morphogenesis, and cardiac phenotypes in null mutants, members of the ErbB family of receptor tyrosine kinases represent potential ligands to act coordinately with hyaluronan to mediate this process. ErbB2, ErbB3 and ErbB4 are upstream activators of Ras and are important in heart development.30,31 We recently demonstrated that the induction of AV endothelial cushion EMT by hyaluronan involves ErbB2 and ErbB3. The ErbB3 receptor is activated in response to exogenous hyaluronan rescue in Has2-/- AV canals explants, and EMT is rescued in Has2-/- AV canals explants by the ErbB receptor ligand heregulin. In addition, soluble ErbB3 and ErbB2 inhibitors block these signaling events. These studies demonstrate a costimulatory pathway involving hyaluronan and a tyrosine kinase receptor-ligand family are required for AV endocardial cushion EMT.28
One approach to explore the process of EMT, was the production of a polyclonal antiserum against the inductive ECM fraction (ES antibodies). This antiserum recognized a number of ECM components and was capable of blocking EMT in the collagen gel cultures.32 Due to the success of this approach, the ECM extract was used as an antigen for monoclonal antibody production. Clones were tested for activity against EMT cultures. One of these antibodies was particularly effective and as it recognized a 130 KD protein called ES/130.33 By a variety of criteria, the antigen recognized by ES/130 appeared to be a critical mediator of EMT.34 However analysis of the ES/130 sequence suggests that it is most likely an intracellular protein that participates in regulated secretion (Krug, personal communication). The presence of the ES/ 130 antigen in the ECM of the heart may have to do with the extensive secretory activity of the myocardium during the development of the cardiac cushions. Under the conditions of specimen preparation utilized by these investigators, packets of secreted material may remain associated with the ES/130 antigen. The basis for EMT inhibition produced by ES/130 is unclear. Perhaps the antibody perturbs interaction between the antigen and one or more components of the ECM. This immune approach towards the identification of critical components of the extracellular matrix also resulted in the identification of transferrin as a mediator of cardiac cushion development.35 An additional molecule identified by this approach, hLAMP1, remains under investigation.36
An alternative approach, to identification of mediators of EMT was to test a series of growth factors against competent endothelium (AV endothelial cell cultures with the myocardium removed prior to induction of EMT). There were few differences observed between the ECM of the ventricle and the AV canal in two dimensional protein gels but those that were observed were in a low molecular weight area consistent with growth factors.10 A series of growth factors, including EGF, FGF2, and TGFβ were tested.27 Alone, none of these growth factors produced EMT. However, when cultured in combination with a ventricular explant, TGFβproduced EMT.27 (For a review of TGFβsee chapter 16; Vignais and Fafet, and also Roberts et al37. EGF and FGF2 had no effect on ventricular cultures. The original experiment utilized the TGFβ1 isoform but it is clear that the exogenous delivery of any of the three TGFβisoforms was sufficient to produce EMT.27,38 Confirmation that TGFβwas a mediator of EMT was provided by the observation that a pan-specific antibody against all of the TGFbeta;isoforms blocked EMT in intact AV canal explant cultures.27
TGFβas a Mediator of EMT
The demonstration of TGFβas a mediator of EMT resulted in a series of experiments to resolve the parameters of the interaction. RNase protection assays were undertaken to define the TGFβisoforms present in the chick heart. These experiments showed a greater concentration of TGFβ3 in the AV canal than in the adjacent ventricle but approximately equal amounts of TGFβ2 mRNA in the AV canal and the ventricle. TGFβ4 (the avian equivalent of mammalian TGFβ139 was not found in the heart during early valve development.40 In contrast, studies by Akhurst and colleagues41,42 in the mouse heart showed that TGFβ1 was present largely in the endothelium and that TGFβ1 was present in the myocardium and in the mesenchyme of the cardiac cushions after EMT. Though these investigators found little TGFβ3 in the mouse heart, a recent study shows this isoform appears in the mouse heart cushions after EMT.43 In situ hybridization studies by Barnett et al44 and Boyer et al45 have produced the following pattern of TGFβtranscription in the chick heart. Prior to EMT, TGFβ2 mRNA is present throughout the myocardium and endothelium of the heart. In contrast, TGFβ3 transcripts are only present in the myocardium. Subsequent to endothelial activation, TGFβ3 is detected in the endothelium and mesenchyme of the AV canal. Migratory mesenchymal cells continue to produce TGFβ2 as they migrate in the cushion. Considering the preservation of specific TGFβ isoform homology between species, the differences between chick and mouse hearts in terms of isoform distribution and utilization (below) are not completely explained.
Functional utilization of TGFβisoforms in the AV canal was first explored by the use of antisense oligonucleotides designed against each of the published TGFβsequences. Modified antisense oligonucleotides were delivered to explant cultures and the effect on EMT was observed. Oligonucleotides directed against TGFβ3, alone, were able to perturb EMT in the explant cultures.46 Though it was noted that the antisense oligonucleotides against TGFβ3 did not completely recapitulate the phenotype produced by the pan-specific anti-TGFβantibody it was accepted that TGFβ3 was the critical member of this family involved in cardiac EMT.
Studies by Doetschman and colleagues47-49 were focused on the production of null mice for each of the TGFβisoforms. The surprising observation in the mouse was that only the TGFβ2 null mouse had heart defects. Defects in this mouse include structures derived from the cardiac cushions and had a similarity to defects seen in the human as the Tetrology of Fallot as well as an atrial septal defect. The TGFβ3 null mouse had defects in EMT in the palate but its heart was normal.
