

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

“It is no longer just sufficient to synthesise and test; experiments are played out in silico with prediction, classification, and visualisation being the necessary tools of medicinal chemistry.” (L. B. Kier)1
Drug Design Is an Optimization Task
Pathological biochemical processes within a cell or an organism are usually caused by molecular interaction and recognition events. One major aim of drug discovery projects is to identify bioactive molecular structures that can be used to systematically interfere with such molecular processes and positively influence and eventually cure a disease. Both significant biological activity and specificity of action are important target functions for the molecular designer. In addition, one has to consider absorption, distribution, metabolic (ADME), and potential toxic properties during the drug development process to ensure that the drug candidate will have the desired pharmacokinetic and pharmacodynamic properties. Usually the initial “hit”, i.e., a bioactive structure identified in a high-throughput screen (HTS) or any other biological test system, is turned into a “lead” structure. This often includes optimization/minimization towards ADME/Tox parameters. Further optimization is carried out as the compound enters the next phases of drug discovery, namely lead optimization and development, nonclinical and finally clinical trials (Fig. 1). Only if all of the required conditions are met can the molecule become a trade drug. The target function in drug design clearly is multi-dimensional.2 Typically several rounds of optimization must be performed to eventually obtain a clinical candidate.
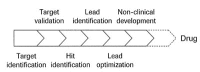
Figure 1
Early drug development stages.
Before describing particular optimization strategies in more detail, we shall define what we mean by an “optimization problem” (Definition 1.1) (adapted from Papadimitriou and Steiglitz).3
Definition 1.1
An optimization problem is a set I of instances of an optimization problem. (Note: Informally, in an instance we are given the “input data” and have enough information to obtain a solution). An instance of an optimization problem is a pair (F, q), where F is the domain of feasible points, and q is the quality function. The problem is to find an □ F for which
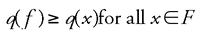
The virtually infinite (judged from a practical, synthesis-oriented perspective) set of chemically feasible, “drug-like” molecules has been estimated to be in the order of magnitude of approximately 10100 (a “googol”) different chemical structures.4,5 This inconceivable large number gives hope that there exists an appropriate molecule for almost every type of biological activity. The task is to find such molecules, and the molecular designer is confronted with this problem. As time and resources prohibit systematic synthesis and testing of each of these structures—and thus perform an exhaustive search—more intelligent search strategies are needed. Random search is one workable approach to solve the task. However, due to the extremely large search space its a priori probability of success is very low. HTS is a practical example of this random approach to drug discovery.6,7 Several technological breakthroughs during the past years have validated this strategy. By means of current advanced ultra-HTS methods it is now possible to test several hundred thousand molecular structures within a few days.2,8 Typically, the hit rate with respect to low micromolar activity in these experiments is 0.1–2%.9,10 This means that for many drug targets hits are actually being identified by HTS. Why is that? Wouldn't one expect rarely any hit at all? One possible explanation is that the compounds tested contain a relatively large number of “biologically preferred”, drug-like structures already.2 Very often the corporate compound collections (“libraries”) used in the primary HTS have been shaped by many experienced medicinal chemists over several decades already, therefore inherently representing accumulated knowledge about biochemically meaningful structural elements. In addition, many of the “new” drug targets belong to some well-studied protein families (e.g., kinases, GPCRs). In other words, corporate compound collections and “historical” collections represent activity-enriched libraries compared to a truly random set. As a consequence the actual a posteriori probability of finding active molecules by random guessing is sometimes higher than expected.
HTS provides an entpoint—often the only possible—to drug discovery in the absence of knowledge about a desired bioactive structure. There are additional complementing possibilities available through virtual screening techniques, and the field of virtual screening has been extensively reviewed over the past years.4,11,12 The design of activity-enriched sets of molecules that are specifically shaped for some desired quality represents one of the major challenges in rational drug design and virtual screening. In the hit-to-lead optimization phase following the primary HTS, smaller-sized compound libraries are generated and tested, typically containing between some ten and a few hundred molecules per optimization cycle. These compound collections are more and more focused towards a desired bioactivity, which is reflected by a steadily decreasing structural diversity yet simultaneously increasing hit rates at higher activity levels. In other words, molecular diversity is an adaptive (lat. ad-aptare, “to fit to”) parameter in drug design (Fig. 2).13,14 The cover illustration “Development I” by M.C. Escher illustrates pattern formation that can be interpreted as resulting from an adaptive process (see book cover). Starting with only little knowledge about molecular structures exhibiting a desired biological activity—the “twilight zone”, patterns begin to emerge in subsequent synthesis-and-test cycles. Knowledge about essential, function-determining structures grows with the time resulting from an adaptive variation-selection process.
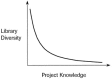
Figure 2
Idealized course of adaptive library diversity with increasing knowledge in a drug discovery project.
The term “adaptive system” must be defined before starting on the survey of adaptation with a focus on molecular feature extraction, pattern formation, and simulated molecular evolution. Frank15 gave a comprehensive definition that highlights the main aspects discussed here (Definition 2).
Definition 1.2
“An adaptive system is a population of entities that satisfy three conditions of natural selection: the entities vary, they have continuity (heritability), and they differ in their success. The entities in the population can be genes, competing computer programs, or anything else that satisfies the three conditions.”15
Adaptation involves the progressive modification of some entity or structure so that it performs better in its environment. “Learning” is some form of adaptation for which performance consists of solving a problem. Koza16 summarized the central elements of artificial adaptive systems:
- Structures that undergo adaptation
- Initial structures (starting solutions)
- Fitness measure that evaluates the structures
- Operations to modify the structures
- State (memory) of the system at each stage
- Method for designating a result
- Method for terminating the process
- Parameters that control the process
In the context of drug design, the “entities” mentioned in Definition 2 are small organic molecules; successful structures or substructural elements are kept and passed on in subsequent design rounds (“heritability”); and the compounds that are synthesized definitely differ in their success (biological activity, pharmacokinetic and pharmacodynamic properties). The cyclic interplay between synthesis and testing of compound libraries can be interpreted as a simplified evolutionary variation-selection process.13,17–20 In fact, this conceptual view helps to turn the more or less blind screening by HTS—also termed “irrational” design—into a guided search, and to perform systematic focused screening. An important feature of adaptive systems is that they use “history”, i.e., they “learn” from experience and use this knowledge for future decision making. They are able to consider several conditions or target variables at a time—multidimensional optimization—which makes them parallel processing systems. Furthermore, their behavior is characterized by relative tolerance to noise or disturbance.21,22 Dracopoulos22 described adaptive systems from a more technically oriented, system control point of view, taking into account the work of Narendra and Annaswamy21 and Luenberger23 Definition 3.
