

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Statistical analysis of aligned DNA sequences, both among and within species, has proven to be a valuable tool for inferring the evolutionary history of genetic loci. Of particular interest are cases where the observed data depart from the neutral expectation and suggest adaptive evolution due to positive natural selection. In this chapter, we use the Drosophila janusA, janusB and ocnus genes to demonstrate methods of evolutionary inference from both inter- and intraspecific DNA sequence data. Interspecific comparisons suggest that these three paralogous, testes-expressed genes have diverged in function following duplication and have evolved under different selective constraints. The three genes show the increased rate of between-species amino acid replacement common to genes with reproductive function, which may be the result of recurrent positive selection. Intraspecific comparison of D. simulans alleles provides evidence for more recent positive selection in this region of the genome. There are two divergent haplotype groups segregating in the worldwide population, one of which has risen to high frequency within the past 5000 years. The observed pattern of within-species variation may best be explained by a selective sweep that has not gone to completion.
Detecting Selection by Inter- and Intraspecific DNA Sequence Comparison
Recent advances in DNA sequencing technology have lead to an enormous increase in the amount of DNA sequence available for testing evolutionary hypotheses. This wealth of data includes collections of homologous gene sequences from different species as well as multiple sequences of particular genes sampled from different individuals within a single species. These data clearly indicate that there are abundant changes in DNA sequence between species as well as large amounts of DNA sequence polymorphism within species. Kimura's1 neutral theory of molecular evolution explains this observation by assuming that the vast majority of nucleotide polymorphisms within species are the result of neutral mutations that have risen to detectable frequency due to random genetic drift in finite populations. Under Kimura's model, sequence differences between species reflect neutral polymorphisms that have drifted to fixation in one or the other species. Because evolutionary geneticists are primarily interested in the molecular basis of adaptive evolution, they focus largely on cases where the data depart from the neutral model. Such departures from neutrality may be detected through both interspecific and intraspecific DNA sequence comparison. Because divergence and polymorphism have a simple relationship under the neutral theory, the power to detect departures from neutrality is often increased by combining both inter- and intraspecific studies.
Interspecific DNA sequence comparisons are useful for determining the evolutionary history of particular genes and for identifying functionally important regions of genes and genomes. For example, interspecific comparisons can be used to estimate the order and timing of gene duplication events. They may also be used to identify functionally important protein motifs or gene regulatory sequences, which are expected to be highly conserved among species. One parameter that can be estimated from interspecifc DNA sequence data is the ratio of the nonsynonymous substituton rate (Ka) to the synonymous substitution rate (Ks). The Ka/Ks ratio (sometimes designated as dN/dS or ω) can reflect the selective constraints on a gene, particularly those acting to remove amino acid replacement substitutions. Ka/Ks = 1 is expected for genes evolving neutrally, where selection neither favors nor disfavors changes in the amino acid sequence. Ka/Ks < 1 is the commonly observed situation and suggests negative (purifying) selection acting to remove amino acid replacements.Ka/Ks > 1 indicates positive selection favoring the fixation of amino acid replacements. Ka/Ks > 1 is a strict criterion for the detection of positive selection and is rarely observed.2 Notable exceptions include the antigenic proteins of some pathogens,3,4 which are under strong selection to evade the host's immune response, and some male reproductive proteins that may be subject to sexual selection.5-7 Recently, a number of maximum likelihood-based approaches for estimating Ka/Ks ratios for particular protein regions or amino acid positions have been introduced4,8 that have increased statistical power to detect signatures of positive selection from interspecific data, particularly when a wide sampling of species with a known phylogenetic relationship is available.