In light of these findings, the role of TGFβ2 in the chick was reexamined. As only one antisense oligonucleotide was tested against TGFβ2 in the previous study, it was possible that the lack of inhibition seen in the earlier study was due to an ineffective oligonucleotide sequence. Studies were undertaken with isoform-specific, blocking antibodies in collagen gel cultures. This approach uncovered separate and sequential activities for TGFβ2 and TGFβ3 during EMT in the chick heart. As seen before with antisense TGFβ3 oligonucleotides, anti-TGFβ3 antibodies blocked EMT after cell separation by preventing invasion of the collagen gel matrix. However, anti-TGFβ2 antibodies blocked the initial step of cell-cell separation similar to the effect seen with the pan-specific anti-TGFβantibody used in a previous study.27,45 This apparent difference between mice and chicks was tested using mouse heart explants in collagen gel cultures. As seen in the TGFβnull mice, inhibition of TGFβ2, not TGFβ3 blocked EMT in vitro. TGFβ3 levels in the AV canal of the mouse rise only after EMT.43
TGFβReceptors
There are three principal TGFβreceptors in most biological systems (reviewed by50). A signaling complex is formed between the TGFβType II receptor and the Type I receptor after binding TGFβ The Type II receptor has higher affinity for TGFβ1 and TGFβ1 than for TGFβ2.51 The Type I receptor does not appear to bind TGFβ in the absence of the Type II receptor but provides the serine-threonine kinase activity that initiates signal transduction into the cell.52 A third receptor, betaglycan or the TGFβType III receptor, has a large extracellular domain and a small cytoplasmic domain. This receptor has a somewhat greater affinity for TGFβ2 compared to the other isoforms and has been proposed to present this isoform to the Type II receptor.53
Studies examining the TGFβreceptors in the heart were made possible by the efforts of Barnett and colleagues who cloned the avian homologues of the TGFβreceptors.54-56 Initial studies with antibodies towards the TGFbeta;Type II receptor showed that this receptor was expressed throughout the endothelium lining the developing heart and the blood vessels of the embryo. The antibodies were shown to block receptor function and EMT in collagen gel culture was inhibited.55 Similar studies were performed after the cloning and production of antibodies to the Type III receptor.56 Unlike the Type II receptor, immunostaining for the Type III receptor in the endothelium was limited to the developing cardiac cushions. This antibody proved to block EMT in collagen gel cultures as well.56 Examination of antibody-treated cultures revealed some differences between the two receptors.57 Cultures treated with antibody towards TGFβType III receptor had a close similarity with cultures treated with a blocking antibody towards TGFβ2.45 In both cases, the endothelial outgrowth of the cardiac explants remained cohesive and “unactivated”. In contrast, cultures blocked with antibody towards the Type II receptor demonstrated a separated, “activated” endothelium where there were numerous fusiform cells on the cell surface. This was similar to the inhibition previously seen with either antisense oligonucleotides or antibodies against TGFβ2.45,46 These data suggest that although ligand and receptor affinity differences do not appear to be significant in vitro, there is specificity between ligand isoforms and receptor types that can be distinguished in situ.
TGFβActivation
One aspect of TGFβbiology in the heart that is difficult to assess is the role of ligand activation. Each of the TGFβisoforms is normally secreted in an inactive form with its amino terminal sequence attached and functional as a block to receptor binding.58 Both pH and proteolytic treatment can be performed in vitro to activate the ligand. In vivo, it appears that that TGFβisoforms associate with a latent TGFβbinding protein and that this association is critical for either activation or presentation of the active ligand to its receptor.59 Brauer et al60 demonstrated that ninety five percent of the TGFβ3 found in the extracellular matrix of the heart outflow tract is un-activated. Similar levels of unactivated TGFβs were found by McCormick.61 Nakajima and colleagues showed that inhibition of Latent TGFβbinding Protein-1 (LTBP-1) by a blocking antibody could prevent EMT in mouse heart explants in culture. 62 Utilization of both antisense oligonucleotides and antibodies against LTBP-1 in chick heart cultures confirms this finding and shows that the TGFβ2-mediated endothelial separation is particularly sensitive to these reagents (Berkompas and Runyan, in preparation). These data show that regulation of the TGFβmediated embryonic induction could be accomplished by secretion or activation of proteases into the ECM between the endothelium and the myocardium. Studies by McGuire and colleagues63-65 have identified several proteases, including urokinase and matrix metalloproteinases 2 and 9, in the heart that could mediate TGFβactivation. While the work of these authors has focused largely on the role of these molecules in cell migration, inhibitory antibodies towards urokinase will block EMT in vitro (Romano and Runyan, unpublished).
Extracellular Matrix in EMT
The structure of a cardiac cushion prior to EMT is essentially a large area of extracellular matrix (ECM) bounded by the myocardium and the endothelium. This ECM is rich in hyaluronan (HA), chondroitin sulfate proteoglycan, type VI collagen, fibulins, and fibrillins.66,67 In addition to serving as primitive valves, the cushion matrix is remodeled to form the mature valves and septum. Molecular genetic studies in the mouse demonstrate that both hyaluronan (HA) and versican, a HA binding proteoglycan, are required for formation of the cardiac cushions. Animals lacking these matrix molecules die at E9.5 to 10.5, shortly after the time of normal development of the AV and outflow tract cushions. Evidence for a critical role of versican in heart development was provided by the Heart Defect (hdf) transgenic mouse line.68,69 This line resulted from the transgenic insertion of a putative enhancer-lacZ reporter gene construct into the region encoding the glycosaminoglycan attachment domain of versican. Hdf mice do not survive past E10.5, and the AV and outflow tract cushions are absent.
The data support both space filling and ligand type roles for HA. While deficits in vivo correspond to a loss of space-filling HA, many of the in vitro studies suggest that a ligand type of role is equally important. For example, the removal of HA by hyaluronidase digestion of chick heart ECM produced an incomplete EMT in culture.10 ECM extracts were fully inductive when extracted without hyaluronidase.25 In whole rat embryo cultures, hyaluronidase blocked cardiac looping, cushion development and resulted in collapsed cushions.70 The addition of streptococcus hyaluronidase to normal E9.5 AV canal cushion mouse explants blocked endothelial transformation into mesenchyme where, presumably, space-filling is less important.71 Gene targeting of the principle hyaluronan synthase, Has2, also results in embryonic lethality ~E9.5 in part due to endocardial cushion defects both of the AV canal and OFT.72 Embryonic cushion cultures established from Has2-/- embryos in the collagen gel invasion assay lack mesenchyme formation which can be rescued by adding HA or reintroducing the Has2 gene into the explanted cultures.72 Finally, it was recently demonstrated that HA cooperates with growth factors to signal specified endocardium to transform into cushion mesenchyme.28 Together, these data suggest that both HA and versican are essential for formation of the acellular premigratory endocardial cushion cardiac jelly. It is not yet clear if other matrix components in this matrix also play essential roles, although collagen VI has been implicated in the cardiac defects common in trisomy 21 (Down Syndrome).73
In addition to space-filling and ligand roles, components of the ECM provide a motility substrate for the cell migration component of EMT. The effect of adhesion signals on cellular behavior is complex. Cell migration shows a biphasic dependence on adhesion, with maximal cell movement occurring at intermediate adhesion levels.74 This balance requires exquisite control of cellular activities with the ECM. In this regard, integrins are a well-characterized family of cell surface receptors capable of mediating these activities. Integrins are heterodimers composed of α and β subunits that function as bidirectional signaling molecules.75 Variation in levels of specific matrix molecules has been demonstrated to govern integrin-mediated cell migration.76 Modulation of integrin-ECM binding affinity,77-79 and changes in levels of integrins expressed on the cell surface can alter cell migration rates.80 Integrin binding of the ECM is well known to initiate signals that are transmitted into the cell altering cell adhesion, migration, proliferation, differentiation, and cell survival.81 Loeber et al82 used the collagen gel assay to directly examine the effects of disrupting integrin-ECM binding during AV canal EMT. Function blocking β1 integrin antibodies, RGD peptides against FN, and YIGSR peptides against laminin blocked EMT and cell migration. These observations demonstrate that β1 integrin binding to the ECM is essential for AV canal EMT. Studies are now investigating distinct integrin heterodimer modulation of AV canal EMT.
