

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Mucosal surfaces represent the major interface between host and environment. They constitute the point of entry of most infectious pathogens, and are in contact with potentially injurious antigens present in the normal mucosal microflora and in ingested or inhaled substances. To deal appropriately with this challenge, the host immune system rely on both cell-mediated and humoral responses. Whereas cell-mediated responses involve a range of different effector cells,1,2 the humoral immune defense at mucosal level is mediated predominantly by antibodies of the immunoglobulin (Ig) A isotype. The mucosal immune system contains more than 80% of all Ig-producing cells in the body, and the major product of these cells in normal individuals is IgA.3 In the circulation, IgA is the second most abundant Ig class, its concentration (˜2 mg/ml) being surpassed only by that of IgG (˜12 mg/ml). Considering the distribution in various body fluids of the major Ig isotypes and their catabolic rates, IgA is clearly synthesized in quantities (˜66 mg/kg body weight/day) that exceed by far the combined daily synthesis of all other isotypes.4,5
The mucosal immune system has a unique anatomical and functional organisation. IgA is present in several molecular forms and displays biological properties not shared by other immunoglobulin classes. IgA antibodies perform their protective functions discretely without interfering with the physiological activities of mucosal membranes. Moreover, IgA is believed to mitigate, when appropriate, the activity of phlogistic, potentially injurious immune defence mechanisms and to contribute in this way to mucosal homeostasis. However, a major question in mucosal immunity remains: how does IgA, and the mucosal immune system in general, discriminate between innocuous dietary components or commensal bacteria, and pathogens that represent a threat to the individual's health.
Mucosal IgA antibody responses have been comprehensively studied, particularly with respect to a potential exploitation for immune prophylaxis. Less is known of the immune-modulating properties of IgA, and few studies have directly addressed the relationship of IgA antibodies to the concept of oral (or mucosal) tolerance. In this chapter, we will describe fundamental aspects of the mucosal IgA system and discuss current conceptions regarding the biological functions of IgA antibodies. A final section will review briefly some aspects of oral tolerance related to B cell reactions and IgA antibody responses.
Structure of IgA
IgA is the most heterogeneous immunoglobulin isotype. It is present in a variety of molecular forms, subclasses and allotypes, with patterns of heterogeneity that vary between different species of mammals and birds.6 Early studies revealed that the variation in molecular form is intimately related to the fact that IgA is not only present in the circulation and tissues, but is also the predominant immunoglobulin in external secretions, in the form of secretory IgA (S-IgA).7,8 Secretory IgA exerts biological activities different from those of IgA in the circulation and tissues. The functional significance of structural variation in terms of IgA subclasses and allotypes, however, is poorly understood.
In the serum of adults, IgA is found in concentrations ranging from 0.7 to 3.4 mg/ml,9 and is mainly present (> 80%) in the form of monomeric IgA (mIgA) composed of two α (Mr ˜53,000) and two κ or λ (Mr ˜22,500) chains. Human α chains are glycoproteins (6 to 10% carbohydrate) consisting of one variable and three constant domains. The remaining part of serum IgA is in a polymeric, mainly dimeric form, with monomers connected by disulphide bonds and linked to an additional polypeptide called J chain (Mr ˜16,000; 8% carbohydrate). The J chain is present also in IgM (pentameric) and appears to play a regulatory—though not indispensable—role in the polymerisation of these isotypes.10
S-IgA is a molecule of dual cellular origin. It consists of a polymeric, J chain-containing IgA disulphide-linked to another glycoprotein, secretory component (SC; Mr ˜70,000). Whereas polymeric IgA is produced by local plasma cells, SC originates from secretory epithelial cells. SC consists of five immunoglobulin-like domains and is highly glycosylated (22%). It is a fragment of the polyimmunoglobulin receptor (pIgR), which serves as a vehicle for the transport of polymeric, J chain-containing immunoglobulins (polymeric IgA and IgM) through the polarized epithelial cells that line mucosal surfaces and constitute the secretory cells of mucosal glands. pIgR-mediated IgM transport is quantitatively insignificant in normal, IgA-producing individuals. In most IgA-deficient individuals, though, secretory IgM (S-IgM) substitutes for S-IgA in the secretions, which may explain why many IgA-deficient individuals remain healthy.11
The perpetual regulated transfer of pIgR through secretory epithelial cells is a key element of humoral mucosal immunity (for review, see ref. 12). Briefly, pIgR, synthesized as an integral membrane protein in the rough endoplasmic reticulum, is delivered to the baso-lateral surface of the epithelial cell where it binds pIgA produced by the plasma cell in the underlying lamina propria. pIgR is endocytosed and travels by vesicular transport to the apical plasma membrane. There, the extracellular ligand-binding portion of pIgR (the future SC) is cleaved off and released with its potential Ig ligand. The binding of pIgR (and SC) to immunoglobulins appears to depend on the presence of the J chain, although SC and J-chain are not mutually linked by covalent bonds (both polypeptides are disulphide-attached to the C-terminal half of the α chain). In addition to its role in IgA secretion, SC confers functional advantages to the S-IgA molecule.
Secretions collected at some mucosal surfaces, particularly the airways and the female genital tract, contain significant proportions of mIgA.13,14 Like other systemic immunoglobulins, mIgA reach the secretion by passive transudation, which reflects the degree of mucosal inflammation.
IgA Subclasses and Allotypes
In humans, hominoid primates, and lagomorphs (including rabbits), several IgA subclasses have been described on the basis of their α chain isotype. Within a given species, the α chains belonging to each subclass are highly homologous in amino acid sequence. Two subclasses, IgA1 and IgA2, have been identified in human serum and secretions. A major difference between the two subclasses is found in the hinge region: IgA2 molecules lack a 13 amino acid segment, which is present in the hinge region of IgA1 and consists exclusively of prolyl, seryl, and threonyl residues. Four to five of the seryl and threonyl residues carry O-linked glycans.15,16 This extended hinge region confers greater segmental flexibility on IgA1 molecules, but renders this isotype susceptible to the activity of certain post-proline endopeptidases, called IgA1 proteases, which are produced by bacterial pathogens and commensals colonizing the mucosa (see below). These enzymes, by their ability to cleave IgA1 molecules into intact Fc (or Fc.SC) and monovalent Fab fragments, seem to constitute means of evading IgA antibody-mediated defence (reviewed by Kilian et al17). For this particular reason, IgA2 has been hypothesized to be the phylogenetically youngest of the IgA subclasses, selected for its resistance to bacterial IgA1 protease activity. This hypothesis, however, was proven wrong when phylogenetical studies of the IgA subclasses in hominoid primates and humans demonstrated that IgA1 evolved subsequently to IgA2 through events of gene-duplication and conversion.18 The human IgA subclasses also differ at 14 amino acid positions in the α chain sequence, and in N-linked glycans.
The two IgA subclasses are unequally distributed in the systemic and mucosal lymphoid compartments. IgA1 is vastly predominant in the serum (>85%), consistently with a similar predominance of IgA1-producing cells in the bone marrow, where most serum IgA is produced.19,20,21 In secretions, however, proportions of IgA2 vary from 10% in the upper respiratory tract secretions, to 30-40% in the saliva and the small intestine, and more than 50% in the secretions of the large intestine and female genital tract, reflecting the subclass distribution of IgA-producing plasma cells in the local mucosa.19,21,22 The local distribution of IgA1- and IgA2-producing cells may be the result of subclass-specific clonal expansion induced by certain types of antigens.10,23 However, the factors that determine the subclass of IgA antibodies are still partly unknown.24
The human IgA2 subclass exists in two allotypic forms designated A2m(1) and A2m(2).25,26 The A2m(1) allotype is unconventional in that the heavy (H) and light (L) chains are not covalently linked, and therefore can be separated by non-reducing dissociating agents. Population studies of the IgA2 allotypes have revealed a characteristic racial and ethnic distribution. The A2m(1) allotype is highly predominant in caucasians whereas A2m(2) dominates in people of African origin.25,26
Glycan Moiety
The glycan moiety of α chains, which is of significance to the function of IgA antibodies, has been analysed in detail. In addition to the contrasting presence of glycans in the α1 and α2 hinge regions mentioned above, early studies reviewed by Mestecky et al10 found a N-glycosylation at two Asn residues located in CH2 and in the very C-terminal “tail-piece” of IgA1 and IgA2. In IgA2, two or three additional sites of N-glycosylation have been identified in the Fc. A recent study reported considerable O- and N-glycosylation in Fab of polyclonal serum IgA1.16 While the short O-linked glycans in the hinge of IgA1 contain GalNAc, Gal, and frequently terminal NeuNAc, N-linked glycans in Fc of IgA are of the complex, multiantennary type. Unlike the corresponding N-linked glycans in IgG1, confined to the space between the two heavy chains, those of IgA are directed away from the protein backbones and are highly sialylated.16 With this make up, the glycans presumably confer hydrophilicity and negative net charge to the IgA molecule. Also at variance with the situation for IgG, IgA glycans are not involved in interactions with the isotype-specific Fc receptor (CD89 in the case of IgA) that triggers IgA-mediated defence reactions in granulocytes and macrophages.16,27,28 N-linked glycans of the high-mannose type have been identified in both IgA subclasses. IgA molecules carrying such glycans may interact through their terminal mannose residues with corresponding receptors (type 1 fimbriae) on certain bacteria and inhibit their adherence to epithelial cells.29,30
The total carbohydrate content of S-IgA is considerably higher than that of serum IgA because both J chain and SC are rich in carbohydrates. Moreover, glycans of both components are sialylated.31,32 Accordingly, these components can be expected to add significantly to the negative charge and hydrophilicity of S-IgA molecules.
Induction of IgA Responses
Antigen Handling and Reactions in Mucosa-Associated Lymphoid Tissue (MALT)
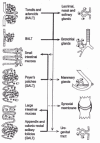
Any site on mucosal membranes may under various circumstances permit the entry of exogenous material, including potential immunogens. Nevertheless, mucosal membranes are equipped with lymphoepithelial structures specialized in the sampling of microbial and molecular antigens by way of particular epithelial cells highly efficient in transcytosis (microfold or M cells). In these structures, collectively denoted mucosa-associated lymphoid tissue (MALT), the sampled material is immediately brought into contact with the cells and molecular environment required for an appropriate immune response. The lymphoepithelial structures of MALT are distributed strategically at multiple mucosal sites. In the gut-associated lymphoid tissue (GALT), they are the Peyer's patches, which contain several lymphoid follicles. Analogous small solitary follicles are dispersed throughout the intestine in humans and some other species.33 In the airways, bronchus-associated lymphoid tissue (BALT) has a similar architecture and function. BALT is not constitutively present but develops in response to infections by respiratory pathogens and to persisting allergic reactions. MALT also includes the organized lymphoid tissues of the Waldeyer's pharyngeal ring, which includes the palatine tonsils and the nasal-associated lymphoid tissue (NALT), such as the adenoids.
In response to an antigen uptake by the elements of MALT, germinal centers develop in the follicles, triggering the affinity maturation of antibodies and the development of memory cells, much like during parenteral antigen challenge in the lymph nodes and spleen. Unlike these organs, however, MALT provides a unique microenvironment that promotes the development of B cells producing antibodies in the form of dimeric, J chain-containing IgA, although switching to other non-IgM isotypes also occurs.34 Apparently, the induction of IgA antibody responses at these sites occurs irrespectively of the local Th1/Th2 balance, which may vary depending on the activity of mucosal adjuvants, such as substances derived from the mucosal microflora.35 The mechanisms responsible for the preferential expression of IgA are not clear, but the effects of specialized antigen-presenting cells (APC), such as dendritic cells,36,37 and the local expression of cytokines, such as transforming growth factor-β (TGF- β),38 appear to be important. A cytokine-independent factor, activation-induced cytidine deaminase, has also been implicated as a regulator of switching from IgM to IgA.39 Studies of Peyer's patches indicate that TGF-β and cytokines that regulate subsequent differentiation of B cells into antibody-secreting plasma cells (IL-5, IL-15, IL-6, IL-10) may be produced by several cell types including T cells, epithelial cells, and dendritic cells.40,41 The mechanisms that regulate the differentiation of human B cells into IgA1- or IgA2-producing cells have only been sparsely elucidated.42
Mucosal Lymphocyte Trafficking in MALT
Contrary to other peripheral lymphoid tissues, the immune induction sites of MALT lack afferent lymphatics, but do have efferent lymphatics. Naive B and T lymphocytes enter MALT through high endothelial venules. Upon stimulation they exit as lymphoblasts via efferent lymphatics to regional lymph nodes, where they may undergo further division and differentiation.43 From there, the cells travel via the thoracic duct to the blood stream, and subsequently migrate extensively to the lamina propria of the inductive and the remote mucosal membranes and glands, where they terminate as effector cells. A small fraction of the stimulated IgA-producing effector cells also migrate to the systemic compartment, including the bone marrow.24 The preferential migration (homing) of mucosally stimulated lymphocytes to the mucosal compartment may be linked to the expression of adhesion molecules that are complementary to endothelial addressins specifically expressed within mucosal tissues.44 Accordingly, stimulation of e.g., GALT B cells (fig. 1) by an intestinal antigen may lead to the presence of IgA antibodies in intestinal secretions, and also in the secretions of the respiratory tract and the lacrimal, salivary, and lactating mammary glands. In contrast, antibodies of any isotype are barely detectable in serum. Thus, through functional specialization of its afferent and efferent limbs, MALT is partially independent of the systemic immune apparatus. This integrated common mucosal immune system (CMIS) was originally reported in rodents, and convincingly documented in humans.45-47 Production and transport of S-IgA represent a major physiological investment. In humans, the intestine alone receives 3 to 5 g of S-IgA each day, testifying to the biological significance of this system.

Recent studies indicate a partial compartmentalization within the mucosal immune system, especially a dichotomy between the gut and the upper respiratory and digestive tracts with respect to migration of stimulated B cells (fig. 2).46,48,49 The mechanisms involved in the preferential homing to selected mucosal effector sites have not been revealed, but tissue-specific homing receptors or local chemotactic factors are probably critical. The patterns of migration have important implications for the design of vaccine protocols that could provide protective immunity at mucosal surfaces.
Uptake and Handling of Antigens by Enterocytes
Whereas stratified epithelia, present in the oral cavity, oesophagus, and vagina, are impermeable to molecules larger than ˜40,000 Da,50 the polarized cells of the pseudostratified, simple epithelia lining the small and large intestines (enterocytes) actively sample luminal macromolecules by endocytosis. Although most endosomal constituents are sorted for the degradative pathway,51,52 enterocytes have been shown to transport small amounts of intact proteins and peptides across the epithelium by transcytosis.53 This mechanism probably explains why dietary proteins can be detected in the circulation after a protein-rich meal.54,55 Low levels of antibodies to dietary substances are found in the secretions (S-IgA) and the circulation (IgG and IgA) of most healthy individuals (reviewed by Husby56). How and where these antibodies are induced is not clear. IgA responses in particular may involve priming in the organized GALT, followed by restimulation of primed B cells in the lamina propria by antigens transferred through intestinal epithelial cells.24 The capacity of M cells to bind and take up innocuous dietary antigens is a matter of controversy, mainly because experimental feeding with such antigens without adjuvant generally results in low and transient secretory and systemic responses, if any at all.57,58 The remarkably low levels of antibodies generally found against ubiquitous food substances59,56 are likely to reflect a downregulation of responses by the mechanisms of oral tolerance, as discussed below.
Enterocytes may display antigen-presenting properties under specific circumstances in vitro.60,61 However, there is no evidence that they exhibit similar properties in vivo.
Role of the Mucosal Microflora in the Development and Function of the Mucosal Immune System
The alimentary tract (particularly the proximal and distal parts), upper respiratory tract, and distal female reproductive tract are permanently colonized by communities of commensal organisms. There is considerable site-dependent variation in the load and composition of the commensal flora, presumably due to the selective effects of local ecological factors, such as supply of nutrients, spectrum of mucosal receptors for microbial adhesins, and synergistic versus antagonistic intermicrobial and commensal-host relationships.62-66 Selected in this way, communities of commensal bacteria are stable in terms of the species (though not necessarily the individual clones) represented, and may interfere efficiently with the colonization by pathogens.67
At the same time, however, the commensal flora represents an enormous antigenic challenge to the mucosal immune system. It has been estimated that the number of microbial cells in the body (most of them in the large intestine) exceeds by far the total number of cells of the body.68 The large number of plasma cells and other stimulated immune cells permanently present throughout the intestine and in the tonsils, indicate that the mucosal immune system is in a constant state of stimulation, which some investigators interpreted as a state of physiological inflammation. Exactly how the immune system deals with the load of microbial antigens is not clear, but observations in humans,70,71 and comparisons between germ-free and conventionally reared animals (reviewed by Cebra et al69) have revealed essential elements of this interaction, two of which will be discussed here.