Intraspecific DNA sequence comparisons can allow the detection of recent positive selection, that is, selection acting much more recently than the time of the last speciation event. For example, one can infer selection by a departure from the neutral expectation in the average number of nucleotide differences between two sampled alleles, also known as nucleotide heterozygosity. Positive directional selection is expected to cause a reduction in nucleotide heterozygosity in the genomic region linked to the selected site. This is because as the selected variant increases in frequency in the population and eventually goes to fixation, it will drag linked neutral variants to fixation along with it. Thus this phenomenon, known as genetic hitchhiking, or a selective sweep, leads to a decrease in the standing level of DNA polymorphism.9 The extent of the chromosomal region affected by a selective sweep depends on the strength of selection and the local recombination rate.10,11 The lower the recombination rate, or the stronger the selection, the larger the region of the genome that will be affected. The observation that chromosomal regions with little or no recombination show reduced levels of polymorphism when compared to regions of normal recombination in Drosophila12-15 is consistent with genetic hitchhiking having acted in these areas. However, it has also been proposed that this observation can be explained by recurrent purifying selection removing linked neutral variants from the population. This mechanism, known as background selection,16 is also expected to be stronger in regions of reduced recombination.
A number of statistical tests have been proposed to detect departures from neutrality using only intraspecific polymorphism data. For example, the test of Tajima17 compares the observed frequencies of variants at polymorphic sites to the frequencies expected under the neutral theory. Other tests, commonly referred to as haplotype tests, compare the distribution of variants at segregating sites among chromosomes within a population sample to the neutral expectation.18,19 Significant departures from neutrality detected by either Tajima's or the haplotype tests may be attributable to various forms of natural selection or to demographic factors reflecting the historical size and geographic distribution of the population. For example, an excess of low frequency variants detected by Tajima's test can result from either a recent selective sweep or from recent population expansion. Typically, it is impossible to distinguish among these possibilities from analysis of a single locus, and data from additional loci are required before a conclusion can be reached.
The combination of inter- and intraspecific DNA sequence comparisons can be used for more powerful statistical methods of detecting departures from neutrality. In addition to DNA sequence polymorphism data from within a single species, the presence of at least one sequence from a closely related species can be of great value for two reasons. First, the DNA sequence from the related species can be used to classify the within-species DNA polymorphisms as derived or ancestral. In this case, the ancestral variant at the polymorphic site is assumed to be the one that matches the outgroup sequence. Examples of neutrality tests that consider the frequency of derived variants include those of Fu and Li,20 Fay and Wu,21 and Kim and Stephan.22 The latter two tests are particularly relevant to genetic hitchhiking, because hitchhiking with a positively selected variant is expected to increase the frequency of derived variants in a population sample. The second benefit of having at least one sequence from a closely related species is that the divergence data can be used to eliminate variation in mutation rates or selective constraints as a cause for between-locus (or within-locus) differences in intraspecific polymorphism. For example, the low levels of DNA sequence polymorphism observed in regions of low recombination in Drosophila could be explained in theory by relatively low mutation rates in these regions of the genome. The observation that genes in regions of low recombination do not show a correlated reduction in interspecific divergence eliminates this possibility.23 The expected correlation between divergence and polymorphism has lead to the development of several statistical tests of neutrality, including the HKA test24 and the MK test.25 The latter test compares ratios of polymorphism and divergence between synonymous and nonsynonymous sites from within a single protein-encoding gene. An excess of nonsynonymous changes between species can occur as a result of positive selection for amino acid replacements, although there may be other causes for this pattern as well.
In this chapter, we use the Drosophila janus and ocnus genes to illustrate the utility of inter- and intraspecific DNA sequence comparison for inferring evolutionary history. Several features of these genes make them interesting for evolutionary studies. First, they are a group of paralogous genes that have been created by several gene duplication events, with apparent specialization and functional divergence following duplication. Second, they are male-specific, testis-expressed genes that show the increased rate of molecular evolution characteristic of many Drosophila genes with reproductive function. Finally, analysis of DNA sequence polymorphism within D. simulans suggests the recent action of positive selection in this region of the genome, resulting in a selective sweep of sequence variation.