Definition 1.3
“An adaptive system is a system which is provided with a means of continuously monitoring its own performance in relation to a given figure of merit or optimal condition and a means of modifying its own parameters by a closed loop action so as to approach this optimum. Adaptive systems are inherently nonlinear.”22
One may regard drug discovery as driven by an adaptive process that is guided by a complex, nonlinear objective function (also called “fitness function” in the following Chapters).18 It is evident that the classical experimental synthesize-and-test approach to drug design is a practicable and sometimes very successful implementation of an adaptive system: A team of scientists decides to synthesize a set of molecular structures which they believe to be active based on a hypothesis that was generated from their knowledge about the particular project. Then, these compounds are tested for their biological activity in a biochemical assay. The results provide the basis for a new hypothesis, and the next set of molecules will be conceived taking these facts into account. Along the course of the optimization the molecular diversity will drop, and the quality of the compounds will increase. This drug discovery procedure has been successful in the past, so why should we bother about any additions or modifications? There are several obvious caveats and limitations to this classical, entirely experimental approach.2,24
- The problem of large numbers. The numbers of molecules that can be examined by a human is limited; a scientist cannot compete with the capacity throughput of virtual, computer-based storage, modeling, and screening systems;
- The problem of multidimensional optimization. Often no real parallel optimization of bioactivity (e.g., as measured in a ligand binding assay) and ADME/Tox properties is performed;
- The cost of experiments. Screening assays, in particular when they are used in HTS, can be very expensive. This is especially true when difficult biochemical assays or protein purification schemes are employed; also, blind screening can be wasted time and resources if the screening library is biased towards inactive structures or no hit is found (this statement is not entirely correct, as much can be learned from “negative” examples, i.e., inactive structures).
Certainly many of the above limitations can be alleviated by a good experimental design during lead optimization. This book provides several examples of adaptive virtual screening systems that can help to overcome some of these problems and complement classical experimental drug discovery and HTS efforts. It must be stressed that virtual screening is meant to support and complement the experimental approach. Real-life chemical and biological bench experiments are always needed to validate theoretical suggestions, and they are an indispensable prerequisite for any model building and formulation of a hypothesis. The aim is to enhance the lead discovery process by rationalizing the library shaping process through adaptive, “self-learning” modeling of quantitative structure-activity relationships (QSAR) or structure-property relationships (SPR) and molecular structure generation (de novo design, combinatorial design), thereby enabling focused screening and data mining. The main objective with such “intelligent” control is to obtain acceptable performance characteristics over a wide range of uncertainty, computational complexity, and nonlinearity with many degrees of freedom.25,26
The presence of uncertainty and nonlinear behavior in lead optimization and throughout the whole drug development process is evident. For example, the data generated by HTS and ultra HTS assays—which often provides the knowledge base for a first semiquantitative SAR attempt—is inherently noisy and sometimes erroneous due to limitations of assay accuracy and experimental setups. One can rarely expect highly accurate predictions from models that are grounded on very fuzzy data. The response of a linear system to small changes in its parameters (or to external stimulation) is usually in direct proportion to the stimulation (“smooth” response). For nonlinear systems, however, a small change in the parameters can produce a large qualitative difference in the output. There are many observations of such behavior in drug design. For example, considering chemical substructures being the free parameters of a lead optimization process, the presumably “small changes” made in the molecules shown in Figure 3 led to significant modification of their biological activity. Exchange of the phenolic hydroxy groups of isoproterenol 1 with chlorine results in the conversion of an agonist (β-adrenergic, isoproterenol) to an antagonist (β-adrenergic blocker, dichloro-isoproterenol 2).27 The closely related chemical structures 3, 4, and 5 reveal very different biological activity. Close structural relationships of gluco- and corticosteroids, e.g., also reflect the observation that apparently small structural variations can lead to signifcant loss of or change in bioactivity. Obviously in these cases essential parts of the function-determining pharmacophore pattern were altered, resulting in qualitatively different activities. Had other non-essential atom groups been changed, added or removed, the effect on bioactivity would likely have been much less dramatic. As we do not know a priori which parts of a molecule are crucial determinants of bioactivity, we tend to believe that any “small” change of structure will only slightly affect molecular function, i.e., the active analog similarity principle. This way of thinking is often not appropriate. This example demonstrates that there probably is no generally valid definition of a “small” structural change of a molecule. The definition and quantitative description of chemical similarity is a critical issue in medicinal chemistry.27 This definition is context-dependent, i.e., it depends on the particular structural class of the molecule to be optimized, the drug target (receptor, enzyme, macromolecule etc.) or the particular property of interest, and the assay system.
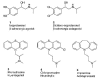
Figure 3
Seemingly “minor” chemical differences can have a dramatic effect on biological activity (adapted from ref. ). These examples illustrate the difficulty of defining and quantitatively describing molecular similarity.
Our knowledge about the underlying SAR or SPR drives the design process at a given time and is important to constantly re-formulate the prediction model or hypothesis during the process to achieve convergence. At the start of a drug discovery project one hardly knows which substructural groups of a molecule are crucial for a desired bioactivity, simply because our knowledge base is extremely small or not existent at all. Virtual screening is thought to provide the medicinal chemist with ideas, assist in molecular feature extraction, and cope with large amounts of real structural and assay data and virtual data (predicted properties and virtual compound structures).