(i) The establishment of the mucosal flora after birth represents a major developmental and functional stimulus to the immune system.72 Accordingly, germ-free neonates show a profound hypotrophy of mucosal and systemic lymphoid tissues and respond poorly to most antigens. 73,74 In animals maintained germ-free through adulthood, systemic responses are normalized but mucosal lymphoid tissues are poorly developed and IgA responses to mucosal antigens remain weak.75 Germ-free animals kept on a hydrolyzed “antigen-free” diet display only limited additional hypotrophy of intestinal lymphoid tissues,69 indicating that non microbial antigens in food and drink are not a major stimulus to the immune system. Interestingly, the repertoire as well as the level of mucosal immune responses in formerly germ-free animals may be normalized by colonization with selected bacterial taxa belonging to the intestinal commensal flora.69,76-78 The mechanisms by which the mucosal flora stimulates the development and reactivity of the immune system in the early stages of lymphocyte development may involve specific bacterial components, such as lipopolysaccharides.69 The local load of microorganisms is also important, as indicated by the poor development of BALT compared to GALT in the absence of respiratory infections. Interestingly, the commensal flora may also influence the immune reactions to mucosally applied antigens that lead to a state of mucosal tolerance (see below). Thus, interactions with the commensal flora seem to be a fundamental element of mucosal immune homeostasis.
(ii) Specific immune responses to the commensal flora, including IgA antibody production, do not normally eliminate the target organisms. Colonization by commensals starts right after birth by transfer of bacteria, particularly from the mother. Nevertheless, breast-feeding does not significantly interfere with the establishment of the commensal flora, although the mother's early milk contains abundant S-IgA, including antibodies that may inhibit the adherence of the relevant bacteria in vitro.6 Individual clones of commensal bacteria may persist for years at mucosal sites in immunocompetent infants and adults,70,71,79,80 apparently unaffected by local clone-reactive S-IgA antibodies (Reinholdt et al, unpublished data). While some studies suggest that the generation of IgA antibodies to commensal bacteria is driven by specific responses involving organized MALT,81 others, involving natural transfer of oral bacteria into gnotobiotic animals, demonstrate that IgA antibodies are present at low levels even prior to acquisition of the bacteria.82,83 These conflicting data remain unexplained. However, the latter result, along with observations that S-IgA antibodies against individual commensal bacteria are maintained at constant and relatively low levels in secretions,84 corroborates the hypothesis that S-IgA antibodies reacting with the commensal flora derive largely from the B1 category of B cells believed to be a major source of so-called natural antibodies.85-88
IgA Produced by B1 cells
In mammals, B cells are physiologically heterogenous,89 and their ontogeny seems to reflect their evolution. The B cells generated from progenitors in the liver and the omentum during fetal and early postnatal life display certain characteristics of B cells in lower vertebrates. This early B variant, denoted B1, is maintained lifelong as a subset distinct from the population of conventional B cells (B2) that expand to predominance through infancy and adulthood (for reviews, see 90-92). B1 and B2 cells differ in the expression of several markers, e.g., contrary to B2, some but not all B1 cells are CD5+.91
The early B1 cells express mainly self-reactive, germ-line encoded, low-affinity antibodies, many of which show high interconnectivity and anti-idiotype activity.93 B1 network interactions largely determine the B1 repertoire94 and may, together with an additional mechanism of B1 feedback regulation around the time of weaning,95 explain that the B1 population from infancy throughout life consists of a restricted and fixed set of clones. Contrary to B2 cells, which are continuously replenished by the progeny of stem cells in the bone marrow, B1 cells persist as a self-renewing population not appreciably augmented by immigrants from the bone marrow.96 This is possible because B1 cells constitutively display partly activated, yet tightly controlled intracellular signalling pathways, allowing them to be maintained, and even secrete small amounts of antibodies, in response to low affinity engagement of their antigen receptors with self epitopes.97-100 Thus, the autologous immunoglobulins of the fetus and the newborn, mainly IgM but also IgA and IgG, are the products of B1 cells.101
After birth, despite their reactivity with self epitopes, B1 antibodies contribute to the defence against exogenous antigens. Studies of B1 antibodies produced by hybridoma technique showed that individual B1 monoclonals frequently reacted with several apparently unrelated epitopes, including epitopes of ubiquitous microorganisms (polyreactivity or multispecificity).93,102-104 Microbial and self epitopes identified as targets of the B1 antibody repertoire are listed in reviews.105,106 The phenomenon of polyreactivity may be explained by structural characteristics of the antibody CDR3 region.107-109
Although B1 cells, like other lymphocytes, are maintained by moderate signalling through their antigen receptors, extensive cross-linking of their receptors leads to apoptosis.110-114 This explains why the mere presence of autoreactive B1 cells does not normally lead to autoimmune disease. If stimulated by antigens in combination with polyclonal activators (LPS) or certain cytokines, B1 cells can conversely be driven into differentiation and increased antibody secretion.115 V-region somatic mutations leading to affinity maturation may also occur.106 Because of the controlled signalling in B1 cells, however, their response to stimulation may differ from that of B2 cells. Apparently, B1 cells may increase antibody production without dividing, without becoming permanently committed to antibody production, and without losing their self-renewal capacity, i.e.,their future capacity to reconstitute a portion of the B1 repertoire.91 Stimulated B1 responses are suspected to cause certain autoimmune conditions.116,117 Nevertheless, B1 antibodies are normally not pathogenic, and may even inhibit the effect of pathogenic autoantibodies via anti-idiotype activity.118,119
B1 cells produce a large proportion of the “natural” low-affinity antibodies (mainly IgM, but also IgA and IgG) that are present lifelong in normal individuals and have been detected in small amounts also in germ-free and antigen-free animals.120-123 It seems that B1 cells have evolved to recognize mainly a limited spectrum of common (phylogenetically conserved) microbial and self structures with the characteristics of T-independent epitopes.124 Such epitopes seem distinct from virulence-associated, genetically variable microbial structures such as the ligand-binding domains of microbial adhesins.125 B1 cells express MHC class II and B7.1, but their maturation into antibody-secreting cells does not require cognitive T cell help. Accordingly, stimulation may occur outside the follicles of organized lymphoid tissues and does not generate classical B cell memory. B1 cells, together with marginal zone B cells, appear to be largely responsible for the initial T-independent phase of systemic IgM antibody responses to invading pathogens, which play an important role in the early phase of a protective antimicrobial immune response.126-128 Notably, complement-activating IgM antibodies produced by B1 cells may also facilitate the induction of T-mediated responses, as demonstrated in the case of hapten-induced contact sensitivity.129 The capacity of B1 cells for T-independent stimulation and isotype switch recombination may explain why athymic (nude, nu/nu) mice are only partially deficient in cells producing IgA and some other non-IgM isotypes.130
B1 cells, together with marginal zone B cells, γδ T cells, and certain NK cells, represent a phylogenetically old strategy of innate immune recognition distinct from adaptive lymphocyte responses. They are probably involved in the elimination of damaged tissue and in antimicrobial defence.124 The physiology and functional significance of B1 and other B cell subsets have been recently reviewed.124,131,132
In adult animals, B1 cells are present in the pleural and peritoneal cavities and rarely in follicular lymphoid tissues such as pheripheral lymph nodes, spleen, and Peyer's patches.133,134 B1 cells migrate from the cavities to mucosal membranes via regional lymph nodes while maturing into antibody-producing cells (fig. 1). In the case of the peritoneal cavity, this process is stimulated by the intestinal flora.87,122 Studies in an irradiated, Ig-allotype-chimeric mouse model have shown that a fraction of peritoneal B1 cells seed to the gut lamina propria where they produce IgA antibodies reacting with the intestinal flora, as B2 cells do upon stimulation in the lymphoid follicles of MALT.85,87,135,136 In a transgenic mouse model, the migration and stimulation of peritoneal B1 cells involved non-cognitive help from locally activated γδ T cells,i.e.,another innate-type lymphocyte.137 Interestingly, γδ T cell receptor knock-out mice display a partial deficiency in mucosal IgA and mucosal IgA responses to antigens.138 Whether this deficiency selectively affects B1-derived IgA is unknown. Further support for a dual origin of mucosal IgA antibodies comes from observations that the presence of IgA-producing B1 cells in the gut, contrary to IgA-producing B2 cells, depends largely on IL-5 and IL-15.112,134,139 Recent studies suggested that T-independent priming of mouse B cells (presumably B1), and subsequent switch to the IgA isotype can take place in the gut lamina propria without involvement of Peyer's patches or solitary lymphoid follicles, the appropriate mediators being delivered by local stromal or dendritic cells.140-142
A study involving TCRβ-/-δ-/- mice suggested that B1 cells produce 20-30% of intestinal IgA antibodies to normal flora in a T-independent fashion, and that these antibodies cover a range of antimicrobial specificities similar to that of intestinal IgA in normal animals.88 This suggests that B1-derived intestinal IgA have biological effects on the flora similar to those of IgA antibodies from B2 cells stimulated in mucosal follicles. However, this interpretation has been questioned.143 Studies in various mouse models, some of which involved IgA-producing B1 cell back-pack tumors (see below), suggested that B2-derived IgA antibodies were more effective than B1-derived IgA antibodies in excluding intestinal commensals and opportunistic pathogens from the mucosal tissues.87 A difference in the roles of B1 and B2 cells was also suggested by a study of immunity to influenza virus,144 and by the observation that B2, but not B1 cells in reconstituted SCID mice may contribute to the defence against rotavirus by specific IgA antibody production and cooperation with CD4+ T cells.145
In mice, B1 cells are a source of mucosal IgA distinct from B2 cells, which are stimulated in lymphoid follicles of the common mucosal immune system (CMIS). B1 cells are believed to constitute a CMIS-independent source of IgA antibody induction.134 A validation of this term should take into account that B cells able to produce polyreactive IgA antibodies have been isolated from Peyer's patches,146,147 and that receptors for homing to mucosal membranes have been detected on peritoneal B1 cells.148
In humans, the significance of B1 cells is less clear. B1 is the major B cell subset in umbilical cord blood and constitutes 20-35% of the total B cell population in late adolescence.148,149 The CD5+ subset of B1 cells is present mostly in the peritoneal cavity,148and only sparsely in the gut lamina propria.150 However, B1-like antibodies apparently account for a significant part of salivary S-IgA antibodies to the oral microflora.151 Antigens of non-microbial origin (e.g., foodstuffs, and allergens) seem to be inefficiently taken up by the M cells of follicle-associated epithelium.152 The extent to which human B1 cells are responsible for IgA antibodies against these innocuous antigens is unknown. The subclass distribution of B1- versus B2-derived IgA antibodies in humans also remains to be examined.
IgA Antibody Functions
The presence of antibodies in external secretions was first suggested by the detection of local (not serum-derived) immunity in animals orally immunized with intestinal pathogens (reviewed by Mestecky et al153). This observation stimulated intensive research in mucosal immunity to infection, with the prospect of exploiting this mechanism for vaccination. Thus, experimental studies of mucosal immune responses to microbial antigens, often involving potent adjuvants such as cholera toxin, have contributed largely to the current knowledge of IgA biological activities. The significance of IgA responses to natural infections, and to innocuous antigens from the commensal flora, has received less attention.
Because immune responses to microbial antigens are heterogeneous, experiments involving passive immunization with purified poly- or monoclonal antibodies have been designed to identify the specific activities of IgA versus those of other Ig classes. Antibodies have been delivered either directly, often mixed with the infectious inoculum, or indirectly as in the elegant “back-pack” tumor model. In this model, hybridoma cells producing a relevant monoclonal IgA antibody in a polymeric form are injected subcutaneously in the back of a mouse. As the tumor enlarges, the IgA antibody appears in both plasma and gut mucosal secretions due to efficient transport of polymeric IgA from the circulation into the bile in this species.154 Other activities have been ascribed to IgA mainly on the basis of observations in simplified models in vitro. Presumably, evaluation of such activities may require the involvement of gene knockout animal models to control for effects of other factors that may obscure IgA-mediated effects in vivo.
In addition to being the major Ig at mucosal surfaces, IgA is also abundant in the circulation and tissues, including the lamina propria. The host defense problems in these two compartments are by nature essentially different. The presence of a microorganism in the blood or tissues represents a potentially life-threatening invasion, which must be met with a forceful reaction. By contrast, S-IgA and the mucosal immune system in general must keep a balance with the normal microbiota, while maintaining the ability to respond vigorously to potential pathogens. Mucosal immune reactions, irrespective of the nature of the antigen, should not result in inflammatory reactions that might jeopardize the physiological functions of the mucosal membranes. The two forms of IgA, functioning in their respective compartments, appear to meet these requirements.6,155
Functions of IgA in Relation to Mucosal Membranes
IgA functions in mucosal membranes are often defined collectively as “immune exclusion”. Whereas systemic defence aim at the ultimate destruction of intruding antigens, mucosal IgA prevent the attachment and penetration of microorganisms and molecular antigens, blocking their potential effects on the host. In secretions, S-IgA antibodies are well suited to these tasks because of their molecular characteristics. Their four or more antigen binding sites allow them to block adherence determinants, including microbial,156-158 neutralize microbial enzymes and toxins, and agglutinate microorganisms,159 thereby facilitating their disposal by muco-ciliary flow or peristalsis. The abundant and outward directed glycans of the Fc.Sc part of the S-IgA molecule contribute to these effects by conferring hydrophilicity and negative charge to the resulting immune complex.160 The glycans of the highly glycosylated SC component are largely responsible for the marked resistance of S-IgA to degradation by nonspecific microbial and digestive proteases.161,162 Structural elements of the Fc.SC part also facilitate the entrapment of immune complexes containing S-IgA in the mucus blanket covering mucosal surfaces, which leads to their disposal.163-165 Elegant experiments have recently shown that entrapment in the mouse airways mucus depends largely on SC glycans.164 In the human female genital tract, Ig affinity to secretory leukocyte proteinase inhibitor, a nonspecific defence protein present in most external secretions, seems to be important.166 The capacity of bacterial IgA1 proteases to cleave IgA (including S-IgA) molecules in the hinge region has been exploited to analyse the defence mechanisms mediated by the Fc (or Fc.SC) part of IgA antibodies.17
Support for the notion that IgA antibodies inhibit the penetration of antigens comes from studies involving passive or active immunization,167,169,170 and observations that IgA- and pIgR-deficient individuals often have elevated plasma levels of dietary antigen-containing immune complexes.55,171 A report of allergen-specific IgA antibody deficiency in type 1 allergic patients is frequently cited in this context,172 but was never confirmed. Allergy seems to be associated with elevated levels of allergen-reactive antibodies of not only IgE, but also IgG and IgA (including S-IgA) classes, whereas such antibodies are barely detectable in nonallergic individuals.173-176 The reason for the apparent functional impairment of allergen-specific S-IgA in allergic individuals is unknown, and may involve an IgE-mediated uptake of allergens by the mucosal epithelial cells in these individuals.177-179
S-IgA antibodies were found to inhibit also the uptake of microbial antigens through the specialized epithelium that cover mucosal lymphoid follicles.180 This might explain the self-limiting nature of S-IgA responses to intestinal bacteria observed in certain animal experiments.81 However, other studies suggest that coating of antigens with enteric S-IgA antibodies facilitates their uptake through M cells.181,182 Receptor-mediated uptake of S-IgA by dendritic cells has also been observed.183,184 The potential consequences of S-IgA coating for the quality of the immune reactions induced, if any, are unknown. This might be a rewarding topic for research, particularly in the view of recent reports of reduced immune responsiveness to microbial antigens in IgA-deficient animals.185
The protective functions of mucosal IgA antibodies are not restricted to the activities of S-IgA antibodies in secretions. In vitro studies have shown that immune complexes containing polymeric IgA (pIgA) antibodies can be transported intact across polarized epithelial cell monolayers expressing pIgR, according to the same process that allows free pIgA to migrate to the apical surface.186 As an in vivo correlate, pIgA mediate the excretion by liver cells of antigens into the bile in mice (though not significantly in humans).187 This excretion also takes place over the intestinal epithelium.188 Furthermore, there is evidence that pIgA antibodies during transfer through the epithelium may neutralize viruses by interfering specifically with the viral life cycle.189 Thus, polymeric IgA antibodies, acting in concert during epithelial transfer and in the secretions, can provide for the neutralization and exclusion of potentially harmful substances without disturbing the mucosal physiology.190
Functions of IgA in Tissues
The two distinct environments in which IgA antibodies operate differ with respect to the presence or absence of ancillary factors. In the mucosal secretions, there is no evidence of a biologically active complement system or of significant numbers of live phagocytes. Conversely, in the circulation and tissues, both of these components are prominent defense factors.