Molecular Evolution of the janus and ocnus Genes in the D. melanogaster Species Subgroup
In D. melanogaster, janusA (janA), janusB (janB) and ocnus (ocn) are located in a gene-dense region near the telomeric end of the right arm of chromosome 3. The genomic organization of this region is shown in (fig. 1). The janA and janB transcriptional units are adjoining, with the 3' end of janA overlapping with the 5' end of janB (fig. 1).26 Despite this overlap, janA and janB produce separate transcripts that are under the control of independent promoters.27 The ocn transcriptional unit begins approximately 250 bp downstream from the janB polyadenylation site, and there is no overlap between the janB and ocn transcripts (fig. 1). Phylogenetic comparison of janA, janB, and ocn sequences among species of the D. melanogaster species subgroup, as well as janA and janB sequences from the more distantly related D. pseudoobscura, suggests two separate duplication events have occurred in this region of the genome (fig. 2).28,29 The janB and ocn genes share greater homology with each other than either does with janA, indicating that they are the result of a more recent duplication. Consistent with this interpretation, janB and ocn are equally divergent from janA. Since janB and ocn are found in all members of the D. melanogaster species subgroup, the duplication event that produced them must have occurred at least 10 million years ago (mya). The original duplication that produced janA and janB must predate the divergence of the D. melanogaster and D. obscura group lineages, placing this duplication event at a minimum of 25 mya. A single gene with greatest homology to janA was found in the C. elegans genome30 suggesting that the ancestral gene was most similar in sequence to janA.

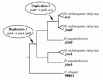
The evolution of these genes appears to have included increasingly restrictive function following duplication, as evidenced by their expression pattern. Experimental studies indicate that janB and ocn produce testis-specific transcripts.26,28 In addition, translation of janB mRNA is restricted to the postmeiotic stages of sperm development through control elements located in the 5' UTR.29 Conserved control elements are also found in the ocn 5' UTR, suggesting that it is under similar post-transcriptional regulation.28 In contrast, janA produces two alternatively-spliced transcripts, one that is specific to testes and another that is found in various tissues and in both sexes.26 The two janA transcripts differ in their 5' UTRs and their translation begins at different start codons, with initiation of the sperm-specific polypeptide occurring 48 bp downstream of the general initiation site.26 As janA appears to be the most ancestral in sequence, the general expression pattern observed for janA is likely the ancestral state, with specialization to the testis-specific expression of janB and ocn occurring after duplication.
Additional support for the functional divergence of the janA, janB, and ocn genes comes from an analysis of Ka/Ks ratios of the three genes within the D. melanogaster species subgroup. If the three genes have diverged in function, then they are expected to differ in their selective constraints and potentially in their Ka/Ks ratios. To test this hypothesis, two evolutionary models were compared using a maximum likelihood approach.28 The null model (no functional divergence) predicts that the three genes should not differ significantly from each other in their level of selective constraint, and so similar Ka/Ks ratios are expected for all three genes over all branches of the phlyogenetic tree. The alternative model predicts that following duplication each gene was subject to unique selective constraints and thus the three genes should differ in their Ka/Ks ratios. For this model, three distinct Ka/Ks ratios are expected, one each for janA, janB, and ocn. Maximum likelihood analysis indicates that the observed data are much more likely under the alternative model than under the null model.28 Thus, there is strong evidence for functional and selective divergence of the three genes following duplication.
The Ka/Ks ratios for janA, janB, and ocn are all well below one, so there is no evidence for positive selection from the interspecific data using this strict criterion. However, all three genes have significantly higher Ka/Ks ratios than other genes (all encoding metabolic enzymes) that have been sequenced in species of the D. melanogaster species subgroup.28 This observation is consistent with a general pattern of increased evolutionary rate in Drosophila genes with a sex-related function.31 This increased rate of molecular evolution could have two very different explanations. One possibility is that positive selection has favored an increased fraction of amino acid replacements in reproductive genes relative to genes with other functions. This may be the result of selection on reproductive traits such as male fertility or sperm competition. The other possibility is that selective constraints are relaxed in reproductive genes, and that these genes accept more neutral amino acid changes than nonreproductive genes. Except in rare cases where the Ka/Ka ratio is significantly greater than one, such as in the accessory protein gene Acp26Aa,5 it is generally not possible to distinguish between these two explanations solely through interspecific comparison of protein-encoding sequences. Additional helpful information may be gained from intraspecific studies, i. e. from analysis of DNA sequence polymorphism.