Other Growth Factors in EMT
As described in chapter 19: Klymkovsky, in this volume, Wnt proteins are associated with several EMTs during embryonic development. A recent report of the expression of the soluble Wnt inhibitor, FrzB, showed expression in the cardiac cushions.83 While such expression may indicate a role in EMT for Wnts, it is conceivable that Wnt activity mediates other developmental events in the heart. To test the activity of Wnts in cardiac EMT, several antisense oligonucleotides were prepared against FrzB. Collagen gel experiments with these oligonucleotides showed a two-fold increase in the number of mesenchymal cells compared to controls. Conversely, the application of mouse FRP-3, a homologue of FrzB, to collagen gel cultures blocked EMT (Person, Klewer and Runyan, in preparation). Preliminary experiments have identified 6 different Wnt proteins in the heart at the time of EMT. Experiments are underway to identify which of these are involved in EMT (unpublished data).
The TGFβisoforms are not the only members of the TGFβsuperfamily that appear to play a role in cardiac EMT. Lyons et al84 observed that BMP4 (formerly BMP2A) was expressed in a collar of myocardium around the AV canal in the mouse embryo at the time of EMT. While it was tempting to suggest a functional role in EMT in the mouse, null animals for BMP4 died around the time of EMT and it was difficult to determine whether normal EMT had taken place. More recently, expression in the AV canal of the mouse heart and the chick heart that suggested both BMP2 and BMP4 might be involved. Nakajima and coworkers85,86 found that BMP2 was synergistic with TGFβ3 in promoting EMT in chick AV canal tissue cultures. Further, BMPs 5, 6 and 7 have all been found in the heart.87 However, experiments utilizing misexpression of the BMP inhibitor, Noggin, and mutations in either BMP receptors or BMPs 6 and 7 have consistently shown stronger effects in the outflow tract than the AV canal.87,88 These data suggest that EMT in the outflow tract of the heart may be somewhat differently regulated.
Signal Transduction During EMT
Studies of signal transduction during EMT began with the observation that pertussis toxin was a potent blocker of EMT. Pertussis toxin ADP-ribosylates the alpha subunit of G proteins in the Gi or Gq class of G proteins. This inactivates and inhibits the functions of these molecules. 89 This study also showed that EMT was also inhibited by inhibitors of protein kinase C and serine-threonine kinase (and that activation of endothelial cells was accompanied by a flux in intracellular calcium.89 Subsequent studies of signal transduction in the heart focused on the TGFβ receptors described above. Evidence for ErbB and Ras function during EMT suggests that these signal transduction mechanisms are also involved see also chapter 17: Boyer.28 Though not yet integrated into a adequate picture of EMT in the heart, a modulator of Ras signaling, NF-1, is known to be present and critical in this system for normal valve development.90
Cell-cell Regulation of EMT
Early in cardiac development distinct cadherin subtypes mediate adhesion and segregation of endocardial and myogenic precursors into the heart forming fields.91,92 Following fusion of the paired precardiac mesoderm, the primary heart tube consists of an endocardium lining the lumen, and an outer myocardium. Adhesion between neighboring cells of the endocardium is mediated by homophilic interaction of VE-cadherin and PECAM-1D`and in the myocardium by N-cadherin.93 VE-cadherin/catenin complex is found in adherens junctions of vascular endothelium and has been show to inhibit migration and proliferation.94 Disruption of the cadherin/catenin complex is a critical step in the transformation of epithelium to mesenchyme.
During the transformation of endothelium to mesenchyme in the cardiac cushions, endothelial cells repress endothelial genes and activate expression of mesenchymal genes. The majority of endocardial cells will remain as VE-cadherin positive endothelial, and go on to express adult endothelial markers, such as factor vWF.95 A subset of cells will display a loss of cell-cell contact and will undergo an EMT to invade the underlying extracellular matrix. VE-cadherin mRNA expression is lost by endocardial cells that have undergone cell transformation in the endocardial cushion (Heimark, unpublished). PECAM-1 mRNA down regulation has been described in endocardial cells that have migrated to form mesenchymal cells in the AV canal (E11.5) during cell transformation.70 Regulation of the balance of multiple cell-cell adhesion molecules is likely to play a role in control of cell invasion during cell transformation.