The aim is to design a robust drug optimization process that is able to cope with uncertainty, computational complexity including multidimensional objective functions, and nonlinearity. To do so, a QSAR hypothesis generator (inference system) is needed, a molecular structure generator, and a test system (Fig. 4). In a feedback loop, QSAR hypotheses are derived from a knowledge base, e.g., experimental assay results or predicted activities. Then new molecular structures are generated based on the QSAR model either by de novo design, combinatorial assembly, or by systematically changing already existing structures. Finally, activity measurements or predictions of these new structures establish new facts, thereby augmenting the knowledge base. For an adaptive system it is important to use the error between the observed and expected behavior (e.g., biological activity) to tune the system parameters (feedback control). Otherwise no adaptation will take place.
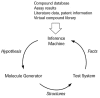
In this volume emphasis is put on two kinds of virtual adaptive systems that have proven their usefulness for drug design: evolutionary algorithms for molecular optimization, and artificial neural networks (ANN) for flexibly modeling structure-activity relationships. These techniques seem to be methods of choice especially when the receptor structure or an active site model of the drug target are unavailable, and thus more conventional computer-based molecular design approaches cannot be applied.28–30 This situation is given for over 50% of the current drug targets since the majority of drug targets belongs to the family of integral membrane proteins (Fig. 5). Until today only some few high-resolution structures of such proteins are available, but hopefully novel crystallization and structure determination methods will lead to progress in this field in the near future.31–33 Today, the most promising—and often the only possible—approach to rational drug design in these cases is ligand optimization based on small molecule structures and assay data as the only sources of information.
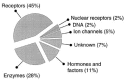
Figure 5
Overview of current drug targets (adapted from ref. ). More than 50% belong to protein classes (membrane proteins) for which structure determination by X-ray or NMR methods is extremely complicated and only some rare solved structures exist today.
Principles of Adaptive Evolutionary Systems
A big challenge recurs in all adaptive systems, namely to balance exploration versus exploitation,34 i.e., the balance between costly exploration for improved efficiency and the comparably cheap exploitation of already known solutions.15 Adaptive design is a process of discovery that follows cycles of broad exploration and efficient exploitation. Greater variability of molecular structures in a library improves the chances that some of the molecules will have properties that match unpredictable challenges (which may be caused, e.g., by the nonlinear nature of a multimodal and multidimensional fitness function). In an entirely experimental drug design approach the price for broad exploration is high because many different chemical structures must be synthesized and tested. In contrast, the combinatorial chemistry approach aiming at the variation of successful schemes provides one possibility to exploit or fine-tune an already successful solution. In artificial, computer-based drug design both of these two extremes can be efficiently managed. In particular, the exploration of chemical space can be much broader by considering all kinds of virtual “drug-like” structures, and the exploitation of successful solutions by systematic variation of a structural framework can be extremely efficient following a modular building-block approach (see Chapter 5 for details). Again, a central task is to operate at the right level of structural diversity at the different stages of molecular design (Fig. 2).18
Adaptive optimization cycles pursued in drug design projects can be mimicked by evolutionary algorithms (EA), in particular evolution strategies (ES)35–37 and genetic algorithms (GA).34,38–41 These algorithms perform a local stochastic search in parameter space, e.g., a chemical space. During the search process, good “building blocks” of solutions are discovered and emphasized. The idea here is that good solutions to a problem tend to be made up of good building blocks, which are combinations of variable values that confer higher “fitness” on the solutions in which they are present. Building blocks are often referred to as “schemata” according to the original notation of Holland,34 or as “hyperplanes” according to Goldberg's notation.38
In the past decades many families of heuristics for optimization problems have been developed, typically inspired by metaphors from physics or biology, which follow the idea of local search (sometimes these approaches are referred to as “new age algorithms”).3,42 Among the best known are simulated annealing,43 Boltzmann machines44, and Tabu search.45,46 There are of course many additional mathematical optimization techniques known (for an overview, see ref. 3). Why should EAs be particularly appropriate for drug design purposes? To answer this question, let us consider the following two citations, where the first one is taken from Frank,15 and the second from Globus and coworkers:47
- “A simple genetic algorithm often performs reasonably well for a wide range of problems. However, for any specific case, specially tailored algorithms can often outperform a basic genetic algorithm. The tradeoffs are the familiar ones of general exploration versus exploitation of specific information.”48,49
- “The key point in deciding whether or not to use genetic algorithms for a particular problem centers around the question: what is the space to be searched? If that space is well understood and contains structure that can be exploited by special-purpose search techniques, the use of genetic algorithms is generally computationally less efficient. If the space to be searched is not so well understood, and relatively unstructured, and if an effective GA representation of that space can be developed, then GAs provide a surprisingly powerful search technique for large, complex spaces.”50,51
These two excerpts address two critical points for any optimization or control system: First the structure of search space (parameter space), and second the knowledge that is available about this space. If not much is known about the structure of the search space, and the fitness landscape is rugged with possible local discontinuities, then an EA mimicking natural adaptation strategies can still be a practicable optimization method.52 The molecular designer faces exactly this situation: optimization in the presence of noise and nonlinear system behavior.53 As soon as more information about the (local) search space becomes available—e.g., by a high-resolution X-ray structure of the receptor protein that can guide the molecular design process, or sets of active and inactive molecules to derive a preliminary SAR—more specialized optimization strategies can be applied that may, for example, rely on an analytical description of the error surface. There is no general best optimization or search technique. In a smooth fitness landscape, advanced steepest descent or gradient search represents a reasonable choice to find the global optimum, and several variations of this technique have been described to facilitate navigation in a landscape with some few local optima.54 Typically, virtual screening is confronted with a high-dimensional search space and convoluted fitness functions that easily lead to a rugged fitness landscape. Robust approximation algorithms must be employed to find practicable (optimal or near-optimal) solutions here.55,56 Stochastic search methods sometimes provide answers that are provably close to optimal in these cases. In fact, several systematic investigations demonstrated that a simple stochastic search technique might be comparable or sometimes superior to sophisticated gradient techniques, depending on the structure of fitness landscape or the associated error surface.57,58