DNA Sequence Polymorphism in the janus-ocnus Region of D. simulans
The pattern of intraspecific DNA sequence polymorphism in the jan-ocn region of D. simulans provides evidence for the recent action of positive selection in this region of the genome. 32 A graphical representation of the polymorphic nucleotide sites in the janA, janB, and ocn genes of 36 D. simulans chromosomes sampled from a worldwide distribution is shown in (fig. 3). In this figure, each vertical column represents a segregating site and each horizontal row represents a different chromosome. At each site, the derived variant (inferred from the D. melanogaster outgroup sequence) is shown in black. The unusual arrangement of variation in this sample is immediately apparent. Many alleles are identical or nearly identical in their DNA sequence, while a few are quite different. This pattern is strongest over the region containing the 3' end of janA and the entire janB gene. Here there are 16 chromosomes that are identical in their combination of variants at segregating sites (haplotype), and 9 more chromosomes that differ at only a single site. We refer to this group of 25 chromosomes as haplotype group 1 and the remaining chromosomes as haplotype group 2. Polymorphism within haplotype group 2 is in the range typically observed in D. simulans,33 while haplotype group 1 shows a marked reduction in diversity. This observation, along with genealogical reconstruction of the alleles, suggests that haplotype group 2 represents a diverse collection of ancestral alleles and that haplotype group 1 represents a collection of more recently derived alleles.32 Within haplotype group 1 there is evidence for two distinct recombination events occurring on either side of janB. Over the first 31 segregating sites in janA there are five alleles that are identical to each other, but differ from the other haplotype group 1 alleles at seven sites (fig. 3). Similarly, there are seven ocn alleles that are identical to each other, but differ from the other haplotype group 1 alleles at 12 sites (fig. 3). In both cases, the differing alleles contain many ancestral variants, indicating recombination between an allele of haplotype group 1 and an allele of haplotype group 2.

A number of statistical tests reject the neutral evolution model for the jan-ocn polymorphism data (fig. 4). For example, two haplotype tests indicate that the structure of variation observed in each of the three genes differs significantly from the neutral expectation. This is due to the large number of haplotype group 1 alleles that contain very little polymorphism. The deviation is strongest for janA and janB (fig. 4). The janB gene departs significantly from neutrality by several additional statistical tests, including those of Tajima17 and Fu and Li.20 This indicates an excess of low frequency variants and is caused primarily by the large number of singleton polymorphisms occurring within haplotype group 2. janB also produces a significant result for the MK test.25 D. simulans and D. melanogaster differ at 11 synonymous sites and seven nonsynonymous sites at janB. Within D. simulans, there are 11 synonymous polymorphisms and zero nonsynonymous polymorphisms. Thus the deviation is in the direction of an excess of interspecific nonsynonymous fixations. Given that at present the amino acid sequence of janB in D. simulans appears to be under strong purifying selection, this observation suggests either relaxed selective constraint or positive selection for amino acid replacement in janB soon after the D. simulans/D. melanogaster split. For janA, janB, and ocn combined, there are 22 synonymous differences and 12 nonsynonymous differences between species. Within D. simulans there are 36 synonymous polymorphisms and one nonsynonymous polymorphism. This is a highly significant departure from the neutral expectation, which suggests that positive selection may have acted not only on janB, but perhaps also on another gene(s) in this region since the time of the D. melanogaster/D. simulans divergence.