Transcriptional Regulation of EMT
It is clear that EMT in the heart is a product of multiple inductive and permissive influences. One approach undertaken in this system has been to explore the transcriptional regulators required for EMT. The zinc finger transcription factor slug is required for EMT during neural crest migration and gastrulation in the chick embryo.96 Studies exploring a potential role for slug during endocardial cushion formation were undertaken to determine whether it is functional in this EMT as well. Antibody and antisense oligonucleotide studies showed that slug was located in the atrioventricular canal and that it was required for cell separation. Loss of slug expression produced a polygonal endothelial morphology consistent with that previously obtained by treatment with antiTGFβ2.97 Subsequent experiments showed that expression of slug in collagen gel cultures could overcome inhibition by anti-TGFβ2 antibody.98 These data suggest that regulation of slug expression is the major function of TGFβ2 in the atrioventricular canal. While slug is likely to transcriptionally regulate several molecules, it has been implicated in the loss of cadherins during cell transformation. Slug and several related molecules, snail, zeb1 and zeb2, bind to the Ebox elements to repress E-cadherin see also chapter 11: Berx et Van Roy.99 It has recently been shown that snail is also a positive regulator of matrix metalloprotease-2 (MMP-2) expression during snail-induced EMT in carcinoma cells.100 This enzyme is required for mesenchymal cell migration in the AV canal and may be concomitantly regulated by Slug.64
A second transcription factor, Mox-1 (Meox-1) was first identified in the mouse as a marker of mesenchymal cells throughout the embryo (including in the outflow tract cushions.101 The avian version of this molecule was cloned and its distribution and function was explored in the chick heart (Huang and Runyan, submitted; Wendler, Klewer and Runyan, submitted). Unlike Mox-1 in the mouse, we found this molecule distributed in the endothelium and the myocardium of the chick. Antisense oligonucleotides towards Mox-1 specifically blocked EMT in the collagen gel assays after cell separation but prior to invasion. As this pattern was similar to the effects of anti-TGFβ3 we determined that Mox-1 expression could be blocked by anti-TGFβ3. However, overexpression of Mox-1 in endothelial cells was not sufficient to cause EMT.
Several additional transcription factors critical to EMT are less well characterized but likely involved. It was observed that large T mouse mutants had defects in AV canal development.102 The T gene was subsequently identified as brachyury, the prototype of the Tbx family of transcription factors.103 In situ hybridization shows that cells undergoing EMT in the heart express brachyury (Huang and Runyan, unpublished) but experiments with antisense techniques have not yet identified the specific role played by brachyury in the AV canal. NFATc1 was identified as an expressed transcription factor in the AV canal and Outflow tract cushions of the mouse.104,105 However, there is some divergence of opinion as to whether NFATc1 plays a functional role in AV canal development. This transcription factor interacts with calcineurin in gene regulation106 but experiments to date with cyclosporine as an inhibitor of this pathway have shown no effect on AV canal EMT (Runyan, unpublished data). Additional transcription factors expressed in the AV canal but without a clear understanding of a role in EMT include the bHLH repressor, Id2, and GATA4.107,108
Clinical Significance of EMT in the Heart
The formation of valves in the heart requires precise interactions among multiple cell types. The high frequency of congenital heart defects (CHD) reflects the complexity of these developmental events. One of the most common, significant forms of CHD are atrioventricular septal defects (AVSD). AVSD consist of a deficiency of the inlet ventricular septum, a common AV valve, and an incomplete atrial septum primum. This anatomy is reminiscent of a 3-4 month human embryonic heart, and suggests that a developmental perturbation of normal morphogenetic pathways might be responsible for this CHD. AVSD communications between the right and left sides of the heart lead to symptoms of heart failure shortly after birth; surgery is usually required by 6 months of age. AVSD account for approximately 10% of all CHD and are the second most common CHD repaired in the first year of life.109,110 Post-operatively, these patients require close follow-up with a likelihood of additional surgeries for AV valve insufficiency or pulmonary hypertension.
Nearly 70 percent of AVSD are diagnosed in infants with trisomy 21 (Down syndrome).111 Likewise, the majority of CHD diagnosed in trisomy 21 infants are AVSD.110,112 This association has led to speculation that chromosome 21 genes are important in AV valve development. While most trisomies include an entire extra chromosome 21, partial trisomies have been useful in identifying a region of this chromosome that corresponds specifically to CHD in these patients.111 What is striking is that the candidate genes in this region include both cell adhesion molecules (CAMs) and ECM molecules (Collagen VI chains). This observation fits with the observations of Kurnit and colleagues113 that fibroblasts from trisomy 21 patients are more adhesive and that increased adhesiveness could account for altered morphology in the AV canal.114
There are likely additional forms of CHD attributable to perturbation of EMT in the heart. Sheffield and colleagues115 identified a large family with inherited endocardial cushion defects. While identification of the specific basis for this problem has eluded detection, the defect has been mapped to a region of chromosome 1 in the vicinity of the TGFβType III receptor. The power of genetics to identify candidate molecules and our ability to test for function in AV canal cultures should be productive in identifying additional mediators of CHD.
Questions and Future Directions
There are several questions raised by these studies and those of our colleagues in other chapters of this work. Among these, what are the common elements of EMT and are there specific mechanisms or molecules that are common to all or many of the EMTs observed in the embryo. As perusal of this volume suggests, there appear to be a variety of different regulators and it is clear that not all EMTs are identical. We have begun to systematically test a number of molecules found in other embryonic EMTs as found in a survey of embryonic gene expression (http://geisha.biosci.arizona.edu). To date, it appears that some of these molecules will be functional in the heart and others will not be. The characterization of these molecules may enable the clustering of EMTs into classes and shed light on pathologic EMTs in postnatal life.
While we have a basic understanding of TGFβmediated EMT in the chick heart, there are some significant differences between the mouse and the chick. We are very interested in understanding the basis for this difference and whether the human uses TGFβisoforms more like the chick or the mouse. Confounding analysis of EMT in the AV canal is evidence in both species that there are a great number of ligands, receptors and signal transduction mechanisms functional in the heart. The complexity appears to increase on a daily basis. We have embarked on a microarray analysis of transcriptional regulation for many of the identified signal transduction processes in the heart. It is our hope that a bioinformatics approach can help identify the intersection or independence of the various pathways.
Together, these approaches should be relevant to the entire field of EMT as represented in this volume and the usefulness of the heart in resolving aspects of this field will be validated.
Acknowledgements
The authors thank Shannon Shoemaker for drawing the illustrations used in figures 1-3. Original research in each of the author's laboratories was supported by a program project grant from the National Heart, Lung and Blood Institute of the National Institutes of Health (P01 HL63926).
References
- 1.
- Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77(1):1–6. [PubMed: 7788867]
- 2.
- Barry A. The functional significance of the cardiac jelly in the tubular heart of the chick embryo. Anat Rec. 1948;102:289–298. [PubMed: 18098957]
- 3.
- Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148(1):85–120. [PubMed: 842477]
- 4.
- Markwald RR, Fitzharris TP, Smith WN. Structural analysis of endocardial cytodifferentiation. Dev Biol. 1975;42(1):160–180. [PubMed: 1112439]
- 5.
- Manasek F. Glycoprotein synthesis and tissue interactions during establishment of the functional embryonic chick heart. J Mol Cell Cardiol. 1976;8:389–402. [PubMed: 945841]
- 6.