The question remains of whether or not IgA activates complement.6 Clearly, IgA is unable to activate complement by the classical pathway.191 To the contrary, IgA antibodies to capsular polysaccharides may inhibit IgG or IgM antibody-dependent complement-mediated lysis of meningococci under specific circumstances,192 and similar effects have been observed for IgA antibodies in vitro.193,194 However, IgA molecules denatured by various procedures, including partial deglycosylation by streptococcal glycosidases, activate complement by the alternative pathway.195 This may explain why IgA antibodies to the capsule of glycosidase-producing pneumococci mediate opsonophagocytosis by resting human phagocytes only in the presence of complement.196 Purified human serum IgA immobilized on plastic surfaces is capable of activating complement by the mannan-binding lectin pathway.197 However, this capacity may be restricted to IgA molecules rich in the high-mannose type of N-linked glycans (Reinholdt and Jensenius, unpublished data). Overall, it seems that human IgA antibodies have a poor or no complement-activating ability when bound physiologically to antigens, except for some degree of alternative pathway activation when the IgA is abnormally glycosylated or otherwise denatured.
Early studies indicated that IgA antibodies were relatively inefficient mediators of opsonophagocytosis when compared to IgG.198 IgA was also reported to inhibit the mobilization of phagocytes,199-201 and the release of inflammatory cytokines by LPS-stimulated human monocytes.202 It is now clear that these results do not reflect the full potential of IgA cooperation with phagocytes. An Fc-specific IgA receptor (FcαR, designated CD89) is variably expressed on human granulocytes and monocytes/macrophages.203,204 Several groups have shown that neutrophils activated by treatment with inflammatory cytokines display an enhanced phagocytosis of IgA-coated particles consistent with an enhanced surface expression of FcαR.27,205-207 FcαR binds both subclasses of IgA and S-IgA, but pIgA is generally more effective than mIgA in mediating phagocytosis. Eosinophils also express cytokine-regulated FcαR, and easily degranulate upon reaction with pIgA-containing immune complexes.208 Eosinophils also express a receptor for SC, which may explain why immobilized S-IgA are particularly efficient in eosinophil degranulation.209,210 Whereas IgA-mediated degranulation of eosinophils may be important in the defense against parasites, it is suspected to contribute to the detrimental effects of eosinophils during the late phase of atopic reactions in the respiratory mucosa.174,210 Conversely, IgA has been found to inhibit induced IgE-mediated hypersensitivity. 211 Taking into account the elevated levels of allergen-reactive IgA antibodies in most allergic individuals, and the inconclusive body of published data on the prevalence of allergy in IgA-deficient versus normal humans,212-215 the overall effect of allergen-reactive IgA antibodies in relation to atopy remains unknown.
In the view of this information, the plasma and tissue forms of IgA appear to be biomolecules of high functional adaptability, able to function according to local demands. In the absence of invading pathogens, IgA antibodies may modulate the inflammatory responses to complement and phagocytic cell activation, and possibly the IgE-mediated reactions. Conversely, in reaction to alarm signals from microorganisms and the innate immune system, IgA antibodies may promote a forceful inflammatory response.
Mucosal Tolerance
Mucosal (or oral) tolerance is the suppression or down-regulation of immune effector cell responses (T and B) to an antigen by prior administration of the antigen by mucosal (e.g., oral) route. Mucosal tolerance probably evolved in order to prevent irrelevant and potentially injurious reactions to food substances and other harmless antigens encountered at mucosal surfaces. By reference to the IgA activities discussed above, mucosal tolerance may be viewed as a collaborator of IgA in the maintenance of mucosal homeostasis through peaceful neutralisation of innocuous antigens. Like other forms of immunological tolerance, mucosal tolerance also provides mechanisms to suppress pathologic reactivity against self. 216,217
Oral tolerance has raised considerable research interest over the last 20 years, but it has been difficult to reach definite mechanistic explanations. Essential inductive events can be
- the development of regulatory T cells, which mediate active suppression,
- the inactivation of T cells (Th-1, Th-2, T-CTL), that may be brought about by cellular anergy or clonal deletion,
- the generation of antibodies which mediate suppression, possibly as part of an anti-idiotype network.
Generally, several tolerance mechanisms such as suppression and anergy may operate sequentially or simultaneously (for reviews, see refs. 217, 218).
Mucosal tolerance, even when it involves clonal anergy or deletion, represents an active immune response and not a simple absence of antigen recognition.218-220 Accordingly, the induction of tolerogenic reactions depends largely on the properties of the antigen-presenting cells (APC) involved. Whereas several types of mucosal cells may display antigen-presenting capacity, certain phenotypes of dendritic cells (DC) in mucosal tissues and the draining lymph nodes seem to be particularly significant in the induction of tolerogenic reactions.221-228 The performance of DC and other APC is highly dependent on the quality of the signals received through Toll-like and other receptors.229 These signals are mediated largely by molecular components of the mucosal flora. As mentioned, the development of a commensal flora not only represents a physiological stimulus to the immune system, but also conditions the mucosal immune system for the induction of tolerogenic reactions, which suggests that the commensal flora and the immune system have evolved to cooperate in the maintenance of mucosal homeostasis. Thus, germ-free mice develop short-lived tolerance and no tolerance at all, when fed with ovalbumin and sheep red blood cells, respectively.230-232 The capacity for development of oral tolerance to certain antigens may be reconstituted in germ-free animals by treatment with LPS, a prominent cell wall component of gram-negative bacteria,232,233 whereas in antigen-fed conventionally reared animals LPS may promote immune responses.69,234,235 The tolerization of IgE antibody responses to mucosal allergens is also deficient in germ-free mice.236 Interestingly, this deficiency can be reversed by neonatal colonization of the animals with Bifidobacterium infantis, a gram-positive bacterium of value as an intestinal probiotic in human.236,237 Remarkably, immune responses to the commensal flora appear to be down-regulated by certain mechanisms of mucosal tolerance in healthy individuals.238-240 However, the hyporesponsive, yet activated state of the immune system towards the commensal flora may break down in some individuals, in which case strong T cell-mediated and humoral responses against a subset of bacterial antigens may develop, much like a response against pathogenic bacteria. Such breakdown possibly underlies inflammatory bowel diseases such as Crohn's disease and ulcerative colitis.238,240,241 Furthermore, changes in the flora caused by mucosal infection with certain pathogens may adversely affect the induction of tolerance to non-microbial substances like dietary antigens and allergens.242,243 As an exploitation of this principle, administration of antigens to mucosal surfaces together with cholera toxin as an adjuvant prevents the induction of tolerance and triggers systemic as well as mucosal immune responses.244 The contrasting effects of pathogens versus commensals on mucosal immune regulation probably reflect a different quality of alarm signals to APCs, either directly245 or via cytokines produced by the mucosal epithelium.64-66 The distinct effects of different mucosal bacteria on the immune system may explain why mechanisms of mucosal tolerance identified in inbred animals kept under pathogen-free conditions do not always apply in humans.35,246
The dose of mucosally applied antigen influences the type of tolerogenic mechanism involved. 247 High doses of antigen (10-100 mg in repeated doses in mice) induced anergy.248,249 The antigens cross the mucosa in significant amounts, circulate either in a native form or as immune aggregates, and presumably anergize antigen-specific T cells by reacting with the cells under conditions different from those required for T cell activation.228 Anergy in T cells is characterized by an induced defect in IL-2 transcription, while the expression of IL-2 receptor is preserved.250 High dose mucosal tolerance may be detected 1-2 days after antigen is fed to the animal.251 It is not clear whether persistence of antigen is required for maintenance of tolerance, but repeated tolerogenic stimuli cause a reduction in T cell responses.252
The immune responses to common dietary antigens appear to be controlled by mechanisms of oral tolerance that involve T cell anergy, as shown in a comprehensive study of humoral and cellular immunity to common antigens in healthy humans.253 Consistent with the inactivation of T cells, serum levels of both IgG and IgA antibodies to individual antigens were low. Within each of these two isotypes, individuals with higher titers of antibodies to one kind of antigen had higher titers of antibodies of the same isotype to other dietary antigens. In contrast, antibodies of the T-independent IgM isotype were present at slightly higher levels, with no correlation between antigens.253
Low doses of antigen applied to mucosal surfaces may lead to active suppression.254-256 Antigen-specific regulatory T cells develop after presentation by APC in organized lymphoid follicles or in the epithelium. Studies in experimental allergic encephalomyelitis with myelin-basic protein (MBP) as a model antigen257 identified CD8+ T cells as the major regulatory cells, acting primarily via secretion of TGF-β.258 The same research group observed that when ovalbumin feeding is followed by parenteral immunization with ovalbumin together with MBP and adjuvant, there is a suppression of responses not only to ovalbumin but also to MBP.259 This effect, denoted antigen-driven bystander suppression, involves the secretion of antigen-nonspecific suppressive cytokines by regulatory cells stimulated by the oral antigen. Subsequent studies indicated that T cells stimulated by distinct suppressor epitopes of a protein mediate epitope-driven bystander suppression of responses to non-suppressor epitopes within the same protein.260 The fact that tolerogenic reactions to fed antigens/epitopes may induce bystander suppression of concurrent pathogenic, or beneficial responses to other antigens/epitopes is of potential clinical and therapeutical relevance.261 Several studies have suggested that certain phenotypic variants of CD4+ T cells, some of which may be in a state of anergy, are important regulatory cells in mucosal tolerance to fed proteins and commensal flora antigens.227,240,262-265 Further complexity arises from observations that γδ T cells, as a result of interactions with mucosal epithelial cells, may suppress certain immune reactions, notably IgE responses, to low doses of mucosal antigen.266,267
B Cells and Antibodies in Mucosal Tolerance
Stimulated B cells are one of several types of APC involved in the induction of mucosal immune responses.268 Conversely, resting B cells, which express MHC class II but no costimulatory molecules, do not activate naive T cells and can mediate T cell anergy and tolerance.269,270 However, B cells are not essential for the development of peripheral T cell tolerance by parenterally administered antigens.271 The same seems to apply to oral tolerance, since genetically B cell deficient mice are still able to develop T cell tolerance upon oral administration of a single high dose or repeated low doses of a protein antigen.272 These mice, as a corollary of their genetic defect, have involuted Peyer's patches, which suggests that these structures are not obligatory for the induction of oral tolerance. However, other investigators found that B cells273and Peyer's patches274 were of significance for tolerance induction, at least against low oral doses of protein antigens. Furthermore, oral tolerance cannot be induced in mice lacking both Peyer's patches and mesenteric lymph nodes.275,276
Early studies indicated that B cells can be involved in the induction of oral tolerance by way of antibody production. Ad libitum feeding of sheep red blood cells to mice resulted in specific suppression of IgM, IgG, and IgA responses upon subsequent parenteral immunization with that antigen, and the suppressive effect could be transferred across MHC barriers by a serum Ig factor or antigen-antibody complexes from fed animals.277,278 The transfer of the factor did not result in suppression of delayed-type hypersensitivity in the recipient.279 The factor (IgG or IgA) could not always be absorbed with the fed antigen.278 Such tolerance-mediating antibodies may be part of an anti-idiotype regulatory network,278,280,281 and appear not to be involved in the induction of oral tolerance by feeding highly T-dependent protein antigens such as ovalbumin.235,282 However, there is evidence that rodent mothers may transfer IgE-suppressive IgG antibodies to their offspring transplacentally or by milk.283 It is possible that such presumably anti-idiotypic antibodies confer tolerance to the neonate at a time when susceptibility to atopic priming is high. No similar allergy-preventive role of maternal IgG antibodies was observed in human infants at risk of allergy development.284 Human neonates may take up small amounts of maternal S-IgA from colostrum in the immediate neonatal period.285 The physiological significance of this, if any, is not known.
Conceivably, mucosal IgA antibodies, either passively acquired by the neonate or actively produced by children and adults, may prevent or arrest immune responses by excluding mucosal antigens from the tissues. Indirect support for this assumption comes from the observation that polyimmunoglobulin receptor-deficient mice, which lack S-IgA and S-IgM, show signs of mucosal inflammation suggesting an undue triggering of systemic immunity.171 In addition, as mentioned before, IgA-deficiency has been associated with an increased prevalence of atopic diseases. However, apart from the documented protection by S-IgA antibodies against certain pathogens (discussed earlier), the role of mucosal IgA-mediated immune exclusion in the limitation of immune responses in normal individuals is not clear. The presence of low levels of systemic and mucosal antibodies reacting with innocuous environmental antigens in healthy subjects does not necessarily reflect a previous failure by mucosal antibodies to prevent the presence and immune recognition of these antigens in the tissues. Theoretically, the antibodies represent cross- or polyreactive antibodies mainly of B1 origin. On the other hand, antigen-exclusion by mucosal IgA must be limited in time or space because a state of immunological tolerance to dietary antigens and other ubiquitous antigens seems to be present in healthy individuals, reflecting previous handling of the antigens by cells of the immune system. Particular attention has been paid to the possibility that colostral S-IgA antibodies might reduce the risk of atopic priming of the newborn to environmental antigens. Increased prevalence of cow's milk allergy was found in children who were formula-fed, as compared to breast-fed, as neonates.286,287 However, extensive studies involving additional colostral components indicated that not S-IgA antibodies, but TGF-β and certain fatty acids in colostrum were responsible for the allergy-prophylactic effect of breast feeding.288,289 Yet other studies provided inconsistent data on the role of breast-feeding in prevention of allergies and asthma.290 Disturbance of S-IgA-mediated immune exclusion by IgA1 protease-producing mucosal bacteria has been suggested to be a possible factor for the development or perpetuation of atopy in infancy.291,292 However, subsequent observations suggested that IgA1 protease-producing nasopharyngeal bacteria, if present, do not cause widespread cleavage of S-IgA1 molecules in the mucosal secretion.13,176 Future studies on the significance of mucosal IgA in immune regulation should take into account the role of M cells and dendritic cells in receptor-mediated uptake of S-IgA immune complexes,181-184 and the possible consequences of S-IgA binding for the stimulatory or tolerogenic reactions induced.
Early experiments indicated that the B cell system was more difficult to tolerize by primary oral administration of antigen than the T cell system.293,294 Once antibody responses had been established, B cells were also more difficult to tolerize than T cells, and they recovered more rapidly.295,296 An exception to this pattern seems to be IgE responses, which in rodents are exquisitely sensitive to oral tolerance.283 As mentioned above, oral tolerization of mouse IgE antibodies (and other Th2-dependent products such as IgG1 and IL-4), is conditioned by postnatal colonization by commensal bacteria.236 Since this does not apply to Th1-assisted antibody responses including IgG2a,236 and because Th1 and Th2 cells appear to be equally susceptible to oral tolerance in conventionally reared mice,262 there is no clear implication of the mucosal flora in the differential susceptibility of T cell and non-IgE B cell responses to oral tolerization.