Distinguishing between Demographics and Selection
The age of a recently derived haplotype group can be estimated based on the D. melanogaster/D. simulans divergence and on the number of mutations observed within D. simulans.34 For the janB sample, where two mutations are observed within 25 alleles, the age of haplotype group 1 is estimated to be ≈ 5000 years (95% confidence interval, 1,000-15,000 years). This indicates a rapid increase in the frequency of haplotype group 1 alleles, which currently represent 70% of a worldwide sample. Such a rapid increase in allele frequency may be the result of positive selection, but could also be explained by population demographics. For example, a recent founder event followed by rapid population expansion could also explain the observed haplotype structure. The key to distinguishing between these two possibilities is the pattern of variation at other loci on the same chromosome. Demographic factors should affect the entire chromosome in the same way, while selection should affect only particular regions of the chromosome. A survey of 19 other loci located on chromosome 3R indicates that the pattern observed in the janA-ocn region is highly unusual.32,35 None of the other 19 loci shows a significant departure from neutrality by the tests described above. In addition, a survey of polymorphism in the rp49 gene, which lies ≈ 7 kb proximal to janA on chromosome 3, revealed a low polymorphism haplotype group present at nearly equal frequency in D. simulans populations from both Europe and Africa.34 Equal frequencies of alleles in two different populations, one presumably ancestral (Africa) and one derived (Europe), is unlikely if allele frequency is the result of founder effects.34
Can the pattern of variation in the janA-ocn region be explained by a selective sweep? Some features of the data are inconsistent with alternative explanations. First, the level of polymorphism is reduced in this region relative to other loci on the same chromosome. This reduction cannot be explained by a low mutation rate or unusually high selective constraint in this region of the genome, because there is no corresponding decrease in interspecific divergence in the janA-ocn region.32 The reduced polymorphism is also unlikely to be explained by background selection, because this region of the genome does not appear to have an unusually low recombination rate.34,36,37 Beyond the haplotype structure, another aspect of the data that is consistent with the selective sweep model is the high frequency of derived variants, which is a unique feature of genetic hitchhiking.21,22 Although an original sample of eight D. simulans alleles showed a significant excess of derived variants by Fay and Wu's test,32 the test result is not significant when applied to the larger sample of 36 alleles. This is due to the large number of low-frequency, derived variants within haplotype group 2 counteracting the high-frequency, derived variants within haplotype group 1. However, the maximum likelihood-based hitchhiking test of Kim and Stephan38 produces a highly significant result when applied to the complete janA-ocn dataset (Y. Kim, pers. comm.), supporting the hypothesis of a recent selective sweep in this region of the genome.
A complete selective sweep is expected to eliminate variation and to drive derived variants to high frequency or fixation; however it is not expected to produce two divergent haplotype groups like those observed in the janA-ocn region. The observed pattern can only be explained if the sweep is incomplete. A diagram of the selective sweep model is shown in (fig. 5). In this figure, a sample of 10 chromosomes is shown at three different time-points during the course of a selective sweep. The solid rectangles represent derived, neutral variants. In the first panel, these variants show an arrangement expected under neutrality. They are in low frequency and are randomly distributed among chromosomes. A new, positively-selected mutation is represented by an open rectangle. As this new variant increases in frequency in the population, linked neutral variants also increase in frequency (panel 2). As this process continues, the selected variant and the linked neutral variants reach a high frequency as a single haplotype (panel 3). If this haplotype does not become fixed in the population, then some ancestral alleles may still remain present. These ancestral alleles should differ from the common haplotype at a number of sites and also differ among themselves at a level expected for a group of neutrally-evolving alleles. This pattern of variation is exactly what is observed for the janA-ocn region. A common haplotype group with little polymorphism and many derived variants is at high frequency, while an ancestral haplotype group with much greater variation is at lower frequency. Thus the observed data may best be explained by a selective sweep that has not gone to completion.