In short, EAs represent a general method for optimization in large, complex spaces. The fact that EAs are straightforward to implement and easily extended to further problems has certainly added to their widespread use, popularity, and proliferation. The authors do not intend to review the whole field of evolutionary algorithms. Rather it is intended to highlight some of their specific algorithmic features that are particularly suited for molecular design applications. For a detailed overview of EA theory, additional applications in molecular design, and related approaches like evolutionary programming (EP) and combinatorial optimization, see the literature.3,12,20,52,59,60
EAs operate by generating distributions (“populations”) of possible solutions (“offspring”) from a “parent” parameter set, subjecting every “child” to fitness determination and a subsequent selection procedure. The cycle begins again with the selected new parent(s). A schematic of this general algorithm is given in Fig. 6. Offspring are bred by means of two central mechanisms: mutation and recombination (crossover). EAs differ largely in the degree to which each mutation and crossover operator is used to generate offspring and the types of so-called “strategy-parameters” employed. These are additional adaptive variables which can be used to shape the offspring distributions (vide infra). The classical ES is dominated by mutation events to generate offspring—thereby adding new information to a population and tending to explore search space—whereas the classical GA favors crossover operations to breed offspring—thus promoting the exploitation of a population. During the past decades the individual advantages of these approaches were identified and the current advanced implementations are usually referred to as “evolutionary algorithms” to indicate this fact. Historically, Rechenberg35,61 coined the original ES idea over 40 years ago already following ideas of Bremermann.62 It was further developed by Schwefel.36,63 The term “genetic algorithm” appeared soon after the publication of Holland's pioneering text on Adaptation in Natural and Artificial Systems in 1975.34
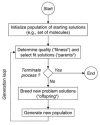
Figure 6
General scheme of an evolutionary algorithm.
It is important to keep in mind that EAs perform a local stochastic search in parameter space, in contrast to a global search. Finding a globally optimal solution to an instance of some optimization problems can be prohibitively difficult. Nevertheless, it is often possible to find a feasible solution f that is best in the sense that there is no better solution in its neighborhood N(f), i.e., it is locally optimal with respect to N. N(f) defines a set of points that are “close” in some sense to the point f. (Note: If F = Rn for a given optimization problem with instances (F, q), the set of points within a fixed Euclidean distance provides a natural neighborhood). The term “locally optimal” shall be defined as given by Definition 4 according to ref. 3):
Definition 1.4
Given an instance (F, q) of an optimization problem and a neighborhood N, a feasible solution f □ F is called locally optimal with respect to N (or simply locally optimal whenever N is understood by context) if
where q is the quality or fitness function.3
A particular example of a one-dimensional optimization task (F, q) is sketched in Fig. 7a Here, the solution space. Obviously the function q has three local optima A, B, and C, but only B is globally optimal. This can be more formally described: Let the neighborhood N be defined by closeness in Euclidean distance for some ϵ > 0, Ne(f) = {x : x □ F and |x−f|≤ ϵ}. Then if e is suitably small, the points A, B, and C are locally optimal. In ordinary local search, a defined neighborhood is searched for local optima (Fig. 7b). A strong neighborhood seems to produce local optima whose average quality is largely independent on the quality of the starting solutions, whereas weak neighborhoods seem to produce local optima whose quality is strongly correlated with that of the starts.3 The particular choice of N may critically depend on the structure of F in many combinatorial optimization problems. A particularly useful aspect of some evolutionary algorithms is that they implement variable choices of N. A further issue is that of how to search the neighborhood. The two extremes are first-improvement, in which a favorable change is accepted as soon as it is found, and steepest descent, where the entire neighborhood is searched. The great advantage of first-improvement is that local optima can generally be found faster. For further details on local search methods and variations of this scheme, see the literature.3,54 An early description of local search addressing some aspects of natural selection can be found in ref. 64. A schematic of some popular neighborhood search methods is shown in Fig. 8.

Figure 7
a) A one-dimensional Euclidean optimization problem. The fitness function c has three local optima in A, B, and C;. b) Schematic representation of ordinary local search with a fixed neighborhood N (adapted from ref. ).
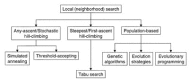
Figure 8
Schematic of neighborhood search methods (adapted from ref. ).
Solution Encoding and Basic Evolutionary Operators
The shape of the fitness surface in parameter space can be modified by choosing an appropriate encoding of the problem solutions. In the case of drug design the “solutions” are molecular structures exhibiting some desired properties or biological functions. This means that without an appropriate, context- and problem-dependent representation of molecules no systematic optimization can be performed. The performance of an optimization process in such a space can be even worse than pure random searching. Therefore, the implementation of any EA begins with the important encoding step. The traditional GAs employ binary encodings, whereas the classical ESs use real number encodings. It is often not trivial to find an appropriate encoding scheme so that an acceptably smooth fitness landscape results that obeys the Principle of Strong Causality18,35 (also known as the Similarity Principle in medicinal chemistry).65 This means that a step in parameter space should lead to a proportional change of fitness, which is identical to the demand for a smooth fitness landscape with respect to the neighborhood N. Actually, this task represents the greatest challenge in rational molecular design. Because it can be regarded as a difficult optimization problem in itself to develop an appropriate encoding scheme, e.g., EAs can be applied to solve this task as detailed in Chapter 3. For scientific problems real number encodings are common, as they generally offer a natural representation of the problem, which is also true for molecular optimization. Many drug design tasks, however, can also be regarded as a combinatorial optimization task, and clever binary encoding schemes of molecular building blocks (e.g., amino acids, building blocks in combinatorial chemistry) can be very appropriate. Usually, the particular encoded representation of a molecule (“solution representation”) is called the genotype, which determines the decoded phenotype (e.g., biological activity).