The differential susceptibility of T- versus B cell responses to tolerization by mucosally administered antigens was confirmed by two studies in humans.297,298 Short term feeding of healthy adults with keyhole limpet hemocyanin (KLH) followed by parenteral challenge with KLH induced the development of tolerance only in the T cell system (skin test reactivity, proliferation assay). Serum antibodies and antibody producing cells as determined by ELISPOT were of IgG, IgM, and IgA classes, and B cell priming by feeding was demonstrated by an accelerated response in the orally fed study group. Furthermore, IgA antibody responses were prominent in saliva and in intestinal secretions.297 Similar results were obtained in parallel experiments involving intranasal instead of oral administration of KLH, except that serum IgA and IgG antibody responses were suppressed.298
Mucosal IgA Antibody Responses As Targets of Mucosal Tolerance
Pioneering studies suggested that oral tolerance is accompanied by mucosal IgA responses.251,254 Conventionally, the induction of tolerance by antigen feeding has been examined by secondary challenge with an antigen injected subcutaneously with an adjuvant. This protocol cannot reveal a potential suppression of mucosal immunity because subcutaneously induced IgA responses do not generally involve S-IgA.24 Studies involving secondary challenge by mucosal or intraperitoneal routes, however, have shown that mucosal IgA responses, like systemic responses, are sensitive to oral tolerance.244,299-304 A few studies on the tolerogenic mechanism indicate the involvement of regulatory T cells, sometimes identified as CD8+.299,302,303 The induction of oral tolerance in the mucosal IgA system is abrogated if innocuous antigens are fed together with cholera toxin as an adjuvant.244,302,303
In two studies, antigen feeding was reported to suppress S-IgA responses to intraperitoneal challenge with antigen and adjuvant.244,301 Because intraperitoneal injection of antigen stimulates the B1 cell population, the suppression observed in these studies suggests that the differentiation of peritoneal B1 cells into IgA-producing cells in the gut wall is controlled by regulatory T cells. T cell-mediated regulation of B1 cells is suggested by additional observations.105 It should be stressed, however, that the mechanisms involved in the stimulation and control of B1 cells in vivo have been poorly explored.131
The susceptibility of the mucosal IgA system to oral tolerance seems to be at odds with the presence of S-IgA antibodies to dietary antigens in most healthy humans.253,305,306 In one study, the saliva levels of S-IgA antibodies to individual dietary antigens (bovine γ-globulin, or casein) were estimated at 0.02—0.46 % of total salivary S-IgA, four to nine fold higher than serum IgA to the same antigens.253 This might indicate a less profound tolerization of mucosal IgA than serum IgA in response to dietary antigens. The maintenance of moderate levels of mucosal IgA antibodies to otherwise tolerogenic dietary antigens may involve complex molecular factors.307 One group has suggested that the phenomenon may result from an interplay between mucosal epithelial cells and two types of intraepithelial T cells, i.e., γδ T cells that interfere with the induction of tolerance, and αβ T cells that provide B cell help.308,309 If so, the apparent variability of this mechanism in different experimental settings needs to be explained.
The susceptibility of the mucosal IgA system to tolerization is relevant to what is known of the biological effects of IgA antibodies. On one hand, IgA is the largest Ig class produced. On the other hand, many IgA-deficient individuals remain healthy. This paradox illustrates the complexity of the issue. Whereas the protective potential of mucosal IgA antibody responses towards pathogenic microorganisms is established, the significance of the interaction of mucosal and systemic IgA antibodies with the commensal flora and innocuous non-living antigens is unclear.6 Hypothetically, IgA against the latter targets might occur at sub-protective concentrations, due to tolerization. This is conceivable in the case of potentially harmful molecular antigens, such as allergens. Concerning commensals, however, the inhibition of bacterial adherence by purified S-IgA antibodies at concentrations within physiological range was reported in vitro.310,157,158 These observations may not reflect the significance of the antibodies in vivo, where other specific or nonspecific factors may substitute for, interfere with, or potentiate the functions of IgA antibodies.311,312 However, oral and intestinal bacteria of healthy individuals were found to be coated with IgA antibodies.313,314 Since this coating does not seem to eliminate the commensals, it remains an attractive, yet unconfirmed hypothesis that the antibodies to beneficial commensals do not block essential colonization determinants and belong to the restricted B1 repertoire.85
The ability to evade host immune defence is a characteristic of successful pathogens as well as commensals. Among the strategies for evading mucosal immunity, IgA1 protease activity has received most attention.17 A hypothesis for IgA1 protease-facilitated infection by the pathogens producing these enzymes was published, based on clinical findings and the effect of potential enzyme-neutralizing antibodies.315 However, because additional non-IgA substrates to IgA1 proteases have been identified, it is not clear to which extent these enzymes impair the protective potential of IgA at mucosal membranes.316-318
Clinical Aspects of IgA Antibodies
The possible clinical significance of systemic antibodies to dietary antigens is a matter of concern. Serum antibodies have been detected in the major immunoglobulin classes IgM, IgG and IgA in the majority of healthy children319 and adults.320 The levels of serum antibodies, but not S-IgA antibodies, seem to decline during adult age.305, 321,322The levels of serum IgG or IgA antibodies to dietary proteins are increased in infants and children with food allergy, but there is considerable overlap between patients and healthy individuals.323 These antibodies may be byproducts rather than mediators of food allergy. However, the presence of serum IgA antibody to gliadin is of diagnostic value in the screening for coeliac disease.324 Serum IgA antibodies against dietary antigens such as gliadin belong predominantly to the IgA1 subclass.325,326
Concluding Remarks
The human body produces IgA in amounts greater than the amounts of all other Ig classes combined. Still, in spite of considerable research efforts, the benefits of this physiological investment are only partly understood. Remarkably, many individuals with IgA deficiency, the most prevalent immunodeficiency of all, remain healthy.
Mechanisms by which S-IgA antibodies neutralize pathogens and molecular antigens at mucosal surfaces have been documented in vitro, and in several cases in vivo. The previously enigmatic functions of IgA antibodies in the circulation and tissues are now better understood. It appears that systemic IgA antibodies may adapt functionally to the quality of the actual target, functioning as powerful opsonins towards pathogens, as opposed to mediating the discrete non-inflammatory neutralization of harmless antigens such as mucosal commensals. The regulation of Fcα receptor expression on phagocytes by inflammatory cytokines seems to be an important determinant of systemic IgA functions. By their functions, S-IgA and systemic IgA antibodies help maintain mucosal homeostasis.
Maintenance of mucosal homeostasis is probably the major evolutionary argument also for the down-regulation of potentially injurious immune responses via mucosal tolerance. In this view, it is remarkable that mucosal as well as systemic IgA antibody responses to innocuous antigens became subject to tolerization by mucosally applied antigens. Nevertheless, mucosal IgA antibodies to innocuous molecular antigens are produced at moderate levels in healthy individuals, as are IgA antibodies to commensal bacteria. The cellular origin, B1 versus B2, and physiological significance of these IgA antibodies are not known. Notably, mucosal IgA responses to pathogenic bacteria, or to innocuous antigens in combination with mucosal adjuvants such as cholera toxin, are not suppressed by mucosal tolerance, presumably testifying to the importance of mucosal IgA antibodies in the protection against pathogens.
Acknowledgements
The authors are grateful to Charles O. Elson for discussions and advice concerning the structure of the manuscript, to Mogens Kilian for discussions, careful reading of the manuscript, and suggestions for its improvement, and to Per Brandtzaeg for providing the two illustrations.
References
- 1.
- Fujihashi K, Ernst PB. A mucosal internet. Epithelial cell-immune cell interactions In: Ogra PL et al, eds.Mucosal Immunology Academic Press,1999619–630.
- 2.
- London SD, Rubin DH. Functional role of mucosal cytotoxic lymphocytes In: Ogra PL et al, eds.Mucosal Immunology Academic Press,1999643–53.
- 3.
- Brandtzaeg P, Farstad IN. The human mucosal B-cell system In: Ogra PL et al, eds.Mucosal Immunology Academic Press,1999439–468.
- 4.
- Mestecky J, Russell MW, Jackson S. et al. The human IgA system: A reassessment. Clin Immunol Immunopathol. 1986;40:105–114. [PubMed: 2424650]
- 5.
- Conley ME, Delacroix DL. Intravascular and mucosal immunoglobulin A: Two separate but related systems of immune defense? Ann Intern Med. 1987;106:892–899. [PubMed: 3579073]
- 6.
- Russell MW, Kilian M, Lamm ME. Biological activities of IgA In: Ogra PL et al, eds.Mucosal Immunology Academic Press,1999225–240.
- 7.
- Hanson LA. Comparative immunological studies of the immunoglobulin of human milk and blood serum. Int Arch Allergy Appl Immunol. 1961;18:227–241. [PubMed: 13711373]
- 8.
- Tomasi TBJr, Tan EM. et al. Characteristics of an immune system common to certain external secretions. J Exp Med. 1965;121:101–124. [PMC free article: PMC2137965] [PubMed: 14253478]
- 9.
- Heremans JF. Immunoglobulin A. In: Sela M, ed. The Antigens. Academic Press, 1974:365-522.
- 10.
- Mestecky J, Moro I, Underdown BJ. Mucosal immunoglobulins In: Ogra PL et al, eds.Mucosal Immunology Academic Press,1999133–552.
- 11.
- Brandtzaeg P, Karlsson G, Hansson G. et al. The clinical condition of IgA-deficient patients is related to the proportion of IgD- and IgM-producing cells in their nasal mucosa. Clin Exp Immunol. 1987;67:626–636. [PMC free article: PMC1542640] [PubMed: 3301101]
- 12.
- Mostov K, Kaetzel C. Immunoglobulin transport and the polymeric immunoglobulin receptor In: Ogra PL et al, eds.Mucosal Immunology Academic Press,1999181–211.
- 13.
- Kirkeby L, Rasmussen TT, Reinholdt J. et al. Immunoglobulins in nasal secretions of healthy humans: Structural integrity of secretory immunoglobulin A1 (IgA1) and occurrence of neutralizing antibodies to IgA1 proteases of nasal bacteria. Clin Diagn Lab Immunol. 2000;7:31–39. [PMC free article: PMC95818] [PubMed: 10618273]
- 14.
- Kutteh WH, Mestecky J. Secretory immunity in the female reproductive tract. Am J Reprod Immunol. 1994;31:40–46. [PubMed: 8166946]
- 15.
- Baenziger J, Kornfeld S. Structure of the carbohydrate units of IgA1 immunoglobulin. II. Structure of the O-glycosidically linked oligosaccharide units. J Biol Chem. 1974;249:7270–7281. [PubMed: 4373463]
- 16.
- Mattu TS, Pleass RJ, Willis AC. et al. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fc alpha receptor interactions. J Biol Chem. 1998;273:2260–2272. [PubMed: 9442070]
- 17.
- Kilian M, Reinholdt J, Lomholt H. et al. Biological significance of IgA1 proteases in bacterial colonization and pathogenesis: Critical evaluation of experimental evidence. APMIS. 1996;104:321–338. [PubMed: 8703438]
- 18.
- Kawamura S, Saitou N, Ueda S. Concerted evolution of the primate immunoglobulin alpha-gene through gene conversion. J Biol Chem. 1992;267:7359–7367. [PubMed: 1559979]
- 19.
- Crago SS, Kutteh WH, Moro et al. Distribution of IgA1-, IgA2-, and J chain-containing cells in human tissues. J Immunol. 1984;132:16–18. [PubMed: 6418796]
- 20.
- Skvaril F, Morell A. Distribution of IgA subclasses in sera and bone marrow plasma cells of 21 normal individuals. Adv Exp Med Biol. 1974;45:433–435. [PubMed: 4213038]
- 21.
- Kett K, Brandtzaeg P, Radl J. et al. Different subclass distribution of IgA-producing cells in human lymphoid organs and various secretory tissues. J Immunol. 1986;136:3631–3635. [PubMed: 3517160]
- 22.
- Kutteh WH, Hatch KD, Blackwell RE. et al. Secretory immune system of the female reproductive tract: I. Immunoglobulin and secretory component-containing cells. Obstet Gynecol. 1988;71:56–60. [PubMed: 3336542]
- 23.
- Mestecky J, Russell MW. IgA subclasses. Monogr Allergy. 1986;19:277–301. [PubMed: 3093850]
- 24.
- Russell MW, Lue C, van den Wall Bake AW. et al. Molecular heterogeneity of human IgA antibodies during an immune response. Clin Exp Immunol. 1992;87:1–6. [PMC free article: PMC1554245] [PubMed: 1733625]
- 25.
- Wang AC, Fudenberg HH. Genetics and evolution of human immunoglobulin A. Adv Exp Med Biol. 1974;45:161–165. [PubMed: 4137684]
- 26.
- van LoghemE, Biewenga J. Allotypic and isotypic aspects of human immunoglobulin. A Mol Immunol. 1983;20:1001–1007. [PubMed: 6196615]
- 27.
- van EgmondM, Damen CA, van Spriel AB. et al. IgA and the IgA Fc receptor. Trends Immunol. 2001;22:205–211. [PubMed: 11274926]
- 28.
- Nose M, Wigzell H. Biological significance of carbohydrate chains on monoclonal antibodies. Proc Natl Acad Sci USA. 1983;80:6632. [PMC free article: PMC391224] [PubMed: 6579549]
- 29.
- Wold AE, Mestecky J, Tomana M. et al. Secretory immunoglobulin A carries oligosaccharide receptors for Escherichia coli type 1 fimbrial lectin. Infect Immun. 1990;58:3073–3077. [PMC free article: PMC313613] [PubMed: 2201644]
- 30.
- Adlerberth I, Ahrne S, Johansson ML. et al. A mannose-specific adherence mechanism in Lactobacillus plantarum conferring binding to the human colonic cell line HT-29. Appl Environ Microbiol. 1996;62:2244–2251. [PMC free article: PMC168005] [PubMed: 8779562]
- 31.
- Niedermeier W, Tomana M, Mestecky J. The carbohydrate composition of J chain from human serum and secretory IgA. Biochim Biophys Acta. 1972;257:527–530. [PubMed: 5022436]
- 32.
- Mizoguchi A, Mizuochi T, Kobata A. Structures of the carbohydrate moieties of secretory component purified from human milk. J Biol Chem. 1982;257:9612–9621. [PubMed: 7107583]
- 33.
- Cornes JS. Peyer's patches in the human gut. Proc R Soc Med. 1965;58:716. [PMC free article: PMC1898873] [PubMed: 5826210]
- 34.
- Craig SW, Cebra JJ. Peyer's patches: an enriched source of precursors for IgA-producing immunocytes in the rabbit. J Exp Med. 1971;134:188–200. [PMC free article: PMC2139023] [PubMed: 4934147]
- 35.
- MacDonald TT, Monteleone G. IL-12 and Th1 immune responses in human Peyer's patches. Trends Immunol. 2001;22:244–247. [PubMed: 11323280]
- 36.
- Spalding DM, Williamson SI, Koopman WJ. et al. Preferential induction of polyclonal IgA secretion by murine Peyer's patch dendritic cell-T cell mixtures. J Exp Med. 1984;160:941–946. [PMC free article: PMC2187398] [PubMed: 6332171]
- 37.
- Iwasaki A, Kelsall BL. Freshly isolated Peyer's patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J Exp Med. 1999;190:229–239. [PMC free article: PMC2195574] [PubMed: 10432286]
- 38.
- Coffman RL, Lebman DA, Shrader B. Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J Exp Med. 1989;170:1039–1044. [PMC free article: PMC2189449] [PubMed: 2788703]
- 39.
- Muramatsu M, Kinoshita K, Fagarasan S. et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. [PubMed: 11007474]
- 40.
- Kelsall BL, Strober W. Gut-associated lymphoid tissue: antigen handling and T-lymphocyte responses In: Ogra PL et al, eds.Mucosal Immunology Academic Press,1999293–317.
- 41.
- McIntyre TM, Strober W. Gut-associated lymphoid tissue: Regulation of IgA B-cell development In: Ogra PL et al, eds.Mucosal Immunology Academic Press,199319–356.
- 42.
- Fayette J, Dubois B, Vandenabeele S. et al. Human dendritic cells skew isotype switching of CD40-activated naive B cells towards IgA1 and IgA2. J Exp Med. 1997;185:1909–1918. [PMC free article: PMC2196343] [PubMed: 9166420]
- 43.
- Scicchitano R, Husband AJ, Clancy RL. Contribution of intraperitoneal immunization to the local immune response in the respiratory tract of sheep. Immunology. 1984;53:375–384. [PMC free article: PMC1454821] [PubMed: 6490090]
- 44.
- Salmi M, Jalkanen S. Regulation of lymphocyte traffic to mucosa-associated lymphatic tissues. Gastroenterol Clin North Am. 1991;20:495–510. [PubMed: 1917024]
- 45.
- Mestecky J, McGhee JR, Michalek SM. et al. Concept of the local and common mucosal immune response. Adv Exp Med Biol. 1978;107:185–192. [PubMed: 742482]
- 46.
- Moldoveanu Z, Russell MW, Wu HY. et al. Compartmentalization within the common mucosal immune system. Adv Exp Med Biol. 1995;371A:97–101. [PubMed: 8526027]
- 47.
- Czerkinsky C, Quiding M, Eriksson K. et al. Induction of specific immunity at mucosal surfaces: prospects for vaccine development. Adv Exp Med Biol. 1995;371B:1409–416. [PubMed: 7502829]
- 48.