- Manasek F. Heart development: Interactions involved in cardiac morphogenesisIn: Poste G, Nicholson G, eds. The Cell Surface in Animal Embryogenesis and Development Elsevier/North-Holland Biomedical Press, 1976545–598.
- 7.
- Kinsella M, Fitzharris T. Origin of cushion tissue in the developing chick heart: Cinematographic recordings of in situ formation. Science. 1980;207:1359–1360. [PubMed: 7355294]
- 8.
- van den Hoff MJB, Moorman AFM, Ruijter JM. et al. Myocardialization of the cardiac outflow tract. Dev Biol. 1999;212(2):477–490. [PubMed: 10433836]
- 9.
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed: 24539719]
- 10.
- Krug EL, Runyan RB, Markwald RR. Protein extracts from early embryonic hearts initiate cardiac endothelial cytodifferentiation. Dev Biol. 1985;112(2):414–426. [PubMed: 3935503]
- 11.
- Markwald RR, Fitzharris T, Bolender DL. et al. Structural analysis of cell: Matrix association during the morphogenesis of atrioventricular cushion tissue. Anat Rec. 1979 [PubMed: 437356]
- 12.
- Bolender DL, Markwald RR. Epithelial-mesenchymal transformation in chick atrioventricular cushion morphogenesis. SEM. 1979;(3):313–321.
- 13.
- Icardo JM, Ojeda JL, Hurle JM. Endocardial cell polarity During the looping of the heart in the chick-embryo. Dev Biol. 1982;90(1):203–209. [PubMed: 7060832]
- 14.
- Markwald RR, Fitzharris T, Bank H. et al. Structural analysis on the matrical organization of glycosaminoglycans in developing endocardial cushions. Dev Biol. 1978;62(2):292–316. [PubMed: 564303]
- 15.
- Klewer SE, Krob SL, Kolker SJ. et al. Expression of type VI collagen in the developing mouse heart. Dev Dyn. 1997;in press [PubMed: 9520112]
- 16.
- De La Cruz M, Gimenez-ribotta M, Saravalli O. The contribution of the inferior endocardial cushion of the atrioventricular canal to cardiac septation and to the development of the atrioventricular valve: Study in the chick embryo. Am J Physiol Regul Integr Comp Physiol. 1983;166:63–72. [PubMed: 6837479]
- 17.
- Elsdale T, Bard J. Collagen substrata for studies on cell behavior. Journal of Cell Biology. 1972;54:626–637. [PMC free article: PMC2200288] [PubMed: 4339818]
- 18.
- Schor S. Cell proliferation and migration on collagen substrata in vitro. Journal of Cell Science. 1980;41:159–175. [PubMed: 7364880]
- 19.
- Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. Journal of Cell Biology. 1982;95:333–339. [PMC free article: PMC2112361] [PubMed: 7142291]
- 20.
- Bernanke DH, Markwald RR. Migratory behavior of cardiac cushion tissue-cells in a collagen-lattice culture system. Developmental Biology. 1982;91(2):235–245. [PubMed: 7095266]
- 21.
- Bernanke DH, Markwald RR. Effects of hyaluronic acid on cardiac cushion tissue cells in collagen matrix cultures. Texas Reports in Biology and Medicine. 1979;39:271–285. [PubMed: 553314]
- 22.
- Overton J. Differential response of embryonic-cells to culture on tissue matrices. Tissue Cell. 1979;11(1):89–98. [PubMed: 451999]
- 23.
- Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: A regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol. 1983;95(1):108–114. [PubMed: 6825921]
- 24.
- Mjaatvedt CH, Lepera RC, Markwald RR. Myocardial specificity for initiating endothelial-mesenchymal cell transition in embryonic chick heart correlates with a particulate distribution of fibronectin. Dev Biol. 1987;119(1):59–67. [PubMed: 3539667]
- 25.
- Krug EL, Mjaatvedt CH, Markwald RR. Extracellular matrix from embryonic myocardium elicits an early morphogenetic event in cardiac endothelial differentiation. Dev Biol. 1987;120(2):348–355. [PubMed: 3556758]
- 26.
- Mjaatvedt CH, Markwald RR. Induction of an epithelial-mesenchymal transition by an in vivo adheron-like complex. Dev Biol. 1989;136(1):118–128. [PubMed: 2509260]
- 27.
- Potts JD, Runyan RB. Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor beta. Dev Biol. 1989;134(2):392–401. [PubMed: 2744239]
- 28.
- Camenisch TD, Schroeder JA, Bradley J. et al. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat Med. 2002;8(8):850–855. [PubMed: 12134143]
- 29.
- Walsh E, Stainier DYR. Novel role for glycosaminoglycans in cell signaling events during heart valve initiation: Cloning of the zebrafish jekyll mutation. Dev Biol. 2001;235(1):229–229.
- 30.
- Gassmann M, Casagranda F, Orioli D. et al. Aberrant neural and cardiac development in mice lacking the Erbb4 neuregulin receptor. Nature. 1995;378(6555):390–394. [PubMed: 7477376]
- 31.
- Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378(6555):386–390. [PubMed: 7477375]
- 32.
- Mjaatvedt CH, Krug EL, Markwald RR. An antiserum (ES1) against a particulate form of extracellular matrix blocks the transition of cardiac endothelium into mesenchyme in culture. Dev Biol. 1991;145(2):219–230. [PubMed: 2040370]
- 33.
- Sinning AR, Krug EL, Markwald RR. Multiple glycoproteins localize to a particulate form of extracellular matrix in regions of the embryonic heart where endothelial cells transform into mesenchyme. Anat Rec. 1992;232(2):285–292. [PubMed: 1546806]
- 34.
- Rezaee M, Isokawa K, Halligan N. et al. Identification of an extracellular 130-kDa protein involved in early cardiac morphogenesis. J Biol Chem. 1993;268(19):14404–14411. [PubMed: 7686155]
- 35.
- Isokawa K, Rezaee M, Wunsch A. et al. Identification of transferrin as one of multiple EDTA-extractable extracellular proteins involved in early chick heart morphogenesis. J Cell Biochem. 1994;54:207–218. [PubMed: 8175895]
- 36.
- Sinning AR. Partial purification of HLAMP-1 provides direct evidence for the multicomponent nature of the particulate matrix associated with cardiac mesenchyme formation. J Cell Biochem. 1997;66(1)(1):112–122. [PubMed: 9215533]
- 37.