An example of binary and real-valued encodings of a molecular structure is given in Fig. 9. In this example, five molecular properties were selected to provide the solution or parameter space: the calculated octanol/water partition coefficient (clogP), calculated molecular weight (cMW), the number of potential hydrogen-bond (H-donors) donors and acceptors, the total number of nitrogen and oxygen atoms (O+N), and the polar surface area (Surfpolar). Some of these properties have been shown to be useful descriptors for predictions of the potential oral bioavailability of drug candidates.66,67 A possible genotype representation contains five “genes”, i.e., either five real-valued numbers or eight three-bit words, respectively. For the binary representation three bits per gene were chosen to encode 23 = 8 different intervals for each property descriptor (Table 1). The offspring solutions for a new generation are generated by genotype variations, mainly by mutation and crossover operations.

Figure 9
Examples of binary and real number encoding of a molecular structure; a) real-valued representation, b) standard binary representation, c) binary Gray code representation.
Table 1
Three-bit codings of standardized property values (x in [0,1]) using a standard binary coding system and a Gray coding system.
The conventional evolution strategies use mutation as their primary operator and crossover as a secondary operator to breed offspring, whereas genetic algorithms use crossover as the primary operator and mutation as a secondary operator. In addition to mutation and crossover, the EA community has developed many additional operators for breeding offspring from one or many parents. These operators are inspired by observations in natural genetics, e.g., insertion, deletion, and death. It should be stressed that EAs do not attempt to provide realistic models of natural evolution. They rather mimic a Darwinian-like evolution process with the idea to solve technical optimization problems. And one such application is evolutionary drug design.
The Mutation Operator
By mutation individual bits of a binary representation are switched from 1 to 0 or from 0 to 1, or real-valued mutation changes a value by a random amount, respectively, to generate offspring vectors ξparentØξchild. The model real-value mutation (5.19,404,5,2,59.9)parentØ(5.19,615,5,2,59.9)child is equivalent to the binary transition (100011100001010)parentØ(100100100001010)child. The degree of mutation, i.e., how many positions of a representation are changed, determines the amount of new information present in a population. Therefore, mutation is essential for exploration of solution space. As we have discussed above, the appropriate degree of exploration must be allowed to change during an optimization experiment. ESs apply a simple but powerful adaptive control mechanism for mutation by the introduction of one or several so-called strategy parameters.35,68,69 The general idea is to give an accompanying variable si to each of the real-valued vectors of the candidate solutions ξl comprising a population. Offspring solutions are generated by using Gaussian mutation of the parental values (Eq. 1), where the strategy variable σi denotes the standard deviation (the width of the Gaussian), and g is a Gaussian-distributed random number (Eq. 2):
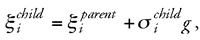
where
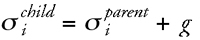
Variation of the standard deviation used to generate offspring distributions is one possibility to determine the size of a move in solution space and is thus sometimes referred to as adaptive (or mutative) step-size control. The parameter s determines the neighborhood N that is considered. In contrast to ordinary local search with a fixed width of N (Fig. 7b), the width of N is an additional adaptive variable in an ES. Basic ESs rely on mutation as the only genetic operator.
A simple “one parent—many offspring” ES, which is termed “(1,λ) ES” according to Schwefel's notation,36 can be formulated as follows using a pseudo-code notation (see Fig. 6):
initialize parent, (Sp, σp, Qp)
repeat for each generation until termination criterion is satisfied:
generate _ variants of the parent, (SV, σV, QV)
select best variant structure, (Sbest, σbest, Qbest)
set (Sp, σp, Qp) = (Sbest, σbest, Qbest)
According to this scheme selection-of-the-best is performed among the offspring only, i.e., the parent dies out. This characteristic helps to navigate in a multimodal fitness landscape because it sometimes allows to escape from local optima a parent may reside on.57,70 As given by Equation 1, the offspring is approximately Gaussian-distributed around the parent. This enables a local stochastic search to be performed and guarantees that there is a finite probability of breeding very dissimilar candidate solutions (“snoopers” in search space). The width of the variant distribution is determined by the variance or standard deviation, s, of the bell-shaped curve reflecting the distance-to-parent probability. The straightforward (1,λ) ES tends to develop large s values if large steps in search space are favorable, and adopts small values if for example fine-tuning of solutions is required. The s value can therefore be regarded as a measure of library diversity. Small values lead to narrow distributions of variants, large values result in the generation of novel structures that are very dissimilar to the parent molecule as judged by a chemical similarity or distance measure. Several such applications are described in Chapter 5.
In the beginning of a virtual screening experiment using ES, σ should be large to explore chemical space for potential optima, later on during the design experiment s will have to be small to facilitate local hill-climbing. This means that s itself is subjected to optimization and will be propagated from generation to generation, thereby enabling an adaptive stochastic search to be carried out. In contrast to related techniques like simulated annealing, there exists no predefined “cooling schedule” or a fixed decaying function for σ. [Note: there are some new SA variants that have “adaptive re-heating”; see Chapter 3]. Its value may freely change during optimization to adapt to the actual requirements of the search. Large s values will automatically evolve if large steps are favorable, small values will result when only small moves lead to success in the fitness landscape. If there is an underlying ordering of local optima this strategy can sometimes provide a simple means to perform “peak-hopping” towards the global optimum.35 Some variations of this adaptive mutation scheme found in ESs have been published.34,71 A more recent addition to the idea are “learning-rule methods”.72–75
Adaptive mutation schemes have also been reported for GAs.74,76,77 For example, in Kitano's scheme the probability of mutation of an offspring depends on the number of mismatches (Hamming distance) between the two parents [Note: GAs need at least two parents to breed offspring, whereas ESs can operate with one parent. The most popular is the (1,λ) ES].76 Low distance results in high mutation, and vice versa. In this way the diversity of individuals in the population can be controlled. In contrast to the strategy parameter s in ESs, however, this scheme does not consider the history of the search, i.e., it does not “learn” from previously successful choices of σ values (step-size control).