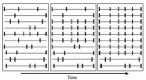
Why is the selective sweep incomplete? It is possible that the sweep is ongoing and that haplotype group 1 alleles will eventually become fixed in the population. This explanation seems unlikely, as the fixation of a strongly selected variant is expected to occur quite rapidly,11 making the observation of a population in mid-sweep hardly probable. Another possibility is that the sweep is incomplete due to population subdivision within D. simulans. For example, haplotype group 2 alleles appear to be more frequent in African populations,32 which are thought to be ancestral. It is possible that there is little migration of derived alleles into the ancestral populations, thus haplotype group 1 alleles do not become fixed in these populations. The problem with this explanation is that alleles from the two haplotype groups cooccur in a number of worldwide populations. Thus a population subdivision model would require a very high, nonsymmetric migration of ancestral alleles from African populations to the rest of the world for them to be present at detectable frequency. Another possibility is that there is some form of balancing selection, such as frequency dependent selection, that prevents alleles of haplotype group 1 from going to fixation. The very low level of polymorphism observed within haplotype group 1 is inconsistent with this being an old, balanced polymorphism. It indicates that if balancing selection is involved, one of the two balanced alleles must be very young and have recently been swept to its equilibrium frequency. A final possibility is that there are multiple positively-selected variants at different sites in the two haplotype groups, and fixation of a single haplotype is delayed until a recombination event brings them together on the same chromosome. This scenario, known as the traffic model,39 will produce a pattern of nucleotide variation similar to that seen for balancing selection until recombination brings the favored variants onto a single chromosome. The high density of genes in this region of the genome,40 along with the general excess of interspecific amino acid replacements in the genes in the region, indicates that there is a high potential for “molecular traffic,” which could result in the observed haplotype structure.
Identifying Specific Targets of Positive Selection
Identification of the particular nucleotide sites that are the target of positive selection will require a combination of population genetic and functional studies. From the intraspecific polymorphism data, the most likely location of the selected site can be inferred to be within the 3' end of janA or within janB. This region is implicated because the newly-derived haplotype is at highest frequency within this region and because there is evidence for recombination with ancestral alleles on either side (fig. 3). If the sweep is incomplete, then the selected variant should still be segregating in the population and should be associated with haplotype group 1 alleles. This narrows the list of candidates to just a few segregating sites. Because all of the polymorphisms in this window are at synonymous or noncoding sites, any phenotypic effect must occur at the level of gene expression. Comparison of expression of genes in this region between alleles of haplotype groups 1 and 2 is therefore the first step in the attempt to elucidate phenotypic differences that may underlie genetic variation in this region. Ultimate proof of a selectively favored genetic variant requires fitness assessment of different genotypes in a controlled genetic background. However, it may be difficult to demonstrate experimentally a clear relationship between genotype and fitness in this and many other instances of putative positive selection. Among the predictable complications are the possibility that balancing selection or molecular traffic may have affected the observed haplotype pattern, the prevalence of male-female interactions affecting genes with reproductive functions, and the likelihood that selective forces operating in nature may be diminished in laboratory conditions. Hence it is gratifying and reassuring to note that statistical analysis of inter- and intraspecific DNA sequence data may have the power to detect past and ongoing natural selection in many cases where direct experimental demonstration of fitness differences is technically complicated or even impossible.
References
- 1.
- Kimura M. The neutral theory of molecular evolution. Cambridge: Cambridge University Press. 1983
- 2.
- Endo T, Ikeo K, Gojobori T. Large-scale search for genes on which positive selection may operate. Mol Biol Evol. 1996;13:685–690. [PubMed: 8676743]
- 3.
- Fitch WM, Bush RM, Bender CA. et al. Long term trends in the evolution of H(3) HA1 human influenza type A. Proc Natl Acad Sci USA. 1997;94:7712–7718. [PMC free article: PMC33681] [PubMed: 9223253]
- 4.
- Yang Z, Nielsen R, Goldman N. et al. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. [PMC free article: PMC1461088] [PubMed: 10790415]
- 5.
- Tsaur SC, Wu CI. Positive selection and the molecular evolution of a gene of male reproduction, Acp26Aa of Drosophila. Mol Biol Evol. 1997;14:544–549. [PubMed: 9159932]
- 6.
- Wyckoff GJ, Wang W, Wu CI. Rapid evolution of male reproductive genes in the descent of man. Nature. 2000;403:304–309. [PubMed: 10659848]
- 7.
- Swanson WJ, Clark AG, Waldrip-Dail HM. et al. Evolutionary EST analysis identifies rapidly evolving male reproductive proteins in Drosophila. Proc Natl Acad Sci USA. 2001;98:7375–7379. [PMC free article: PMC34676] [PubMed: 11404480]
- 8.