- Roberts A, Flanders K, Kondaiah P. et al. Transforming growth factor β: Biochemistry and roles in embryogenesis, tissue repair and remodeling, and carcinogenesis. Recent Prog Horm Res. 1988;44:157–197. [PubMed: 3064207]
- 38.
- Nakajima Y, Krug EL, Markwald RR. Myocardial regulation of transforming growth factor-β-expression by outflow tract endothelium in the early embryonic chick heart. Dev Biol. 1994;165:615–626. [PubMed: 7958426]
- 39.
- Burt DW. Evolutionary grouping of the transforming growth factor-β-superfamily. Biochem Biophys Res Commun. 1992;184(2):590–595. [PubMed: 1575734]
- 40.
- Potts JD, Vincent EB, Runyan RB. et al. Sense and antisense Tgf-Beta-3 messenger-RNA levels correlate with cardiac-valve induction. Dev Dyn. 1992;193(4):340–345. [PubMed: 1511174]
- 41.
- Akhurst RJ, Lehnert S, Faissner A. et al. TGF beta in murine morphogenetic processes: The early embryo and cardiogenesis. Development. 1990;108(4):645–656. [PubMed: 1696875]
- 42.
- Dickson MC, Slager HG, Duffie E. et al. RNA and protein localisations of TFG beta 2 in the early mouse embryo. Development. 1993;117:625–639. [PubMed: 7687212]
- 43.
- Camenisch TD, Molin DGM, Person A. et al. Temporal and distinct TGF beta ligand requirements uring mouse and avian endocardial cushion morphogenesis. Dev Biol. 2002;248(1):170–181. [PubMed: 12142029]
- 44.
- Barnett JV, Moustakas A, Lin W. et al. Cloning and developmental expression of the chick type II and type III TGF beta receptors. Dev Dyn. 1994;199(1):12–27. [PubMed: 8167376]
- 45.
- Boyer AS, Ayerinskas II, Vincent EB. et al. TGF beta 2 and TGF beta 3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Biol. 1999;208(2):530–545. [PubMed: 10191064]
- 46.
- Potts JD, Dagle JM, Walder JA. et al. Epithelial mesenchymal transformation of embryonic cardiac endothelial-cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth-factor beta-3. Proc Natl Acad Sci U S A. 1991;88(4):1516–1520. [PMC free article: PMC51050] [PubMed: 1996351]
- 47.
- Diebold RJ, Eis MJ, Yin M. et al. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc Natl Acad Sci U S A. 1995;92(26):12215–12219. [PMC free article: PMC40327] [PubMed: 8618872]
- 48.
- Proetzel G, Pawlowski SA, Wiles MV. et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11(4):409–414. [PMC free article: PMC3855390] [PubMed: 7493021]
- 49.
- Sanford LP, Ormsby I, Gittenberger-de Groot AC. et al. TGFβ2 knockout mice have multiple development defects that are non overlapping with other TGFβ knockout phenotypes. Development. 1997;124:2659–2670. [PMC free article: PMC3850286] [PubMed: 9217007]
- 50.
- Massague J. TGFβsignaling: Receptors, transducers, and Mad proteins. Cell. 1996;85:947–950. [PubMed: 8674122]
- 51.
- Wrana JL, Attisano L, Wieser R. et al. Mechanism of activation of the TGF-βreceptor. Nature. 1994;370:341–347. [PubMed: 8047140]
- 52.
- Wrana JL, Attisano L, Carcamo J. et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. [PubMed: 1333888]
- 53.
- Lopez-Casillas F, Wrana JL, Massague J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell. 1993;73(7):1435–1444. [PubMed: 8391934]
- 54.
- Barnett JV, Moustakas A, Lin W. et al. Cloning and developmental expression of the chick type II and type III TGF. receptors. Developmental Dynamics. 1994;199:12–27. [PubMed: 8167376]
- 55.
- Brown CB, Boyer AS, Runyan RB. et al. Antibodies to the type II TGF beta receptor block cell activation and migration during atrioventricular cushion transformation in the heart. Dev Biol. 1996;174(2):248–257. [PubMed: 8631497]
- 56.
- Brown CB, Boyer AS, Runyan RB. et al. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science. 1999;283(5410):2080–2082. [PubMed: 10092230]
- 57.
- Boyer AS, Runyan RB. TGFβType III and TGFβType II receptors have distinct activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Dyn. 2001;221(4):454–459. [PubMed: 11500982]
- 58.
- Massague J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597–641. [PubMed: 2177343]
- 59.
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGF beta activation. J Cell Sci. 2003;116(2):217–224. [PubMed: 12482908]
- 60.
- Ghosh S, Brauer PR. Latent transforming growth factor-βis present in the extracellular matrix of embryonic hearts in situ. Dev Dyn. 1996;205(2):126–134. [PubMed: 8834473]
- 61.
- McCormick KM. TGF beta 2 activation status during cardiac morphogenesis. Dev Dyn. 2001;222(1):17–25. [PubMed: 11507766]
- 62.
- Nakajima Y, Miyazono K, Kato M. et al. Extracellular fibrillar structure of latent TGF beta binding protein-1: Role in TGF beta-dependent endothelial-mesenchymal transformation during endocardial cushion tissue formation in mouse embryonic heart. J Cell Biol. 1997;136(1):193–204. [PMC free article: PMC2132455] [PubMed: 9008713]
- 63.
- McGuire PG, Orkin RW. Urokinase activity in the developing avian heart: A spatial and temporal analysis. Dev Dyn. 1992;193:24–33. [PubMed: 1540703]
- 64.
- Alexander SM, Jackson KJ, Bushnell KM. et al. Spatial and temporal expression of the 72-kDa type IV collagenase (MMP-2) correlates with development and differentiation of valves in the embryonic avian heart. Dev Dyn. 1997;209(3):261–268. [PubMed: 9215641]
- 65.
- McGuire PG, Alexander SM. Urokinase production by embryonic endocardial-derived cells: Regulation by substrate composition. Dev Biol. 1993;155(2):442–451. [PubMed: 8432398]
- 66.
- Bouchey D, Argraves WS, Little CD. Fibulin-1, vitronectin, and fibronectin expression during avian cardiac valve and septa development. Anatomical Record. 1996;244(4):540–551. [PubMed: 8694289]
- 67.