Dynamic control of the impact of mutations on a candidate solution is relatively easily done for real-coded representations but not for standard binary encoding. One approach to overcome this limitation of standard binary encoding is to apply a Gray coding scheme (Table 1).78 Gray coding allows single mutations of the genotype to have smaller impact on the phenotype, because here adjacent values differ only by a single bit (neighborhood in space). This can help to fine-tune solutions and avoid unwanted jumps between candidate solutions during optimization. Alpha-numeric coding is another possibility to represent molecular structures, which is especially suited for searching combinatorial chemistry spaces with a GA.79
The Crossover Operator
Crossover is a convenient technique for exploitation of candidate solutions present in a population. It is the main operator for offspring generation by GAs. Consider the two parent “chromosomes” 010011011101 and 000011110100. By crossover, strings of bits are exchanged between these two chromosomes to form the offspring: 010011111101 and 000011010100. Crossover points are usually determined randomly. It must be emphasized that crossover operations—in contrast to mutation events—do not add new information to a population's genotype pool. Therefore, a GA always implements an additional operator for mutation to ensure that innovative solutions can evolve. Without mutation a GA cannot improve on the candidate solutions that are present in a given population of bitstrings. Advanced ESs (so-called “higher-order” ESs) also contain crossover between parents as an additional breeding operator that complements mutation.35
A straightforward GA could be formulated as follows (adapted from ref. 80):
// Initialize a usually random population of individuals (genotypes)
init population P
// Evaluate fitness (quality) of all individuals of the population
for all i: Qi = Quality(Pi)
// iterate over generations
repeat for each generation until a termination criterion is satisfied:
// select a sub-population (parents) for breeding offspring
Pparent = SelectParents(P)
// generate offspring
Poffspring = recombine Pparent
Poffspring = mutate Poffspring
// determine the quality of the offspring chromosomes
for all i: Qi = Quality(Pi offspring)
// select survivors to form the new starting population
P = SelectSurvivors(Poffspring, Q)
In evaluating a population of strings, the GA is implicitly estimating the average fitness of all building blocks (“schemata”) of solutions that are present in the population. Their individual representation is increased or decreased according to the Schema Theorem34,38 (for a detailed discussion, see ref. 77). The driving force behind this process is the crossover operator. The cover illustration of M.C. Escher nicely illustrates the evolution of useful building blocks of the final solution. Simultaneous implicit evaluation of large numbers of schemata in a population is known as the implicit parallelism of genetic algorithms.34 This property of EAs, especially GAs, has proven their value for molecular feature selection and molecular optimization as we shall see in the following Chapters.29,81,82
The Selection Operator
The effect of selection is to gradually bias the sampling procedure towards instances of schemata—and therefore candidate solutions—whose fitness is estimated to be above average.77 EAs implement parent selection mechanisms to pick those individuals of a population that are meant to generate new offspring. The selection process is guided by the fitness values, which might be calculated or experimentally determined. The (1,λ) ES uses plain “selection of the best”, i.e., only the “fittest” survive and become the parent for the next generation (elitism). In its more general form, the m fittest are selected (μλ) ES. GAs usually implement stochastic selection methods. The most popular is roulette wheel selection, where members of the population are selected proportional to their fitness. In this way, an individual with a low fitness can become a parent for the next generation, although with a lower probability than high-fitness individuals. A major problem with such a selection operator can be premature convergence of the search, if single individuals with extremely high fitness values dominate a population. In such cases rank-based selection can be advantageous. A third commonly used selection mechanism is tournament selection, where two or more members of a population are randomly picked, and the best of these is deemed a new parent.83,84
As we have seen, several strategic EA parameters must be controlled to obtain good solutions and avoid premature convergence on local optima or plateaus in the fitness landscape. These parameters also include the optimization time (number of generations), population size, the ratio between crossover and mutation, and the number of parents. There seems to be no generally best set of parameter values, although some rules of thumb have been suggested for classes of problems.35,37,85 In general, GAs have much larger population sizes (recombination dominates) than ESs (mutation dominates).
An example of the dependency between the number of generations and the number of offspring in a simple (1,λ) ES that was used to find the maximum of a multimodal fitness function (in the example a modified Rastrigin function was used, Eq. 3) is given in Fig. 10. In a situation of limited resources (in terms of CPU calls) and a rugged fitness landscape like the one depicted in Fig. 10a, extreme numbers of offspring seem to be disadvantageous.85 Provided this observation is principally valid for experimental drug design, one might speculate that in a situation of limited resources (e.g., manpower, assay material, synthesis capacity) a high-throughput approach is not always appropriate.
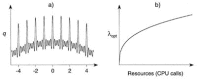
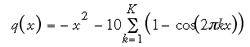
It is evident that a given instance of an optimization problem dictates the appropriate values. However, in drug design tasks we usually do not know the complexity of the optimization problem and the structure of search space in advance, so we must rely on empirical procedures. This also implicates that re-running the EA can easily result in different solutions, especially if the underlying fitness landscape is rugged (multimodal). The results of EA search or optimization runs should therefore not be regarded as the overall best or globally optimal. They represent candidate solutions that have a higher fitness than average. EAs certainly suffer in performance comparison against algorithms which were optimized for particular problems. Nevertheless they have proven their usefulness, mainly due to their ability to cope with challenging real-life search and optimization problems, their straightforward implementation and easy extension to additional problems. Several diverse applications of EAs in drug design are described in the following Chapters of this book.