- Yang Z, Swanson WJ. Codon-substitution models to detect adaptive evolution that account for heterogeneous selective pressures among site classes. Mol Biol Evol. 2002;19:49–57. [PubMed: 11752189]
- 9.
- Yang Z, Swanson>Maynard SmithJ, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed: 4407212]
- 10.
- Kaplan NL, Hudson RR, Langley CH. The “hitchhiking effect” revisited. Genetics. 1989;123:887–899. [PMC free article: PMC1203897] [PubMed: 2612899]
- 11.
- Stephan W, Wiehe THE, Lenz MW. The effect of strongly selected substitutions on neutral polymorphism: Analytical results based on diffusion theory. Theor Pop Biol. 1992;41:237–254.
- 12.
- Aguadé M, Miyashita N, Langley CH. Restriction-map variation at the zeste-tko region in natural populations of Drosophila melanogaster. Mol Biol Evol. 1989;6:123–130. [PubMed: 2566105]
- 13.
- Stephan W, Langley CH. Molecular genetic variation in the centromeric region of the X chromosome in three Drosophila ananassae populations. I. contrasts between the vermilion and forked loci. Genetics. 1989;121:89–99. [PMC free article: PMC1203608] [PubMed: 2563714]
- 14.
- Begun DJ, Aquadro CF. Molecular population genetics of the distal portion of the X chromosome in Drosophila: Evidence for genetic hitchhiking of the yellow-achaete region. Genetics. 1991;129:1147–1158. [PMC free article: PMC1204778] [PubMed: 1664405]
- 15.
- Berry AJ, Ajioka JW, Kreitman M. Lack of polymorphism on the Drosophila fourth chromosome resulting from selection. Genetics. 1991;129:1111–1117. [PMC free article: PMC1204775] [PubMed: 1686006]
- 16.
- Charlesworth B, Morgan MT, Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134:1289–1303. [PMC free article: PMC1205596] [PubMed: 8375663]
- 17.
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. [PMC free article: PMC1203831] [PubMed: 2513255]
- 18.
- Hudson RR, Bailey K, Skarecky D. et al. Evidence for positive selection in the superoxide dismutase (Sod) region of Drosophila melanogaster. Genetics. 1994;136:1329–1340. [PMC free article: PMC1205914] [PubMed: 8013910]
- 19.
- Depaulis F, Veuille M. Neutrality tests based on the distribution of haplotypes under an infinite-site model. Mol Biol Evol. 1998;15:1788–1790. [PubMed: 9917213]
- 20.
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. [PMC free article: PMC1205353] [PubMed: 8454210]
- 21.
- Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405–1413. [PMC free article: PMC1461156] [PubMed: 10880498]
- 22.
- Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;160:765–777. [PMC free article: PMC1461968] [PubMed: 11861577]
- 23.
- Begun DJ, Aquadro CF. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature. 1992;356:519–520. [PubMed: 1560824]
- 24.
- Hudson RR, Kreitman M, Aguad´ M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. [PMC free article: PMC1203113] [PubMed: 3110004]
- 25.
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. [PubMed: 1904993]
- 26.
- Yanicostas C, Vincent A, Lepesant JA. Transcriptional and posttranscriptional regulation contributes to the sex-regulated expression of two sequence-related genes at the janus locus of Drosophila melanogaster. Mol Cell Biol. 1989;9:2526–2535. [PMC free article: PMC362325] [PubMed: 2503707]
- 27.
- Yanicostas C, Lepesant JA. Transcriptional and translational cis-regulatory sequences of the spermatocyte-specific Drosophila janusB gene are located in the 3' exonic region of the overlapping janusA gene. Mol Gen Genet. 1990;224:450–458. [PubMed: 2125114]
- 28.
- Parsch J, Meiklejohn CD, Hauschteck-Jungen E. et al. Molecular evolution of the ocnus and janus genes in the Drosophila melanogaster species subgroup. Mol Biol Evol. 2001;18:801–811. [PubMed: 11319264]
- 29.