- Kitten GT, Markwald RR, Bolender DL. Distribution of basement membrane antigens in cryopreserved early embryonic hearts. Anat Rec. 1987;217(4):379–390. [PubMed: 3592264]
- 68.
- Mjaatvedt CH, Yamamura H, Capehart AA. et al. The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev Biol. 1998;202(1):56–66. [PubMed: 9758703]
- 69.
- Yamamura H, Zhang M, Markwald RR. et al. A heart segmental defect in the anterior-posterior axis of a transgenic mutant mouse. Dev Biol. 1997;186(1):58–72. [PubMed: 9188753]
- 70.
- Baldwin HS, Lloyd TR, Solursh M. Hyaluronate degradation affects ventricular function of the early postlooped embryonic rat heart in situ. Circ Res. 1994;74(2):244–252. [PubMed: 8293563]
- 71.
- Camenisch TD, Biesterfeldt J, Brehm-Gibson T. et al. Regulation of cardiac cushion development by hyaluronan. Exp Clin Cardiol. 2001;6:4–10. [PMC free article: PMC2858958] [PubMed: 20428437]
- 72.
- Camenisch TD, Spicer AP, Brehm-Gibson T. et al. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106(3):349–360. [PMC free article: PMC314332] [PubMed: 10930438]
- 73.
- Klewer SE, Krob SL, Kolker SJ. et al. Expression of type VI collagen in the developing mouse heart. Dev Dyn. 1998;211(3):248–255. [PubMed: 9520112]
- 74.
- DiMilla PA, Stone JA, Quinn JA. et al. Maximal migration of human smooth muscle cells on fibronectin and type IV collagen occurs at an intermediate attachment strength. J Cell Biol. 1993;122(3):729–737. [PMC free article: PMC2119669] [PubMed: 8335696]
- 75.
- McDonald JA. Integrins minireview series. J Biol Chem. 2000;275(29):21783. [PubMed: 10807935]
- 76.
- Palecek SP, Loftus JC, Ginsberg MH. et al. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness.[see erratum] Nature. 1997;388(6638):537–540. [PubMed: 9020360]
- 77.
- Akiyama SK, Nagata K, Yamada K. Cell surface receptors for extracellular matrix components. Biochim Biophys Acta. 1990;1031:91–110. [PubMed: 1689589]
- 78.
- Duband JL, Dufour S, Yamada SS. et al. Neural crest cell locomotion induced by antibodies to beta 1 integrins. A tool for studying the roles of substratum molecular avidity and density in migration. Journal of Cell Science. 1991;98(Pt 4):517–532. [PubMed: 1713595]
- 79.
- Kuijpers TW, Mul EP, Blom M. et al. Freezing adhesion molecules in a state of high-avidity binding blocks eosinophil migration. J Exp Med. 1993;178(1):279–284. [PMC free article: PMC2191076] [PubMed: 7686213]
- 80.
- Keely PJ, Fong AM, Zutter MM. et al. Alteration of collagen-dependent adhesion, motility, and morphogenesis by the expression of antisense alpha 2 integrin mRNA in mammary cells. J Cell Sci. 1995;108(Pt 2):595–607. [PubMed: 7769004]
- 81.
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285(5430):1028–1032. [PubMed: 10446041]
- 82.
- Loeber CP, Runyan RB. A comparison of fibronectin, laminin, and galactosyltransferase adhesion mechanisms during embryonic cardiac mesenchymal cell-migration in vitro. Dev Biol. 1990;140(2):401–412. [PubMed: 2142656]
- 83.
- Ladher RK, Church VL, Allen S. et al. Cloning and expression of the Wnt antagonists Sfrp-2 and Frzb during chick development. Dev Biol. 2000;218(2):183–198. [PubMed: 10656762]
- 84.
- Lyons KM, Pelton RW, Hogan BL. Organogenesis and pattern formation in the mouse: RNA distribution patterns suggest a role for bone morphogenetic protein-2A (BMP-2A). Development. 1990;109(4):833–844. [PubMed: 2226202]
- 85.
- Yamagishi T, Nakajima Y, Miyazono Ketal. Bone morphogenetic protein-2 acts synergistically with transforming growth factor-beta 3 during endothelial- mesenchymal transformation in the developing chick heart. Journal of Cellular Physiology Jul. 1999;180(1):35–45. [PubMed: 10362015]
- 86.
- Nakajima Y, Yamagishi T, Hokari S. et al. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: Roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP). Anat Rec. 2000;258(2):119–127. [PubMed: 10645959]
- 87.
- Kim RY, Robertson EJ, Solloway MJ. Bmp6 and Bmp7 are required for cushion formation and septation in the developing mouse heart. Dev Biol. 2001;235(2):449–466. [PubMed: 11437450]
- 88.
- Allen SP, Bogardi JP, Barlow AJ. et al. Misexpression of noggin leads to septal defects in the outflow tract of the chick heart. Dev Biol. 2001;235(1):98–109. [PubMed: 11412030]
- 89.
- Runyan RB, Potts JD, Sharma RV. et al. Signal transduction of a tissue interaction during embryonic heart development. Cell Regul. 1990;1(3):301–313. [PMC free article: PMC361475] [PubMed: 2129222]
- 90.
- Lakkis MM, Epstein JA. Neurofibromin modulation of ras activity is required for normal endocardial-mesenchymal transformation in the developing heart. Devel Suppl. 1998;125(22):4359–4367.
- 91.
- Garcia-Castro MI, Vielmetter E, Bronner-Fraser M. N-cadherin, a cell adhesion molecule involved in establishment of embryonic left-right asymmetry. Science. 2000;288(5468):1047–1051. [PubMed: 10807574]
- 92.
- Breier G, Albrecht U, Sterrer S. et al. Expression of vascular endothelial growth-factor during embryonic angiogenesis and endothelial-cell differentiation. Development. 1992;114(2):521–532. [PubMed: 1592003]
- 93.
- Linask KK, ImanakaYoshida K, Knudsen KA. N-cadherin/beta-catenin interaction is necessary for chick cardiomyocyte compartmentalization and myofibrillogenesis. Dev Biol. 1997;186(2):B65–B65.
- 94.