Molecular Feature Extraction and Artificial Neural Networks
In the previous section we have briefly introduced evolutionary algorithms as one possible framework to address the problem of adaptive molecular design. In this section we focus on the question of how to calculate the quality or fitness values which drive the EA selection process. The aim of a quality function is to structure the search space of possible solutions into regions of different fitness. A coarse-grained quality function might simply lead to classes of solutions (e.g., active and inactive molecules), and a more complex function introduces gradually different quality levels to the search space. The quality of the model determines the success rate of the multi-dimensional design process. How can we develop a good SAR model? It is apparent that no universal recipe exists; nevertheless some general rules of thumb can be given. One approach is to consider the task as a pattern recognition problem, where three main aspects must be considered: i), the data used for generation of a SAR/SPR hypothesis should be representative of the particular problem; ii), the way molecular structures are described for model generation and its level of abstraction must allow for a reasonable solution for the pattern recognition task; iii), the model must permit non-linear relationships to be formulated since the interdependence between molecular properties and structural entities is generally non-linear. The first point seems to be trivial, but selection of representative data for hypothesis generation is very difficult and often hampered due to a lack of data or populations of data points characterized by abrupt decreases in density functions.53,86
Classification of molecule patterns is based on selected molecular attributes or descriptors (features), which are considered for the classification task: patternsÆfeaturesÆclassification. The aim is to combine pattern descriptors (“input variables”) together so that a smaller number of new variables, features, is found. There are straightforward statistical methods available that can be used to construct linear and nonlinear combinations of features which have good discriminating power between classes.87 These methods are complemented by techniques originating from cognition theory, among them are various artificial neural networks (ANN) and “artificial intelligence” (AI) approaches.88,89 According to this concept the feature selection and extraction process—i.e., the formulation of an SAR/SPR model—can be regarded as an adaptive process and formally divided into several steps:90
- Step 1. Define a set of basic object descriptors (vocabulary); go to Step 2
- Step 2. Formulate “tentative” features (hypothesis) by association of basic descriptors, focusing, and filtering; go to Step 3
- Step 3. Evaluate the descriptors for their classification ability (quality or fitness of the features); go to Step 4
- Step 4. If a useful solution is found then STOP, or else go to Step 2
Usually the features proposed during the early feedback loops are relatively rigid, i.e., sets of raw descriptors and simple combinations of descriptors. During the later stages the features tend to become more flexible and include more complex (“higher-order”) combinations of descriptors. It is important that the feature extraction process is able to focus on certain descriptors and filter-out other descriptors or feature elements.81 Fig. 11 gives an example of a “Bongard-problem” for visual pattern recognition to comprehend this rather crude scheme of adaptive feature extraction. The task is to find a feature that allows for discrimination between Class A and Class B. Six objects belong to each class. These might be six active and six inactive molecules.

Figure 11
A visual classification task. Which feature separates Class A from Class B? This pattern recognition problem is analogous to one of the classical “Bongard-problems” (adapted problem no. 22).
Various types of (ANN) are of considerable value for pharmaceutical research.91 Main tasks performed by these systems are:
- feature extraction,
- function estimation and non-linear modeling,
- classification, and
- prediction.
For many applications alternative techniques exist.92 ANN provide, however, an often more flexible approach offering unique solutions to these tasks. The paradigms offered by ANN lie somewhere between purely empirical and ab initio approaches. Neural networks
- “learn” from examples and acquire their own “knowledge”,93
- are sometimes able to find generalizing solutions,94
- provide flexible non-linear models of input/output relationships,95 and
- can cope with noisy data and are fault-tolerant.96
ANN have found a widespread use for classification tasks and function approximation in many fields of medicinal chemistry and cheminformatics (Table 2). For these kinds of data analysis mainly two different types of networks are employed, “supervised” neural networks (SNN) and “unsupervised” neural networks (UNN). The main applications of SNN are function approximation, classification, pattern recognition and feature extraction, and prediction tasks. These networks require a set of molecular compounds with known activities to model structure-activity relationships. In an optimization procedure, which will be described below, these known “target activities” serve as a reference for SAR modeling. This principle coined the term “supervised” networks. Correspondingly, “unsupervised” networks can be applied to classification and feature extraction tasks even without prior knowledge of molecular activities or properties. A brief introduction to UNN development is provided in Chapter 2. Hybrid systems have also been constructed and successfully applied to various pattern recognition and SAR/SPR modeling tasks.97–99 In Table 2 the main network types and characteristic applications in the life sciences are listed. The description of neural networks in this volume is restricted to the most widely applied in the field of molecular design. More extensive introductions to the theory of neural computation including comparisons with statistical methods are given elsewhere.87,100–103 There are many texts on neural networks covering parts of the subject not discussed here, e.g., the relation of neural network approaches to machine learning,104–106 fuzzy classifiers,96 time series analysis,107 and additional aspects of pattern recognition.108 Ripley provides an in-depth treatment of the relation of neural network techniques to Bayesian statistics and Bayesian training methods.87 A commented collection of pioneering publications in neurocomputing has been compiled by Anderson and Rosenfeld.109 Two texts have become available covering both theory and applications of ANN in chemistry, QSAR, and drug design.97,110
Table 2
Neural network types with a high application potential in pharmaceutical research (adapted from reference ).
Frequently molecular libraries and assay data must be investigated for homogeneity or “diversity”.111–113 UNN are able to automatically recognize characteristic data features and classes of data in a given set based on a rational representation of the compounds and a sensitive similarity measure. The selection of an appropriate measure of similarity is crucial for the clustering results.114–119 UNN systems can be helpful in generating an overview of the distributions of the data sets, and they are, therefore, suited for a first examination of a given data set or raw data analysis. Some types of UNN are able to form a comprehensive model of the data distribution which can be helpful for the identification and an understanding of predominant data features, e.g., molecular structures or properties that are responsible for a certain biological activity.
In contrast to UNN, the structuring of a data set must be already known to apply SNN, e.g., knowledge about bioactivity. SNN are able to approximate arbitrary relationships between data points and their functional or classification values, i.e., they can be used to model all kinds of input-output relationships and classify data by establishing a quality function. The term “supervised” indicates that in contrast to UNN for every data point one or more corresponding functional values must be already known (these might be experimentally measured molecule activities or properties).
Artificial neural networks consist of two elements, i) formal neurons, and ii) connections between the neurons. Neurons are arranged in layers, where at least two layers of neurons (an input layer and an output layer) are required for construction of a neural network. Formal neurons transform a numerical input to an output value, and the neuron connections represent numerical weight values. The weights and the neurons' internal variables (termed bias or threshold values) are free variables of the system which must be determined in the so-called “training phase” of network development. Selected network types and appropriate training algorithms are discussed in the following Chapters. For details on other network architectures and ANN concepts, see the literature.87,101,103 The idea of adaptation of a three-layered network structure to a given SAR/SPC problem is shown in Figure 12. This scheme is thought to illustrate the main idea of “GA-networks” which will be treated in detail in Chapter 3.29,81,120,121 The main advantage of the system is an intelligent selection of neural network input variables by a genetic algorithm. Rather than feeding in all available data descriptors a set of meaningful variables is automatically selected. The GA selects the most relevant variables, and the neural network provides a model-free non-linear mapping device.