- Quiding-Jabrink M, Nordstrom I, Granstrom G. et al. Differential expression of tissue-specific adhesion molecules on human circulating antibody-forming cells after systemic, enteric, and nasal immunizations. A molecular basis for the compartmentalization of effector B cell responses. J Clin Invest. 1997;99:1281–1286. [PMC free article: PMC507943] [PubMed: 9077537]
- 49.
- Wu HY, Russell MW. Nasal lymphoid tissue, intranasal immunization, and compartmentalization of the common mucosal immune system. Immunol Res. 1997;16:187–201. [PubMed: 9212364]
- 50.
- Squier CA, Hall BK. The permeability of skin and oral mucosa to water and horseradish peroxidase as related to the thickness of the permeability barrier. J Invest Dermatol. 1985;84:176–179. [PubMed: 2579163]
- 51.
- Fujita M, Reinhart F, Neutra M. Convergence of apical and basolateral endocytic pathways at apical late endosomes in absorptive cells of suckling rat ileum in vivo. J Cell Sci. 1990;97(Pt 2):385–394. [PubMed: 2277098]
- 52.
- Matter K, Mellman I. Mechanisms of cell polarity: Sorting and transport in epithelial cells. Curr Opin Cell Biol. 1994;6:545–554. [PubMed: 7986532]
- 53.
- Neutra MR, Kraehenbuhl JP. Transepithelial transport of proteins by intestinal epithelial cells In: Audus KL, Raub TJ, eds.Biological Barriers to Protein Delivery Vol. 4. Plenum,1993107–129.
- 54.
- Husby S, Jensenius JC, Svehag SE. Passage of undegraded dietary antigen into the blood of healthy adults. Quantification, estimation of size distribution, and relation of uptake to levels of specific antibodies. Scand J Immunol. 1985;22:83–92. [PubMed: 4023632]
- 55.
- Cunningham-Rundles C. Analysis of the gastrointestinal secretory immune barrier in IgA deficiency. Ann Allergy. 1986;57:31–35. [PubMed: 3729080]
- 56.
- Husby S. Dietary antigens: Uptake and humoral immunity in man. APMIS Suppl. 1988;1:1–40. [PubMed: 3288250]
- 57.
- Dahlgren UI, Wold AE, Hanson LA. et al. Secretory antibody response against bacterial antigens and food proteins. Immunol Res. 1991;10:437–440. [PubMed: 1683357]
- 58.
- McGhee JR, Mestecky J, Dertzbaugh MT. et al. The mucosal immune system: From fundamental concepts to vaccine development. Vaccine. 1992;10:75–88. [PubMed: 1539467]
- 59.
- Korenblat PE, Rothberg RM, Minden P. et al. Immune responses of human adults after oral and parenteral exposure to bovine serum albumin. J Allergy. 1968;41:226–235. [PubMed: 5238598]
- 60.
- Strobel S, Mowat AM. Immune responses to dietary antigens: oral tolerance. Immunol Today. 1998;19:173–181. [PubMed: 9577094]
- 61.
- Hershberg RM, Mayer LF. Antigen processing and presentation by intestinal epithelial cells-Polarity and complexity. Immunol Today. 2000;21:123–128. [PubMed: 10689299]
- 62.
- Freter R, Jones GW. Models for studying the role of bacterial attachment in virulence and pathogenesis. Rev Infect Dis. 1983;5 Suppl 4:S647–S658. [PubMed: 6356286]
- 63.
- Bowden GH, Hamilton IR. Survival of oral bacteria. Crit Rev Oral Biol Med. 1998;9:54–85. [PubMed: 9488248]
- 64.
- Henderson B, Wilson M. Commensal communism and the oral cavity. J Dent Res. 1998;77:1674–1683. [PubMed: 9759664]
- 65.
- Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. [PubMed: 11352068]
- 66.
- Neish AS. The gut microflora and intestinal epithelial cells: A continuing dialogue. Microbes Infect. 2002;4:309–317. [PubMed: 11909741]
- 67.
- van der Waaij D. Colonization resistance of the digestive tract-Mechanism and clinical consequences. Nahrung. 1987;31:507–517. [PubMed: 3657928]
- 68.
- Savage DC. Gastrointestinal microflora in mammalian nutrition. Annu Rev Nutr. 1986;6:155–178. [PubMed: 3524615]
- 69.
- Cebra JJ, Jiang HQ, Sterzl J. et al. The role of mucosal microbiota in the development and maintenance of the mucosal immune system. In: Ogra PLeal, ed. Mucosal Immunology. Academic Press, 1999:267-280.
- 70.
- Caugant DA, Levin BR, Selander RK. Genetic diversity and temporal variation in the E. coli population of a human host. Genetics. 1981;98:467–90. [PMC free article: PMC1214454] [PubMed: 7037535]
- 71.
- Hohwy J, Reinholdt J, Kilian M. Population dynamics of Streptococcus mitis in its natural habitat. Infect Immun. 2001;69:6055–6063. [PMC free article: PMC98734] [PubMed: 11553543]
- 72.
- Crabbe PA, Bazin H, Eyssen H. et al. The normal microbial flora as a major stimulus for proliferation of plasma cells synthesizing IgA in the gut. The germ-free intestinal tract. Int Arch Allergy Appl Immunol. 1968;34:362–375. [PubMed: 4176641]
- 73.
- MacDonald TT, Carter PB. Requirement for a bacterial flora before mice generate cells capable of mediating the delayed hypersensitivity reaction to sheep red blood cells. J Immunol. 1979;122:2624–2629. [PubMed: 448137]
- 74.
- Tlaskalova-Hogenova H, Sterzl J, Stepankova R. et al. Development of immunological capacity under germfree and conventional conditions. Ann NY Acad Sci. 1983;409:96–113. [PubMed: 6347006]
- 75.
- Parrott DM, MacDonald TT. The ontogeny of the mucosal immune system in rodents. In: MacDonald TT, ed. Ontogeny of the Immune System of the Gut. CRC Press. 1990:51–67.
- 76.
- Moreau MC, Ducluzeau R, Guy-Grand D. et al. Increase in the population of duodenal immunoglobulin A plasmocytes in axenic mice associated with different living or dead bacterial strains of intestinal origin. Infect Immun. 1978;21:532–539. [PMC free article: PMC422028] [PubMed: 357289]
- 77.
- Klaasen HL, Van der Heijden PJ, Stok W. et al. Apathogenic, intestinal, segmented, filamentous bacteria stimulate the mucosal immune system of mice. Infect Immun. 1993;61:303–306. [PMC free article: PMC302719] [PubMed: 8418051]
- 78.
- Talham GL, Jiang HQ, Bos NA. et al. Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect Immun. 1999;67:1992–2000. [PMC free article: PMC96557] [PubMed: 10085047]
- 79.
- Alaluusua S, Alaluusua SJ, Karjalainen J. et al. The demonstration by ribotyping of the stability of oral Streptococcus mutans infection over 5 to 7 years in children. Arch Oral Biol. 1994;39:467–471. [PubMed: 8067915]
- 80.
- Caufield PW, Cutter GR, Dasanayake AP. Initial acquisition of mutans streptococci by infants: Evidence for a discrete window of infectivity. J Dent Res. 1993;72:37–45. [PubMed: 8418105]
- 81.
- Shroff KE, Meslin K, Cebra JJ. Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect Immun. 1995;63:3904–3913. [PMC free article: PMC173549] [PubMed: 7558298]
- 82.
- Cole MF, Hsu SD, Sheridan MJ. et al. Natural transmission of Streptococcus sobrinus in rats: Saliva and serum antibody responses to colonization. Infect Immun. 1992;60:778–783. [PMC free article: PMC257554] [PubMed: 1531814]
- 83.
- Cole MF, Bryan S, Evans MK. et al. Humoral immunity to commensal oral bacteria in human infants: salivary secretory immunoglobulin A antibodies reactive with Streptococcus mitis biovar 1, Streptococcus oralis, Streptococcus mutans, and Enterococcus faecalis during the first two years of life. Infect Immun. 1999;67:1878–886. [PMC free article: PMC96541] [PubMed: 10085031]
- 84.
- Riviere GR, Wagoner MA, Freeman IL. Chronic peroral immunization of conventional laboratory rats with mutans streptococci leads to stable acquired suppression of salivary antibodies. Oral Microbiol Immunol. 1992;7:137–141. [PubMed: 1408348]
- 85.
- Kroese FG, de WaardR, Bos NA. B-1 cells and their reactivity with the murine intestinal microflora. Semin Immunol. 1996;8:11–18. [PubMed: 8850294]
- 86.
- Kroese FG, Bos NA. Peritoneal B-1 cells switch in vivo to IgA and these IgA antibodies can bind to bacteria of the normal intestinal microflora. Curr Top Microbiol Immunol. 1999;246:343–349. [PubMed: 10396074]
- 87.
- Bos NA, Cebra JJ, Kroese FG. B-1 cells and the intestinal microflora. Curr Top Microbiol Immunol. 2000;252:211–220. [PubMed: 11125478]
- 88.
- Macpherson AJ, Gatto D, Sainsbury E. et al. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–2226. [PubMed: 10864873]
- 89.
- Hayakawa K, Hardy RR, Parks DR. et al. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J Exp Med. 1983;157:202–218. [PMC free article: PMC2186909] [PubMed: 6600267]
- 90.
- Hardy RR, Hayakawa K. Development and physiology of Ly-1 B and its human homolog, Leu-1 B. Immunol Rev. 1986;93:53–79. [PubMed: 3096878]
- 91.
- Herzenberg LA, Stall AM, Lalor PA. et al. The Ly-1 B cell lineage. Immunol Rev. 1986;93:81–102. [PubMed: 3096879]
- 92.
- Kocks C, Rajewsky K. Stable expression and somatic hypermutation of antibody V regions in B-cell developmental pathways. Annu Rev Immunol. 1989;7:537–559. [PubMed: 2653375]
- 93.
- Vakil M, Kearney JF. Functional characterization of monoclonal auto-anti-idiotype antibodies isolated from the early B cell repertoire of BALB/c mice. Eur J Immunol. 1986;16:1151–1158. [PubMed: 2428627]
- 94.
- Elliott M, Kearney JF. Idiotypic regulation of development of the B-cell repertoire. Ann NY Acad Sci. 1992;651:336–345. [PubMed: 1599129]
- 95.
- Lalor PA, Herzenberg LA, Adams S. et al. Feedback regulation of murine Ly-1 B cell development. Eur J Immunol. 1989;19:507–513. [PubMed: 2785046]
- 96.
- Forster I, Rajewsky K. Expansion and functional activity of Ly-1+ B cells upon transfer of peritoneal cells into allotype-congenic, newborn mice. Eur J Immunol. 1987;17:521–528. [PubMed: 2436924]
- 97.
- Karras JG, Wang Z, Huo L. et al. Signal transducer and activator of transcription-3 (STAT3) is constitutively activated in normal, self-renewing B-1 cells but only inducibly expressed in conventional B lymphocytes. J Exp Med. 1997;185:1035–1042. [PMC free article: PMC2196242] [PubMed: 9091577]
- 98.
- Hippen KL, Tze LE, Behrens TW. CD5 maintains tolerance in anergic B cells. J Exp Med. 2000;191:883–890. [PMC free article: PMC2195862] [PubMed: 10704468]
- 99.
- Gary-Gouy H, Harriague J, Bismuth G. et al. Human CD5 promotes B-cell survival through stimulation of autocrine IL-10 production. Blood. 2002;100:4537–4543. [PubMed: 12393419]
- 100.
- Wong SC, Chew WK, Tan JE. et al. Peritoneal CD5+ B-1 cells have signaling properties similar to tolerant B cells. J Biol Chem. 2002;277:30707–30715. [PubMed: 12070149]
- 101.
- Cukrowska B, Sinkora J, Mandel L. et al. Thymic B cells of pig fetuses and germ-free pigs spontaneously produce IgM, IgG and IgA: detection by ELISPOT method. Immunology. 1996;87:487–492. [PMC free article: PMC1384121] [PubMed: 8778038]
- 102.
- Klinman DM. Analysis of B lymphocyte cross-reactivity at the single cell level. J Immunol Methods. 1992;152:217–225. [PubMed: 1380048]
- 103.
- Kasaian MT, Casali P. Autoimmunity-prone B-1 (CD5 B) cells, natural antibodies and self recognition. Autoimmunity. 1993;15:315–329. [PubMed: 7511005]
- 104.
- Settmacher U, Delvig A, Jahn S. Anti-bacterial specificities in the human fetal B cell repertoire. Hum Antibodies Hybridomas. 1994;5:91–95. [PubMed: 7858188]
- 105.
- Allison AC, Nawata Y. Cytokines mediating the proliferation and differentiation of B-1 lymphocytes and their role in ontogeny and phylogeny. Ann NY Acad Sci. 1992;651:200–219. [PubMed: 1376040]
- 106.
- Casali P, Schettino EW. Structure and function of natural antibodies. Curr Top Microbiol Immunol. 1996;210:167–179. [PubMed: 8565555]
- 107.
- Wedemayer GJ, Patten PA, Wang LH. et al. Structural insights into the evolution of an antibody combining site. Science. 1997;276:1665–1669. [PubMed: 9180069]
- 108.
- Bouvet JP, Dighiero G. Cross-reactivity and polyreactivity: The two sides of a coin. Immunol Today. 2000;21:411–412. [PubMed: 10916145]
- 109.
- Yin J, Mundorff EC, Yang PL. et al. A comparative analysis of the immunological evolution of antibody 28B4. Biochemistry. 2001;40:10764–10773. [PubMed: 11535051]
- 110.
- Rothstein TL, Kolber DL. Anti-Ig antibody inhibits the phorbol ester-induced stimulation of peritoneal B cells. J Immunol. 1988;141:4089–4093. [PubMed: 3264301]
- 111.
- Rott O, Charreire J, Mignon-Godefroy K. et al. B cell superstimulatory influenza virus activates peritoneal B cells. J Immunol. 1995;155:134–142. [PubMed: 7541411]
- 112.
- Bao S, Beagley KW, Murray AM. et al. Intestinal IgA plasma cells of the B1 lineage are IL-5 dependent. Immunology. 1998;94:181–188. [PMC free article: PMC1364203] [PubMed: 9741339]
- 113.
- Murakami M, Tsubata T, Okamoto M. et al. Antigen-induced apoptotic death of Ly-1 B cells responsible for autoimmune disease in transgenic mice. Nature. 1992;357:77–80. [PubMed: 1574128]
- 114.
- Bikah G, Carey J, Ciallella JR. et al. CD5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science. 1996;274:1906–1909. [PubMed: 8943203]
- 115.
- Nawata Y, Stall AM, Herzenberg LA. et al. Surface immunoglobulin ligands and cytokines differentially affect proliferation and antibody production by human CD5+ and CD5- B lymphocytes. Int Immunol. 1990;2:603–614. [PubMed: 1703783]
- 116.
- Murakami M, Honjo T. Involvement of B-1 cells in mucosal immunity and autoimmunity. Immunol Today. 1995;16:534–539. [PubMed: 7495491]
- 117.
- Ray SK, Putterman C, Diamond B. Pathogenic autoantibodies are routinely generated during the response to foreign antigen: A paradigm for autoimmune disease. Proc Natl Acad Sci USA. 1996;93:2019–2024. [PMC free article: PMC39902] [PubMed: 8700878]
- 118.
- Adib M, Ragimbeau J, Avrameas S. et al. IgG autoantibody activity in normal mouse serum is controlled by IgM. J Immunol. 1990;145:3807–813. [PubMed: 2246515]
- 119.
- Melero J, Tarrago D, Nunez-Roldan A. et al. Human polyreactive IgM monoclonal antibodies with blocking activity against self-reactive IgG. Scand J Immunol. 1997;45:393–400. [PubMed: 9105427]
- 120.
- Hayakawa K, Hardy RR, Honda M. et al. Ly-1 B cells: functionally distinct lymphocytes that secrete IgM autoantibodies. Proc Natl Acad Sci USA. 1984;81:2494–2498. [PMC free article: PMC345088] [PubMed: 6609363]
- 121.
- Tlaskalova-Hogenova H, Mandel L, Stepankova R. et al. Autoimmunity: From physiology to pathology. Natural antibodies, mucosal immunity and development of B cell repertoire. Folia Biol.(Praha). 1992;38:202–215. [PubMed: 1426416]
- 122.
- Murakami M, Tsubata T, Shinkura R. et al. Oral administration of lipopolysaccharides activates B-1 cells in the peritoneal cavity and lamina propria of the gut and induces autoimmune symptoms in an autoantibody transgenic mouse. J Exp Med. 1994;180:111–121. [PMC free article: PMC2191544] [PubMed: 8006578]
- 123.
- Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7:812–818. [PubMed: 8679125]
- 124.
- Bendelac A, Bonneville M, Kearney JF. Autoreactivity by design: innate B and T lymphocytes. Nat Rev Immunol. 2001;1:177–186. [PubMed: 11905826]
- 125.
- Bouvet JP, Dighiero G. From natural polyreactive autoantibodies to a la carte monoreactive antibodies to infectious agents: Is it a small world after all? Infect Immun. 1998;66:1–4. [PMC free article: PMC107850] [PubMed: 9423831]
- 126.
- Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. [PubMed: 11371363]
- 127.
- Ochsenbein AF, Fehr T, Lutz C. et al. Control of early viral and bacterial distribution and disease by natural antibodies. Science. 1999;286:2156–2159. [PubMed: 10591647]
- 128.
- Carroll MC, Prodeus AP. Linkages of innate and adaptive immunity. Curr Opin Immunol. 1998;10:36–40. [PubMed: 9523108]
- 129.
- Tsuji RF, Szczepanik M, Kawikova I. et al. B cell-dependent T cell responses: IgM antibodies are required to elicit contact sensitivity. J Exp Med. 2002;196:1277–1290. [PMC free article: PMC2193992] [PubMed: 12438420]
- 130.
- Weisz-Carrington P, Schrater AF, Lamm ME. et al. Immunoglobulin isotypes in plasma cells of normal and athymic mice. Cell Immunol. 1979;44:343–351. [PubMed: 455477]
- 131.
- Martin F, Kearney JF. B1 cells: Similarities and differences with other B cell subsets. Curr Opin Immunol. 2001;13:195–201. [PubMed: 11228413]
- 132.
- Fagarasan S, Watanabe N, Honjo T. Generation, expansion, migration and activation of mouse B1 cells. Immunol Rev. 2000;176:205–215. [PubMed: 11043779]
- 133.
- Hayakawa K, Hardy RR, Herzenberg LA. et al. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J Exp Med. 1985;161:1554–1568. [PMC free article: PMC2187623] [PubMed: 3874257]
- 134.
- Hiroi T, Yanagita M, Iijima H. et al. Deficiency of IL-5 receptor alpha-chain selectively influences the development of the common mucosal immune system independent IgA-producing B-1 cell in mucosa-associated tissues. J Immunol. 1999;162:821–828. [PubMed: 9916704]
- 135.
- Pecquet SS, Ehrat C, Ernst PB. Enhancement of mucosal antibody responses to Salmonella typhimurium and the microbial hapten phosphorylcholine in mice with X-linked immunodeficiency by B-cell precursors from the peritoneal cavity. Infect Immun. 1992;60:503–509. [PMC free article: PMC257656] [PubMed: 1730482]
- 136.
- Bos NA, Bun JC, Popma SH. et al. Monoclonal immunoglobulin A derived from peritoneal B cells is encoded by both germ line and somatically mutated VH genes and is reactive with commensal bacteria. Infect Immun. 1996;64:616–623. [PMC free article: PMC173810] [PubMed: 8550216]
- 137.
- Watanabe N, Ikuta K, Fagarasan S. et al. Migration and differentiation of autoreactive B-1 cells induced by activated gamma/delta T cells in antierythrocyte immunoglobulin transgenic mice. J Exp Med. 2000;192:1577–1586. [PMC free article: PMC2193102] [PubMed: 11104800]
- 138.
- Fujihashi K, McGhee JR, Kweon MN. et al. gamma/delta T cell-deficient mice have impaired mucosal immunoglobulin A responses. J Exp Med. 1996;183:1929–1935. [PMC free article: PMC2192480] [PubMed: 8666951]
- 139.
- Hiroi T, Yanagita M, Ohta N. et al. IL-15 and IL-15 receptor selectively regulate differentiation of common mucosal immune system-independent B-1 cells for IgA responses. J Immunol. 2000;165:4329–4337. [PubMed: 11035068]
- 140.
- Fagarasan S, Kinoshita K, Muramatsu M. et al. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature. 2001;413:639–643. [PubMed: 11675788]
- 141.
- Litinskiy MB, Nardelli B, Hilbert DM. et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–829. [PMC free article: PMC4621779] [PubMed: 12154359]
- 142.
- Brandtzaeg P, Baekkevold ES, Morton HC. From B to A the mucosal way. Nat Immunol. 2001;2:1093–1094. [PubMed: 11725295]
- 143.
- Bos NA, Jiang HQ, Cebra JJ. T cell control of the gut IgA response against commensal bacteria. Gut. 2001;48:762–764. [PMC free article: PMC1728317] [PubMed: 11358892]
- 144.
- Baumgarth N, Herman OC, Jager GC. et al. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proc Natl Acad Sci USA. 1999;96:2250–2255. [PMC free article: PMC26769] [PubMed: 10051627]
- 145.
- Kushnir N, Bos NA, Zuercher AW. et al. B2 but not B1 cells can contribute to CD4+ T-cell-mediated clearance of rotavirus in SCID mice. J Virol. 2001;75:5482–5490. [PMC free article: PMC114260] [PubMed: 11356955]
- 146.
- Rasooly L, Abouzied MM, Brooks KH. et al. Polyspecific and autoreactive IgA secreted by hybridomas derived from Peyer's patches of vomitoxin-fed mice: Characterization and possible pathogenic role in IgA nephropathy. Food Chem Toxicol. 1994;32:337–348. [PubMed: 8206429]
- 147.
- Shimoda M, Inoue Y, Azuma N. et al. Natural polyreactive immunoglobulin A antibodies produced in mouse Peyer's patches. Immunology. 1999;97:9–17. [PMC free article: PMC2326812] [PubMed: 10447709]
- 148.
- Donze HH, Lue C, Julian BA. et al. Human peritoneal B-1 cells and the influence of continuous ambulatory peritoneal dialysis on peritoneal and peripheral blood mononuclear cell (PBMC) composition and immunoglobulin levels. Clin Exp Immunol. 1997;109:356–361. [PMC free article: PMC1904743] [PubMed: 9276533]
- 149.
- Bhat NM, Kantor AB, Bieber MM. et al. The ontogeny and functional characteristics of human B-1 (CD5+ B) cells. Int Immunol. 1992;4:243–252. [PubMed: 1377947]
- 150.
- Farstad IN, Carlsen H, Morton HC. et al. Immunoglobulin A cell distribution in the human small intestine: phenotypic and functional characteristics. Immunology. 2000;101:354–363. [PMC free article: PMC2327091] [PubMed: 11106939]
- 151.
- Quan CP, Berneman A, Pires R. et al. Natural polyreactive secretory immunoglobulin A autoantibodies as a possible barrier to infection in humans. Infect Immun. 1997;65:3997–4004. [PMC free article: PMC175574] [PubMed: 9316998]
- 152.
- McGhee JR, Mestecky J, Dertzbaugh MT. et al. The mucosal immune system: From fundamental concepts to vaccine development. Vaccine. 1992;10:75–88. [PubMed: 1539467]
- 153.
- Mestecky J, Bienenstock J, McGhee JR. et al. Historical aspects of mucosal immunologyIn: Ogra PL et al, eds.Mucosal Immunology Academic Press,1999xxiii–xliii.
- 154.
- Winner L, Mack J. et al. New model for analysis of mucosal immunity: Intestinal secretion of specific monoclonal immunoglobulin A from hybridoma tumors protects against Vibrio cholerae infection. Infect Immun. 1991;59:977–982. [PMC free article: PMC258355] [PubMed: 1705246]
- 155.
- Macpherson AJ, Hunziker L, McCoy K. et al. IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect. 2001;3:1021–1035. [PubMed: 11580989]
- 156.
- Williams RC, Gibbons RJ. Inhibition of bacterial adherence by secretory immunoglobulin A: A mechanism of antigen disposal. Science. 1972;177:697–699. [PubMed: 5054144]
- 157.
- Reinholdt J, Kilian M. Interference of IgA protease with the effect of secretory IgA on adherence of oral streptococci to saliva-coated hydroxyapatite. J Dent Res. 1987;66:492–497. [PubMed: 3040826]
- 158.
- Hajishengallis G, Nikolova E, Russell MW. Inhibition of Streptococcus mutans adherence to saliva-coated hydroxyapatite by human secretory immunoglobulin A (S-IgA) antibodies to cell surface protein antigen I/II: Reversal by IgA1 protease cleavage. Infect Immun. 1992;60:5057–5064. [PMC free article: PMC258277] [PubMed: 1333448]
- 159.
- Liljemark WF, Bloomquist CG, Germaine GR. Effect of bacterial aggregation on the adherence of oral streptococci to hydroxyapatite. Infect Immun. 1981;31:935–941. [PMC free article: PMC351408] [PubMed: 7228408]
- 160.
- Edebo L, Lindstrom F, Skoldstom L. et al. On the physical-chemical effect of colostral antibody binding to Escherichia coli O 86. Immunol Commun. 1975;4:587–601. [PubMed: 770313]
- 161.
- Lindh E. Increased risistance of immunoglobulin A dimers to proteolytic degradation after binding of secretory component. J Immunol. 1975;114:284–286. [PubMed: 1090649]
- 162.
- Crottet P, Corthesy B. Secretory component delays the conversion of secretory IgA into antigen-binding competent F(ab')2: A possible implication for mucosal defense. J Immunol. 1998;161:5445–5453. [PubMed: 9820520]
- 163.
- Bronson RA, Cooper GW, Rosenfeld DL. et al. The effect of an IgA1 protease on immunoglobulins bound to the sperm surface and sperm cervical mucus penetrating ability. Fertil Steril. 1987;47:985–991. [PubMed: 3297809]
- 164.
- Phalipon A, Cardona A, Kraehenbuhl JP. et al. Secretory component: A new role in secretory IgA-mediated immune exclusion in vivo. Immunity. 2002;17:107–115. [PubMed: 12150896]
- 165.
- Biesbrock AR, Reddy MS, Levine MJ. Interaction of a salivary mucin-secretory immunoglobulin A complex with mucosal pathogens. Infect Immun. 1991;59:3492–3497. [PMC free article: PMC258911] [PubMed: 1910004]
- 166.
- Hirano M, Kamada M, Maegawa M. et al. Binding of human secretory leukocyte protease inhibitor in uterine cervical mucus to immunoglobulins: Pathophysiology in immunologic infertility and local immune defense. Fertil Steril. 1999;71:1108–1114. [PubMed: 10360919]
- 167.
- Walker WA, Isselbacher KJ, Bloch KJ. Intestinal uptake of macromolecules: Effect of oral immunization. Science. 1972;177:608–610. [PubMed: 4626070]
- 168.
- Stokes CR, Soothill JF, Turner MW. Immune exclusion is a function of IgA. Nature. 1975;255:745–746. [PubMed: 1169692]
- 169.
- Tolo K, Brandtzaeg P, Jonsen J. Mucosal penetration of antigen in the presence or absence of serum-derived antibody. Immunology. 1977;33:733–743. [PMC free article: PMC1445513] [PubMed: 338476]
- 170.
- Lim PL, Rowley D. The effect of antibody on the intestinal absorption of macromolecules and on intestinal permeability in adult mice. Int Arch Allergy Appl Immunol. 1982;68:41–46. [PubMed: 7076322]
- 171.
- Johansen FE, Pekna M, Norderhaug IN. et al. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory componentdeficient mice. J Exp Med. 1999;190:915–922. [PMC free article: PMC2195652] [PubMed: 10510081]
- 172.
- Stokes CR, Taylor B, Turner MW. Association of house-dust and grass-pollen allergies with specific IgA antibody deficiency. Lancet. 19742:485–488. [PubMed: 4136547]
- 173.
- Platts-Mills TA, von MaurRK, Ishizaka K. et al. IgA and IgG anti-ragweed antibodies in nasal secretions. Quantitative measurements of antibodies and correlation with inhibition of histamine release. J Clin Invest. 1976;57:1041–1050. [PMC free article: PMC436748] [PubMed: 59737]
- 174.
- Reed CE, Bubak M, Dunnette S. et al. Ragweed-specific IgA in nasal lavage fluid of ragweed-sensitive allergic rhinitis patients: Increase during the pollen season. Int Arch Allergy Appl Immunol. 1991;94:275–277. [PubMed: 1937889]
- 175.
- Peebles Jr. RS, Hamilton RG, Lichtenstein LM. et al. Antigen-specific IgE and IgA antibodies in bronchoalveolar lavage fluid are associated with stronger antigen-induced late phase reactions. Clin Exp Allergy. 2001;31:239–248. [PubMed: 11251625]
- 176.
- Benson M, Svensson PA, Carlsson B. et al. DNA microarrays to study gene expression in allergic airways. Clin Exp Allergy. 2002;32:301–308. [PubMed: 11929497]
- 177.
- Campbell AM, Vignola AM, Chanez P. et al. Low-affinity receptor for IgE on human bronchial epithelial cells in asthma. Immunology. 1994;82:506–508. [PMC free article: PMC1414915] [PubMed: 7835911]
- 178.
- Campbell AM, Vachier I, Chanez P. et al. Expression of the high-affinity receptor for IgE on bronchial epithelial cells of asthmatics. Am J Respir Cell Mol Biol. 1998;19:92–97. [PubMed: 9651184]
- 179.
- Yang PC, Berin MC, Perdue MH. Enhanced antigen transport across rat tracheal epithelium induced by sensitization and mast cell activation. J Immunol. 1999;163:2769–2776. [PubMed: 10453020]
- 180.
- Silvey KJ, Hutchings AB, Vajdy M. et al. Role of immunoglobulin A in protection against reovirus entry into Murine Peyer's patches. J Virol. 2001;75:10870–10879. [PMC free article: PMC114667] [PubMed: 11602727]
- 181.
- Weltzin R, Lucia-Jandris P, Michetti P. et al. Binding and transepithelial transport of immunoglobulins by intestinal M cells: demonstration using monoclonal IgA antibodies against enteric viral proteins. J Cell Biol. 1989;108:1673–1685. [PMC free article: PMC2115566] [PubMed: 2541137]
- 182.
- Mantis NJ, Cheung MC, Chintalacharuvu KR. et al. Selective adherence of IgA to murine Peyer's patch M cells: Evidence for a novel IgA receptor. J Immunol. 2002;169:1844–1851. [PubMed: 12165508]
- 183.
- Geissmann F, Launay P, Pasquier B. et al. A subset of human dendritic cells expresses IgA Fc receptor (CD89), which mediates internalization and activation upon cross-linking by IgA complexes. J Immunol. 2001;166:346–352. [PubMed: 11123311]
- 184.
- Heystek HC, Moulon C, Woltman AM. et al. Human immature dendritic cells efficiently bind and take up secretory IgA without the induction of maturation. J Immunol. 2002;168:102–107. [PubMed: 11751952]
- 185.
- Arulanandam BP, Raeder RH, Nedrud JG. et al. IgA immunodeficiency leads to inadequate Th cell priming and increased susceptibility to influenza virus infection. J Immunol. 2001;166:226–231. [PubMed: 11123296]
- 186.
- Kaetzel CS, Robinson JK, Chintalacharuvu KR. et al. The polymeric immunoglobulin receptor (secretory component) mediates transport of immune complexes across epithelial cells: A local defense function for IgA. Proc Natl Acad Sci USA. 1991;88:8796–8800. [PMC free article: PMC52597] [PubMed: 1924341]
- 187.
- Brown TA, Russell MW, Mestecky J. Hepatobiliary transport of IgA immune complexes: Molecular and cellular aspects. J Immunol. 1982;128:2183–2186. [PubMed: 7061858]
- 188.
- Robinson JK, Blanchard TG, Levine AD. et al. A mucosal IgA-mediated excretory immune system in vivo. J Immunol. 2001;166:3688–3692. [PubMed: 11238608]
- 189.
- Mazanec MB, Kaetzel CS, Lamm ME. et al. Intracellular neutralization of virus by immunoglobulin A antibodies. Proc Natl Acad Sci USA. 1992;89:6901–6905. [PMC free article: PMC49612] [PubMed: 1323121]
- 190.
- Mazanec MB, Nedrud JG, Kaetzel CS. et al. A three-tiered view of the role of IgA in mucosal defense. Immunol Today. 1993;14:430–435. [PubMed: 8216720]
- 191.
- Duncan AR, Winter G. The binding site for C1q on IgG. Nature. 1988;332:738–740. [PubMed: 3258649]
- 192.