- Caveda L, Martin-Padura I, Navarro P. et al. Inhibition of cultured cell growth by vascular endothelial cadherin (cadherin-5/VE-cadherin). J Clin Invest. 1996;98(4):886–893. [PMC free article: PMC507501] [PubMed: 8770858]
- 95.
- Coffin JD, Harrison J, Schwartz S. et al. Angioblast differentiation and morphogenesis of the vascular endothelium in the mouse embryo. Dev Biol. 1991;148(1):51–62. [PubMed: 1936575]
- 96.
- Nieto MA, Sargent MG, Wilkinson DG. et al. Control of cell behavior during vertebrate development by slug, a zinc finger gene. Science. 1994;264:835–839. [PubMed: 7513443]
- 97.
- Romano LA, Runyan RB. Slug is a mediator of epithelial-mesenchymal cell transformation in the developing chicken heart. Dev Biol. 1999;212(1):243–254. [PubMed: 10419699]
- 98.
- Romano LA, Runyan RB. Slug is an essential target of TGFbeta2 signaling in the developing chicken heart. Developmental Biology. 2000;223(1):91–102. [PubMed: 10864463]
- 99.
- Cano A, Perez-Moreno MA, Rodrigo I. et al. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2(2):76–83. [PubMed: 10655586]
- 100.
- Yokoyama K, Kamata N, Fujimoto R. et al. Increased invasion and matrix metalloproteinase-2 expression by Snail-induced mesenchymal transition in squamous cell carcinomas. Int J Oncol. 2003;22(4):891–898. [PubMed: 12632084]
- 101.
- Candia AF, Hu J, Crosby J. et al. Mox-1 and Mox-2 define a novel homeobox gene subfamily and are differentially expressed during early mesodermal patterning in mouse embryos. Development. 1992;116(4):1123–1136. [PubMed: 1363541]
- 102.
- Bayna EM, Runyan RB, Scully NF. et al. Cell-surface galactosyltransferase as a recognition molecule during development. Mol Cell Biochem. 1986;72(1-2):141–151. [PubMed: 3102942]
- 103.
- Herrmann BG, Lehrach H. From phenotype to gene: Molecular cloning in the Brachyury (T) locus region. Curr Top Microbiol Immunol. 1988;137:77–81. [PubMed: 3166419]
- 104.
- de la Pompa JL, Timmerman LA, Takimoto H. et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum [see comments] Nature. 1998;392(6672):182–186. [PubMed: 9515963]
- 105.
- Ranger AM, Grusby MJ, Hodge MR. et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392(6672):186–190. [PubMed: 9515964]
- 106.
- Molkentin JD, Lu JR, Antos CL. et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93(2):215–228. [PMC free article: PMC4459646] [PubMed: 9568714]
- 107.
- Evans SM, Obrien TX. Expression of the helix-loop-helix factor id during mouse embryonic-development. Developmental Biology Oct. 1993;159(2):485–499. [PubMed: 8405673]
- 108.
- Kelley C, Blumberg H, Zon LI. et al. GATA-4 is a novel transcription factor expressed in endocardium of the developing heart. Development. 1993;118(3):817–827. [PubMed: 8076520]
- 109.
- Ferencz C, Rubin JD, Mccarter RJ. et al. Congenital heart disease: Prevalence at livebirth; the BaltimoreWashington infant study. Am J Epidem. 1990;121:31–36. [PubMed: 3964990]
- 110.
- Carmi R, Boughman JA, Ferencz C. Endocardial cushion defect: Further studies of “isolated” versus “syndromic” occurrence. American Journal of Medical Genetics. 1992;43(3):569–575. [PubMed: 1534968]
- 111.
- Korenberg JR, Bradley C, Disteche CM. Down Syndrome: Molecular mapping of the congenital heart disease and duodenal stenosis. Am J Hum Genet. 1992;50:294–302. [PMC free article: PMC1682442] [PubMed: 1531166]
- 112.
- Adams MM, Erickson JD, Layde PM. et al. Down's syndrome. Recent trends in the United States. JAMA. 1981;246(7):758–760. [PubMed: 6454794]
- 113.
- Kurnit D, Aldridge J, Matsuoka R. et al. Increased adhesiveness of trisomy 21 cells and atrioventricular canal malformations in down syndrome: A stochastic model. Am J Med Genet. 1985;20:385–399. [PubMed: 3156496]
- 114.
- Jongewaard IN, Lauer RM, Behrendt DA. et al. Beta 1 integrin activation mediates adhesive differences between trisomy 21 and nontrisomic fibroblasts on type VI collagen. Am J Med Genet. 2002;109(4):298–305. [PubMed: 11992484]
- 115.
- Sheffield VC, Pierpont ME, Nishimura D. et al. Identification of a complex congenital heart defect susceptibility locus by using DNA pooling and shared segment analysis. Hum Mol Genet. 1997;6(1):117–121. [PubMed: 9002679]
- Introduction
- Description of EMT In Vivo
- Development of Collagen Gel Culture
- Identification of Components of the EMT Process
- TGFβas a Mediator of EMT
- TGFβReceptors
- TGFβActivation
- Extracellular Matrix in EMT
- Other Growth Factors in EMT
- Signal Transduction During EMT
- Cell-cell Regulation of EMT
- Transcriptional Regulation of EMT
- Clinical Significance of EMT in the Heart
- Questions and Future Directions
- Acknowledgements
- References
- Epithelial-Mesenchymal Transformation in the Embryonic Heart - Madame Curie Bios...Epithelial-Mesenchymal Transformation in the Embryonic Heart - Madame Curie Bioscience Database
- Inferring Evolutionary History through Inter- and Intraspecific DNA Sequence Com...Inferring Evolutionary History through Inter- and Intraspecific DNA Sequence Comparison: The Drosophila janus and ocnus Genes - Madame Curie Bioscience Database
- SELECTIVE NEURODEGENERATION, NEUROPATHOLOGY AND SYMPTOM PROFILES IN HUNTINGTON'S...SELECTIVE NEURODEGENERATION, NEUROPATHOLOGY AND SYMPTOM PROFILES IN HUNTINGTON'S DISEASE - Madame Curie Bioscience Database
- The Restriction Point of the Cell Cycle - Madame Curie Bioscience DatabaseThe Restriction Point of the Cell Cycle - Madame Curie Bioscience Database
- RNA Modification Subsystems in the SEED Database - Madame Curie Bioscience Datab...RNA Modification Subsystems in the SEED Database - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...