- Yanicostas C, Ferrer P, Vincent A. et al. Separate cis-regulatory sequences control expression of serendipity β and janus A, two immediately adjacent Drosophila genes. Mol Gen Genet. 1995;246:549–560. [PubMed: 7700229]
- 30.
- elegans sequencing consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. [PubMed: 9851916]
- 31.
- Civetta A, Singh RS. Sex-related genes, directional sexual selection, and speciation. Mol Biol Evol. 1998;15:901–909. [PubMed: 9656489]
- 32.
- Parsch J, Meiklejohn CD, Hartl DL. Patterns of DNA sequence variation suggest the recent action of positive selection in the janus-ocnus region of Drosophila simulans. Genetics. 2001;159:647–657. [PMC free article: PMC1461828] [PubMed: 11606541]
- 33.
- Moriyama EN, Powell JR. Intraspecific nuclear DNA variation in Drosophila. Mol Biol Evol. 1996;13:261–277. [PubMed: 8583899]
- 34.
- Rozas J, Gullaud M, Blandin G. et al. DNA variation at the rp49 gene region of Drosophila simulans: Evolutionary inferences from an unusual haplotype structure. Genetics. 2001;158:1147–1155. [PMC free article: PMC1461709] [PubMed: 11454763]
- 35.
- Begun DJ, Whitley P. Reduced X-linked nucleotide polymorphism in Drosophila simulans. Proc Natl Acad Sci USA. 2000;97:5960–5965. [PMC free article: PMC18541] [PubMed: 10823947]
- 36.
- Hamblin MT, Aquadro CF. High nucleotide sequence variation in a region of low recombination in Drosophila simulans is consistent with the background selection model. Mol Biol Evol. 1996;13:1133–1140. [PubMed: 8865667]
- 37.
- True JR, Mercer JM, Laurie CC. Differences in crossover frequency and distribution among three sibling species of Drosophila. Genetics. 1996;142:507–523. [PMC free article: PMC1206984] [PubMed: 8852849]
- 38.
- Kim Y, Stephan W. Joint effects of genetic hitchhiking and background selection on neutral variation. Genetics. 2000;155:1415–1427. [PMC free article: PMC1461159] [PubMed: 10880499]
- 39.
- Kirby DA, Stephan W. Multi-locus selection and the structure of variation at the white gene of Drosophila melanogaster. Genetics. 1996;144:635–645. [PMC free article: PMC1207556] [PubMed: 8889526]
- 40.
- Adams MD, Celniker SE, Holt RA. et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. [PubMed: 10731132]
- Detecting Selection by Inter- and Intraspecific DNA Sequence Comparison
- Molecular Evolution of the janus and ocnus Genes in the D. melanogaster Species Subgroup
- DNA Sequence Polymorphism in the janus-ocnus Region of D. simulans
- Distinguishing between Demographics and Selection
- Identifying Specific Targets of Positive Selection
- References
- Inferring Evolutionary History through Inter- and Intraspecific DNA Sequence Com...Inferring Evolutionary History through Inter- and Intraspecific DNA Sequence Comparison: The Drosophila janus and ocnus Genes - Madame Curie Bioscience Database
- SELECTIVE NEURODEGENERATION, NEUROPATHOLOGY AND SYMPTOM PROFILES IN HUNTINGTON'S...SELECTIVE NEURODEGENERATION, NEUROPATHOLOGY AND SYMPTOM PROFILES IN HUNTINGTON'S DISEASE - Madame Curie Bioscience Database
- The Restriction Point of the Cell Cycle - Madame Curie Bioscience DatabaseThe Restriction Point of the Cell Cycle - Madame Curie Bioscience Database
- RNA Modification Subsystems in the SEED Database - Madame Curie Bioscience Datab...RNA Modification Subsystems in the SEED Database - Madame Curie Bioscience Database
- Origin, Recognition, Signaling and Repair of DNA Double-Strand Breaks in Mammali...Origin, Recognition, Signaling and Repair of DNA Double-Strand Breaks in Mammalian Cells - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...