- Griffiss JM. Bactericidal activity of meningococcal antisera. Blocking by IgA of lytic antibody in human convalescent sera. J Immunol. 1975;114:1779–1784. [PubMed: 805178]
- 193.
- Russell MW, Reinholdt J, Kilian M. Anti-inflammatory activity of human IgA antibodies and their Fab alpha fragments: inhibition of IgG-mediated complement activation. Eur J Immunol. 1989;19:2243–2249. [PubMed: 2606139]
- 194.
- Nikolova EB, Tomana M, Russell MW. All forms of human IgA antibodies bound to antigen interfere with complement (C3) fixation induced by IgG or by antigen alone. Scand J Immunol. 1994;39:275–280. [PubMed: 8128187]
- 195.
- Nikolova EB, Tomana M, Russell MW. The role of the carbohydrate chains in complement (C3) fixation by solid-phase-bound human IgA. Immunology. 1994;82:321–327. [PMC free article: PMC1414811] [PubMed: 7927504]
- 196.
- Janoff EN, Fasching C, Orenstein JM. et al. Killing of Streptococcus pneumoniae by capsular polysaccharide-specific polymeric IgA, complement, and phagocytes. J Clin Invest. 1999;104:1139–1147. [PMC free article: PMC408571] [PubMed: 10525053]
- 197.
- Roos A, Bouwman LH, Gijlswijk-Janssen DJ. et al. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol. 2001;167:2861–2868. [PubMed: 11509633]
- 198.
- Fanger MW, Goldstine SN, Shen L. Cytofluorographic analysis of receptors for IgA on human polymorphonuclear cells and monocytes and the correlation of receptor expression with phagocytosis. Mol Immunol. 1983;20:1019–1027. [PubMed: 6646129]
- 199.
- Ito S, Mikawa H, Shinomiya K. et al. Suppressive effect of IgA soluble immune complexes on neutrophil chemotaxis. Clin Exp Immunol. 1979;37:436–440. [PMC free article: PMC1537781] [PubMed: 116785]
- 200.
- Kemp AS, Cripps AW, Brown S. Suppression of leucocyte chemokinesis and chemotaxis by human IgA. Clin Exp Immunol. 1980;40:388–395. [PMC free article: PMC1536983] [PubMed: 7438544]
- 201.
- van EppsDE, Brown SL. Inhibition of formylmethionyl-leucyl-phenylalanine-stimulated neutrophil chemiluminescence by human immunoglobulin A paraproteins. Infect Immun. 1981;34:864–870. [PMC free article: PMC350949] [PubMed: 7333672]
- 202.
- Wolf HM, Fischer MB, Puhringer H. et al. Human serum IgA downregulates the release of inflammatory cytokines (tumor necrosis factor-alpha, interleukin-6) in human monocytes. Blood. 1994;83:1278–1288. [PubMed: 8118031]
- 203.
- Monteiro RC, Kubagawa H, Cooper MD. Cellular distribution, regulation, and biochemical nature of an Fc alpha receptor in humans. J Exp Med. 1990;171:597–613. [PMC free article: PMC2187784] [PubMed: 2137852]
- 204.
- Morton HC, van EgmondM, van de Winkel JG. Structure and function of human IgA Fc receptors (Fc alpha R). Crit Rev Immunol. 1996;16:423–440. [PubMed: 8954257]
- 205.
- Weisbart RH, Kacena A, Schuh A. et al. GM-CSF induces human neutrophil IgA-mediated phagocytosis by an IgA Fc receptor activation mechanism. Nature. 1988;332:647–648. [PubMed: 2451784]
- 206.
- Hostoffer RW, Krukovets I, Berger M. Enhancement by tumor necrosis factor-alpha of Fc alpha receptor expression and IgA-mediated superoxide generation and killing of Pseudomonas aeruginosa by polymorphonuclear leukocytes. J Infect Dis. 1994;170:82–87. [PubMed: 8014525]
- 207.
- Nikolova EB, Russell MW. Dual function of human IgA antibodies: Inhibition of phagocytosis in circulating neutrophils and enhancement of responses in IL-8-stimulated cells. J Leukoc Biol. 1995;57:875–882. [PubMed: 7790770]
- 208.
- Abu-Ghazaleh RI, Fujisawa T, Mestecky J. et al. IgA-induced eosinophil degranulation. J Immunol. 1989;142:2393–2400. [PubMed: 2926137]
- 209.
- Lamkhioued B, Gounni AS, Gruart V. et al. Human eosinophils express a receptor for secretory component. Role in secretory IgA-dependent activation. Eur J Immunol. 1995;25:117–125. [PubMed: 7843220]
- 210.
- Motegi Y, Kita H. Interaction with secretory component stimulates effector functions of human eosinophils but not of neutrophils. J Immunol. 1998;161:4340–4346. [PubMed: 9780211]
- 211.
- Ishizaka K, Ishizaka T. Blocking of Prausnitz-Kustner sensitization with reagin by normal human β2A globulin. J Allergy. 1963;34:395–403. [PubMed: 14066380]
- 212.
- Taylor B, Norman AP, Orgel HA. et al. Transient IgA deficiency and pathogenesis of infantile atopy. Lancet. 1973;2:111–113. [PubMed: 4124040]
- 213.
- Plebani A, Monafo V, Ugazio AG. et al. Comparison of the frequency of atopic diseases in children with severe and partial IgA deficiency. Int Arch Allergy Appl Immunol. 1987;82:485–486. [PubMed: 3570519]
- 214.
- Hanson LA, Bjorkander J, Carlsson B. et al. The heterogeneity of IgA deficiency. J Clin Immunol. 1988;8:159–162. [PubMed: 3292564]
- 215.
- Strober W, Sneller MC. IgA deficiency. Ann Allergy. 1991;66:363–375. [PubMed: 2035899]
- 216.
- Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci USA. 2002;99:351–358. [PMC free article: PMC117564] [PubMed: 11773639]
- 217.
- Weiner HL. Oral tolerance: Immune mechanisms and the generation of Th3-type TGF-beta-secreting regulatory cells. Microbes Infect. 2001;3:947–954. [PubMed: 11564443]
- 218.
- Kagnoff MF. Oral tolerance: Mechanisms and possible role in inflammatory joint diseases. Baillieres Clin Rheumatol. 1996;10:41–54. [PubMed: 8674148]
- 219.
- Sun J, Dirden-Kramer B, Ito K. et al. Antigen-specific T cell activation and proliferation during oral tolerance induction. J Immunol. 1999;162:5868–5875. [PubMed: 10229822]
- 220.
- Meyer AL, Benson J, Song F. et al. Rapid depletion of peripheral antigen-specific T cells in TCR-transgenic mice after oral administration of myelin basic protein. J Immunol. 2001;166:5773–5781. [PubMed: 11313421]
- 221.
- Viney JL, Mowat AM, O'Malley JM. et al. Expanding dendritic cells in vivo enhances the induction of oral tolerance. J Immunol. 1998;160:5815–5825. [PubMed: 9637492]
- 222.
- Holt PG, Stumbles PA, McWilliam AS. Functional studies on dendritic cells in the respiratory tract and related mucosal tissues. J Leukoc Biol. 1999;66:272–275. [PubMed: 10449166]
- 223.
- Akbari O, DeKruyff RH, Umetsu DT. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol. 2001;2:725–731. [PubMed: 11477409]
- 224.
- Brandtzaeg P. Nature and function of gastrointestinal antigen-presenting cells. Allergy. 2001;56 Suppl 67:16–20. [PubMed: 11298000]
- 225.
- Iwasaki A, Kelsall BL. Unique functions of CD11b+, CD8 alpha+, and double-negative Peyer's patch dendritic cells. J Immunol. 2001;166:4884–4890. [PubMed: 11290765]
- 226.
- Jonuleit H, Schmitt E, Steinbrink K. et al. Dendritic cells as a tool to induce anergic and regulatory T cells. Trends Immunol. 2001;22:394–400. [PubMed: 11429324]
- 227.
- Weiner HL. The mucosal milieu creates tolerogenic dendritic cells and T(R)1 and T(H)3 regulatory cells. Nat Immunol. 2001;2:671–672. [PubMed: 11477400]
- 228.
- Bilsborough J, Viney JL. Getting to the guts of immune regulation. Immunology. 2002;106:139–143. [PMC free article: PMC1782714] [PubMed: 12047743]
- 229.
- Janeway CAJr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. [PubMed: 11861602]
- 230.
- Moreau MC, Corthier G. Effect of the gastrointestinal microflora on induction and maintenance of oral tolerance to ovalbumin in C3H/HeJ mice. Infect Immun. 1988;56:2766–2768. [PMC free article: PMC259643] [PubMed: 3417356]
- 231.
- Gaboriau-Routhiau V, Moreau MC. Gut flora allows recovery of oral tolerance to ovalbumin in mice after transient breakdown mediated by cholera toxin or Escherichia coli heat-labile enterotoxin. Pediatr Res. 1996;39:625–629. [PubMed: 8848336]
- 232.
- Wannemuehler MJ, Kiyono H, Babb JL. et al. Lipopolysaccharide (LPS) regulation of the immune response: LPS converts germfree mice to sensitivity to oral tolerance induction. J Immunol. 1982;129:959–965. [PubMed: 6980928]
- 233.
- Michalek SM, Kiyono H, Wannemuehler MJ. et al. Lipopolysaccharide (LPS) regulation of the immune response: LPS influence on oral tolerance induction. J Immunol. 1982;128:1992–1998. [PubMed: 6460815]
- 234.
- Titus RG, Chiller JM. Orally induced tolerance. Definition at the cellular level. Int Arch Allergy Appl Immunol. 1981;65:323–338. [PubMed: 6165688]
- 235.
- Mowat AM, Thomas MJ, MacKenzie S. et al. Divergent effects of bacterial lipopolysaccharide on immunity to orally administered protein and particulate antigens in mice. Immunology. 1986;58:677–683. [PMC free article: PMC1453113] [PubMed: 3488267]
- 236.
- Sudo N, Sawamura S, Tanaka K. et al. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol. 1997;159:1739–1745. [PubMed: 9257835]
- 237.
- Kirjavainen PV, Arvola T, Salminen SJ. et al. Aberrant composition of gut microbiota of allergic infants: a target of bifidobacterial therapy at weaning? Gut. 2002;51:51–55. [PMC free article: PMC1773282] [PubMed: 12077091]
- 238.
- Duchmann R, Kaiser I, Hermann E. et al. Meyer zum Buschenfelde KH. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin Exp Immunol. 1995;102:448–455. [PMC free article: PMC1553362] [PubMed: 8536356]
- 239.
- Khoo UY, Proctor IE, Macpherson AJ. CD4+ T cell down-regulation in human intestinal mucosa: evidence for intestinal tolerance to luminal bacterial antigens. J Immunol. 1997;158:3626–3634. [PubMed: 9103424]
- 240.
- Elson CO, Cong Y. Understanding immune-microbial homeostasis in intestine. Immunol Res. 2002;26:87–94. [PubMed: 12403348]
- 241.
- MacDonald TT. Breakdown of tolerance to the intestinal bacterial flora in inflammatory bowel disease (IBD). Clin Exp Immunol. 1995;102:445–447. [PMC free article: PMC1553355] [PubMed: 8536355]
- 242.
- Shi HN, Ingui CJ, Dodge I. et al. A helminth-induced mucosal Th2 response alters nonresponsiveness to oral administration of a soluble antigen. J Immunol. 1998;160:2449–2455. [PubMed: 9498789]
- 243.
- Schwarze J, Gelfand EW. Respiratory viral infections as promoters of allergic sensitization and asthma in animal models. Eur Respir J. 2002;19:341–349. [PubMed: 11866016]
- 244.
- Elson CO, Ealding W. Cholera toxin feeding did not induce oral tolerance in mice and abrogated oral tolerance to an unrelated protein antigen. J Immunol. 1984;133:2892–2897. [PubMed: 6491278]
- 245.
- Rescigno M, Rotta G, Valzasina B. et al. Dendritic cells shuttle microbes across gut epithelial monolayers. Immunobiology. 2001;204:572–581. [PubMed: 11846220]
- 246.
- Nagata S, McKenzie C, Pender SL. et al. Human Peyer's patch T cells are sensitized to dietary antigen and display a Th cell type 1 cytokine profile. J Immunol. 2000;165:5315–5321. [PubMed: 11046066]
- 247.
- Friedman A, Weiner HL. Induction of anergy or active suppression following oral tolerance is determined by antigen dosage. Proc Natl Acad Sci USA. 1994;91:6688–6692. [PMC free article: PMC44268] [PubMed: 8022835]
- 248.
- Whitacre CC, Gienapp IE, Orosz CG. et al. Oral tolerance in experimental autoimmune encephalomyelitis. III. Evidence for clonal anergy. J Immunol. 1991;147:2155–2163. [PubMed: 1717550]
- 249.
- Melamed D, Friedman A. Direct evidence for anergy in T lymphocytes tolerized by oral administration of ovalbumin. Eur J Immunol. 1993;23:935–942. [PubMed: 8458379]
- 250.
- Schwartz RH. Models of T cell anergy: Is there a common molecular mechanism? J Exp Med. 1996;184:1–8. [PMC free article: PMC2192660] [PubMed: 8691122]
- 251.
- Challacombe SJ, Tomasi Jr. TB. Systemic tolerance and secretory immunity after oral immunization. J Exp Med. 1980;152:1459–1472. [PMC free article: PMC2186016] [PubMed: 7452148]
- 252.
- Melamed D, Friedman A. Modification of the immune response by oral tolerance: Antigen requirements and interaction with immunogenic stimuli. Cell Immunol. 1993;146:412–420. [PubMed: 8174179]
- 253.
- Zivny JH, Moldoveanu Z, Vu HL. et al. Mechanisms of immune tolerance to food antigens in humans. Clin Immunol. 2001;101:158–168. [PubMed: 11683575]
- 254.
- Richman LK, Chiller JM, Brown WR. et al. Enterically induced immunologic tolerance. I. Induction of suppressor T lymphoyctes by intragastric administration of soluble proteins. J Immunol. 1978;121:2429–2434. [PubMed: 82585]
- 255.
- Miller SD, Hanson DG. Inhibition of specific immune responses by feeding protein antigens. IV. Evidence for tolerance and specific active suppression of cell-mediated immune responses to ovalbumin. J Immunol. 1979;123:2344–2350. [PubMed: 90707]
- 256.
- MacDonald TT. Immunosuppression caused by antigen feeding. I. Evidence for the activation of a feedback suppressor pathway in the spleens of antigen-fed mice. Eur J Immunol. 1982;12:767–773. [PubMed: 6216114]
- 257.
- Lider O, Santos LM, Lee CS. et al. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein. II. Suppression of disease and in vitro immune responses is mediated by antigen-specific CD8+ T lymphocytes. J Immunol. 1989;142:748–752. [PubMed: 2464023]
- 258.
- Miller A, Lider O, Roberts AB. et al. Suppressor T cells generated by oral tolerization to myelin basic protein suppress both in vitro and in vivo immune responses by the release of transforming growth factor beta after antigen-specific triggering. Proc Natl Acad Sci USA. 1992;89:421–425. [PMC free article: PMC48249] [PubMed: 1370356]
- 259.
- Miller A, Lider O, Weiner HL. Antigen-driven bystander suppression after oral administration of antigens. J Exp Med. 1991;174:791–798. [PMC free article: PMC2118953] [PubMed: 1717632]
- 260.
- Miller A, al SabbaghA, Santos LM. et al. Epitopes of myelin basic protein that trigger TGF-beta release after oral tolerization are distinct from encephalitogenic epitopes and mediate epitope-driven bystander suppression. J Immunol. 1993;151:7307–7315. [PubMed: 7505026]
- 261.
- Weiner HL. Oral tolerance: Immune mechanisms and treatment of autoimmune diseases. Immunol Today. 1997;18:335–343. [PubMed: 9238837]
- 262.
- Mowat AM, Weiner HL. Oral tolerance, physiological basis and clinical applications In: Ogra et al, eds.Mucosal Immunology Academic Press,1999587–618.
- 263.
- Groux H, O'Garra A, Bigler M. et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. [PubMed: 9338786]
- 264.
- MacDonald TT. Effector and regulatory lymphoid cells and cytokines in mucosal sites. Curr Top Microbiol Immunol. 1999;236:113–135. [PubMed: 9893358]
- 265.
- Singh B, Read S, Asseman C. et al. Control of intestinal inflammation by regulatory T cells. Immunol Rev. 2001;182:190–200. [PubMed: 11722634]
- 266.
- McMenamin C, Pimm C, McKersey M. et al. Regulation of IgE responses to inhaled antigen in mice by antigen-specific gamma delta T cells. Science. 1994;265:1869–1871. [PubMed: 7916481]
- 267.
- Yamamoto M, Kiyono H. Immunoregulatory functions of mucosal gammadelta T cells. Microbes Infect. 1999;1:241–246. [PubMed: 10801236]
- 268.
- Yamanaka T, Helgeland L, Farstad IN. et al. Microbial colonization drives lymphocyte accumulation and differentiation in the follicle-associated epithelium of Peyer's patches. J Immunol. 2003;170:816–822. [PubMed: 12517945]
- 269.
- Eynon EE, Parker DC. Small B cells as antigen-presenting cells in the induction of tolerance to soluble protein antigens. J Exp Med. 1992;175:131–138. [PMC free article: PMC2119092] [PubMed: 1730913]
- 270.
- Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992;258:1156–1159. [PubMed: 1439825]
- 271.
- Vella AT, Scherer MT, Schultz L. et al. B cells are not essential for peripheral T-cell tolerance. Proc Natl Acad Sci USA. 1996;93:951–955. [PMC free article: PMC40165] [PubMed: 8570666]
- 272.
- Hashimoto A, Yamada H, Matsuzaki G. et al. Successful priming and tolerization of T cells to orally administered antigens in B-cell-deficient mice. Cell Immunol. 2001;207:36–40. [PubMed: 11161451]
- 273.
- Gonnella PA, Waldner HP, Weiner HL. B cell-deficient (mu MT) mice have alterations in the cytokine microenvironment of the gut-associated lymphoid tissue (GALT) and a defect in the low dose mechanism of oral tolerance. J Immunol. 2001;166:4456–4464. [PubMed: 11254701]
- 274.
- Fujihashi K, Dohi T, Rennert PD. et al. Peyer's patches are required for oral tolerance to proteins. Proc Natl Acad Sci USA. 2001;98:3310–3315. [PMC free article: PMC30650] [PubMed: 11248075]
- 275.
- Spahn TW, Fontana A, Faria AM. et al. Induction of oral tolerance to cellular immune responses in the absence of Peyer's patches. Eur J Immunol. 2001;31:1278–1287. [PubMed: 11298355]
- 276.
- Spahn TW, Weiner HL, Rennert PD. et al. Mesenteric lymph nodes are critical for the induction of high-dose oral tolerance in the absence of Peyer's patches. Eur J Immunol. 2002;32:1109–1113. [PubMed: 11920578]
- 277.
- Andre C, Heremans JF, Vaerman JP. et al. A mechanism for the induction of immunological tolerance by antigen feeding: Antigen-antibody complexes. J Exp Med. 1975;142:1509–1519. [PMC free article: PMC2190065] [PubMed: 1104748]
- 278.
- Kagnoff MF. Effects of antigen-feeding on intestinal and systemic immune responses. III. Antigen-specific serum-mediated suppression of humoral antibody responses after antigen feeding. Cell Immunol. 1978;40:186–203. [PubMed: 81111]
- 279.
- Kagnoff MF. Effects of antigen-feeding on intestinal and systemic immune responses. II. Suppression of delayed-type hypersensitivity reactions. J Immunol. 1978;120:1509–1513. [PubMed: 659859]
- 280.
- Kagnoff MF. Effects of antigen-feeding on intestinal and systemic immune responses. IV. Similarity between the suppressor factor in mice after erythrocyte-lysate injection and erythrocyte feeding. Gastroenterology. 1980;79:54–61. [PubMed: 7380224]
- 281.
- Karlsson MR, Kahu H, Hanson LA. et al. An established immune response against ovalbumin is suppressed by a transferable serum factor produced after ovalbumin feeding: A role of CD25+ regulatory cells. Scand J Immunol. 2002;55:470–477. [PubMed: 11975758]
- 282.
- Hanson DG, Vaz NM, Maia LC. et al. Inhibition of specific immune responses by feeding protein antigens. III. Evidence against maintenance of tolerance to ovalbumin by orally induced antibodies. J Immunol. 1979;123:2337–2343. [PubMed: 114587]
- 283.
- Jarrett EE, Hall E. The development of IgE-suppressive immunocompetence in young animals: Influence of exposure to antigen in the presence or absence of maternal immunity. Immunology. 1984;53:365–373. [PMC free article: PMC1454820] [PubMed: 6490089]
- 284.
- Falth-Magnusson K, Kjellman NI, Magnusson KE. Antibodies IgG, IgA, and IgM to food antigens during the first 18 months of life in relation to feeding and development of atopic disease. J Allergy Clin Immunol. 1988;81:743–749. [PubMed: 3356852]
- 285.
- Ogra SS, Weintraub D, Ogra PL. Immunologic aspects of human colostrum and milk. III. Fate and absorption of cellular and soluble components in the gastrointestinal tract of the newborn. J Immunol. 1977;119:245–248. [PubMed: 577500]
- 286.
- Jarvinen KM, Laine ST, Jarvenpaa AL. et al. Does low IgA in human milk predispose the infant to development of cow's milk allergy? Pediatr Res. 2000;48:457–462. [PubMed: 11004235]
- 287.
- Machtinger S, Moss R. Cow's milk allergy in breast-fed infants: The role of allergen and maternal secretory IgA antibody. J Allergy Clin Immunol. 1986;77:341–347. [PubMed: 3484762]
- 288.
- Duchen K, Casas R, Fageras-Bottcher M. et al. Human milk polyunsaturated long-chain fatty acids and secretory immunoglobulin A antibodies and early childhood allergy. Pediatr Allergy Immunol. 2000;11:29–39. [PubMed: 10768733]
- 289.
- Saarinen KM, Vaarala O, Klemetti P. et al. Transforming growth factor-beta1 in mothers' colostrum and immune responses to cows' milk proteins in infants with cows' milk allergy. J Allergy Clin Immunol. 1999;104:1093–1098. [PubMed: 10550758]
- 290.
- Sly PD, Holt PG. Breast is best for preventing asthma and allergies—Or is it? Lancet. 2002. pp. 887–888. [PubMed: 12354466]
- 291.
- Sorensen CH, Kilian M. Bacterium-induced cleavage of IgA in nasopharyngeal secretions from atopic children. Acta Pathol Microbiol Immunol Scand [C] 1984;92:85–87. [PubMed: 6369879]
- 292.
- Kilian M, Husby S, Host A. et al. Increased proportions of bacteria capable of cleaving IgA1 in the pharynx of infants with atopic disease. Pediatr Res. 1995;38:182–186. [PubMed: 7478813]
- 293.
- Heppell LM, Kilshaw PJ. Immune responses in guinea pigs to dietary protein. I. Induction of tolerance by feeding with ovalbumin. Int Arch Allergy Appl Immunol. 1982;68:54–59. [PubMed: 7076323]
- 294.
- Mowat AM, Strobel S, Drummond HE. et al. Immunological responses to fed protein antigens in mice. I. Reversal of oral tolerance to ovalbumin by cyclophosphamide. Immunology. 1982;45:105–113. [PMC free article: PMC1555154] [PubMed: 6173311]
- 295.
- Melamed D, Friedman A. In vivo tolerization of Th1 lymphocytes following a single feeding with ovalbumin: Anergy in the absence of suppression. Eur J Immunol. 1994;24:1974–1981. [PubMed: 8088317]
- 296.
- Vives J, Parks DE, Weigle WO. Immunologic unresponsiveness after gastric administration of human gamma-globulin: Antigen requirements and cellular parameters. J Immunol. 1980;125:1811–1816. [PubMed: 6157745]
- 297.
- Husby S, Mestecky J, Moldoveanu Z. et al. Oral tolerance in humans. T cell but not B cell tolerance after antigen feeding. J Immunol. 1994;152:4663–4670. [PubMed: 8157979]
- 298.
- Waldo FB, van den Wall Bake AW, Mestecky J. et al. Suppression of the immune response by nasal immunization. Clin Immunol Immunopathol. 1994;72:30–34. [PubMed: 8020191]
- 299.
- MacDonald TT. Immunosuppression caused by antigen feeding. II. Suppressor T cells mask Peyer's patch B cell priming to orally administered antigen. Eur J Immunol. 1983;13:138–142. [PubMed: 6219883]
- 300.
- Sugita-Konishi Y, Smart CJ, Trejdosiewicz LK. Regulation of intestinal immunoglobulin production in response to dietary ovalbumin. Int Arch Allergy Immunol. 1992;98:64–69. [PubMed: 1624208]
- 301.
- Stok W, Van der Heijden PJ, Bianchi AT. Conversion of orally induced suppression of the mucosal immune response to ovalbumin into stimulation by conjugating ovalbumin to cholera toxin or its B subunit. Vaccine. 1994;12:521–526. [PubMed: 8036826]
- 302.
- Elson CO, Holland SP, Dertzbaugh MT. et al. Morphologic and functional alterations of mucosal T cells by cholera toxin and its B subunit. J Immunol. 1995;154:1032–1040. [PubMed: 7822780]
- 303.
- Grdic D, Hornquist E, Kjerrulf M. et al. Lack of local suppression in orally tolerant CD8-deficient mice reveals a critical regulatory role of CD8+ T cells in the normal gut mucosa. J Immunol. 1998;160:754–762. [PubMed: 9551910]
- 304.
- Kato H, Fujihashi K, Kato R. et al. Oral tolerance revisited: Prior oral tolerization abrogates cholera toxin-induced mucosal IgA responses. J Immunol. 2001;166:3114–3121. [PubMed: 11207263]
- 305.
- Russell MW, Prince SJ, Lidthart GJ. et al. Comparison of salivary and serum antibodies to common environmental antigens in elderly, edentulous, and normal adult subjects. AGING: Immunology and infectious disease. 1990;2:275–286.
- 306.
- Rumbo M, Chirdo FG, Anon MC. et al. Detection and characterization of antibodies specific to food antigens (gliadin, ovalbumin and beta-lactoglobulin) in human serum, saliva, colostrum and milk. Clin Exp Immunol. 1998;112:453–458. [PMC free article: PMC1904991] [PubMed: 9649214]
- 307.
- Ernst PB, Lee ST, Maeba J. et al. A role for isotype-specific binding factors in the regulation of IgA- and IgG-specific responses by the anti/contrasuppressor T cell circuit. J Immunol. 1989;143:1426–1432. [PubMed: 2569491]
- 308.
- Fujihashi K, Taguchi T, Aicher WK. et al. Immunoregulatory functions for murine intraepithelial lymphocytes: Gamma/delta T cell receptor-positive (TCR+) T cells abrogate oral tolerance, while alpha/beta TCR+ T cells provide B cell help. J Exp Med. 1992;175:695–707. [PMC free article: PMC2119151] [PubMed: 1531495]
- 309.
- Iijima H, Takahashi I, Kiyono H. Mucosal immune network in the gut for the control of infectious diseases. Rev Med Virol. 2001;11:117–133. [PubMed: 11262530]
- 310.
- Kilian M, Roland K, Mestecky J. Interference of secretory immunoglobulin A with sorption of oral bacteria to hydroxyapatite. Infect Immun. 1981;31:942–951. [PMC free article: PMC351409] [PubMed: 7014466]
- 311.
- Ogra PL. et al. Mucosal Immunology. Academic Press. 1999:43–64.
- 312.
- Pruitt KM, Rahemtulla B, Rahemtulla F. et al. Innate humoral factors In: Ogra PL et al, eds.Mucosal Immunology Academic Press,199965–88.
- 313.
- Brandtzaeg P, Fjellanger I, Gjeruldsen ST. Adsorption of immunolgobulin A onto oral bacteria in vivo. J Bacteriol. 1968;96:242–249. [PMC free article: PMC252279] [PubMed: 4174058]
- 314.
- van der Waaij LA, Limburg PC, Mesander G. et al. In vivo IgA coating of anaerobic bacteria in human faeces. Gut. 1996;38:348–354. [PMC free article: PMC1383061] [PubMed: 8675085]
- 315.
- Kilian M, Reinholdt J. A hypothetical model for the development of invasive infection due to IgA1 protease-producing bacteria. Adv Exp Med Biol. 1987;216B:1261–1269. [PubMed: 3122532]
- 316.
- Binscheck T, Bartels F, Bergel H. et al. IgA protease from Neisseria gonorrhoeae inhibits exocytosis in bovine chromaffin cells like tetanus toxin. J Biol Chem. 1995;270:1770–1774. [PubMed: 7829513]
- 317.
- Hauck CR, Meyer TF. The lysosomal/phagosomal membrane protein h-lamp-1 is a target of the IgA1 protease of Neisseria gonorrhoeae. FEBS Lett. 1997;405:86–90. [PubMed: 9094430]
- 318.
- Beck SC, Meyer TF. IgA1 protease from Neisseria gonorrhoeae inhibits TNFalpha-mediated apoptosis of human monocytic cells. FEBS Lett. 2000;472:287–292. [PubMed: 10788628]
- 319.
- Firer MA, Hosking CS, Hill DJ. Cow's milk allergy and eczema: patterns of the antibody response to cow's milk in allergic skin disease. Clin Allergy. 1982;12:385–390. [PubMed: 7116615]
- 320.
- Husby S, Oxelius VA, Teisner B. et al. Humoral immunity to dietary antigens in healthy adults. Occurrence, isotype and IgG subclass distribution of serum antibodies to protein antigens. Int Arch Allergy Appl Immunol. 1985;77:416–422. [PubMed: 4018884]
- 321.
- Rothberg RM, Farr RS. Antibodies in rabbits fed milk and their similarities to antibodies in some human sera. J Allergy. 1965;36:450–462. [PubMed: 5212719]
- 322.
- Scott H, Rognum TO, Midtvedt T. et al. Age-related changes of human serum antibodies to dietary and colonic bacterial antigens measured by an enzyme-linked immunosorbent assay. Acta Pathol Microbiol Immunol Scand [C] 1985;93:65–70. [PubMed: 4013749]
- 323.
- Barnes RM. IgG and IgA antibodies to dietary antigens in food allergy and intolerance. Clin Exp Allergy. 1995;25 Suppl 1:7–9. [PubMed: 8542462]
- 324.
- Unsworth DJ, Walker-Smith JA, Holborow EJ. Gliadin and reticulin antibodies in childhood coeliac disease. Lancet. 1983;1:874–875. [PubMed: 6132200]
- 325.
- Kemp M, Husby S, Larsen ML. et al. ELISA analysis of IgA subclass antibodies to dietary antigens. Elevated IgA1 antibodies in children with coeliac disease. Int Arch Allergy Appl Immunol. 1988;87:247–253. [PubMed: 3264546]
- 326.
- Hvatum M, Scott H, Brandtzaeg P. Serum IgG subclass antibodies to a variety of food antigens in patients with coeliac disease. Gut. 1992;33:632–638. [PMC free article: PMC1379292] [PubMed: 1612478]
- Structure of IgA
- Induction of IgA Responses
- Role of the Mucosal Microflora in the Development and Function of the Mucosal Immune System
- IgA Produced by B1 cells
- IgA Antibody Functions
- Mucosal Tolerance
- Mucosal IgA Antibody Responses As Targets of Mucosal Tolerance
- Clinical Aspects of IgA Antibodies
- Concluding Remarks
- Acknowledgements
- References
- IgA and Mucosal Homeostasis - Madame Curie Bioscience DatabaseIgA and Mucosal Homeostasis - Madame Curie Bioscience Database
- Epithelial-Mesenchymal Transformation in the Embryonic Heart - Madame Curie Bios...Epithelial-Mesenchymal Transformation in the Embryonic Heart - Madame Curie Bioscience Database
- Inferring Evolutionary History through Inter- and Intraspecific DNA Sequence Com...Inferring Evolutionary History through Inter- and Intraspecific DNA Sequence Comparison: The Drosophila janus and ocnus Genes - Madame Curie Bioscience Database
- SELECTIVE NEURODEGENERATION, NEUROPATHOLOGY AND SYMPTOM PROFILES IN HUNTINGTON'S...SELECTIVE NEURODEGENERATION, NEUROPATHOLOGY AND SYMPTOM PROFILES IN HUNTINGTON'S DISEASE - Madame Curie Bioscience Database
- The Restriction Point of the Cell Cycle - Madame Curie Bioscience DatabaseThe Restriction Point of the Cell Cycle - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...