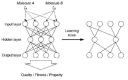
Independent of the choice of network type and architecture applied, the crucial parts of an analysis are appropriate data selection, description and pre-processing. Like any other analysis tool neural networks can only be successful if a solution to a given problem can be found on the basis of the data used. Although this statement seems to be trivial in many real-life applications it can be very difficult to collect representative data and define a comprehensive and useful data description. Sometimes techniques like PCA, data smoothing or filtering can be used prior to network application to facilitate network training (see Chapter 2). In cases where the data description is high-dimensional per se it can be helpful to focus only on a particular sub-space (however, this requires knowledge about essential data features) or to perform a PCA step to reduce the number of dimensions to be considered without significant loss of information. Hybrid network architectures consisting of a pre-processing layer, an unsupervised layer for coarse-grained data clustering, and a supervised layer for fine-analysis were already successfully applied to a variety of tasks and seem to provide very useful techniques for (Q)SAR modeling and molecular design. Both unsupervised and supervised networks have proven their usefulness and applicability to a number of different problems. Nevertheless, it requires significant expertise to apply them effectively and efficiently. For most potential applications of neural networks to drug design conventional techniques exist, and neural networks should be considered as complementary.122,123 However, ANN can sometimes provide a simpler and more elegant, and sometimes even superior solution to these tasks. Of special interest is a combination of evolutionary algorithms for descriptor selection and ANN for function approximation. This conglomerate seems to provide a very useful general approach to QSAR modeling. In connection with other methods the many types of ANN provide a flexible modular framework to help speed up the drug development process and lead to molecules with desired structures and activities.
Conventional Supervised Training of Neural Networks
Neural network architectures with a multi-layered feed-forward architecture have found a widespread application as function estimators and classificators. They follow the principle of convoluting simple non-linear functions for approximation of complicated input-output relationships (Kolmogorov's Theorem, ref. 127). Since in such ANN mainly sigmoidal functions are employed the following brief description of supervised network training will be restricted to this network type. We limit the discussion of SNN to feed-forward networks at this place because of their dominating role in drug design. Other network architectures, e.g., recurrent networks for time-series and sequence analysis, are not considered here.
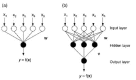
A scheme of a fully connected three-layered feed-forward network is shown in Fig. 13. Input layer neurons receive the pattern vector (molecular descriptor set). They are often referred to as “fan-out units” because they distribute the data vector to the next network layer without any calculation being performed. In the hidden layer neurons “sigmoidal neurons” are used in the majority of supervised ANN. A sigmoidal neuron computes an output value according to Equations 4 and 5:
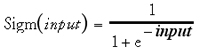
where
Here w is the weight vector connected to the neuron, x is the neuron's input signal, and is the neuron's bias or threshold value. If a single sigmoidal output neuron is used, the overall function represented by the fully connected two-layered feed-forward network shown in Fig. 13a will be (Eq. 6):
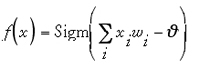
where x represents the input vector (input pattern). The network shown in Fig. 13b with three sigmoidal hidden units and a single sigmoidal output unit represents a more complicated function (Eq. 7):
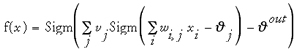
where w are the input-to-hidden weights, v are the hidden-to-output weights, are the hidden layer bias values, and is the output neuron's bias. Even more complicated functions can be represented by adding additional layers of neurons to the network. It has been shown that at most two hidden layers with non-linear neurons are required to approximate arbitrary continuous functions.95,128,129 Depending on the application and the accuracy of the fit the required number of layers and the number of neurons in a layer can vary. Baum and Haussler130 addressed the question which size of a network can be expected to generalize from a given number of training examples. A “generalizing” solution derived from the analysis of a limited set of training data will be valid for any further data point, thus leading to perfect predictions. This is possible only by the use of data that is representative of the problem. Most solutions found by feature extraction from life-like data sets will, however, be sub-optimal in this general meaning (see Chapter 3 for a more detailed discussion of this issue). Determination of a useful architecture by testing the performance of an ANN with independent test and validation data during and after SNN training is essential. If the test performance is bad either the network architecture must be altered, or the data used for training is inadequate, i.e., the problem might be ill-posed. Furthermore, by monitoring test performance the generalization ability can be estimated during the training process and training can be stopped before over-fitting occurs (“forced stop”; see Chapter 3).
During supervised ANN training the weights and threshold values of the network are determined which is—again—an optimization procedure. For training, a set of data vectors plus one or more functional values (“target values”) per data point are required. The optimization task is to adjust the free variables of the system in such a way that the network computes the desired output values. The goal during the training phase is to minimize the difference between the actual output and the desired output; a frequently used error function is the sum-of-squares error as given in Equation 1.8 (here, N is the number of data points (“patterns”) in the training set):
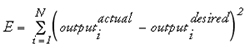
This error function is a special case of a general error function called the Minkowski-R error (Eq. 9):
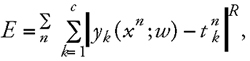
where yk is the actual network output, tk is the desired (target) output vector, x is the input vector, and w is the weight vector. The summation over k will be omitted if only a single output neuron is used. With R = 2 the Minkowski-R error reduces to the sum-of-squares error function, R = 1 leads to the computation of the conditional median of the data rather than the conditional mean as for R = 2. The use of an R value smaller than 2 can help reducing the sensitivity to outliers which are frequently present in a training set, especially if experimental data are used.
Several optimization algorithms can be used, e.g., gradient descent techniques, simulated annealing procedures, maximum likelihood and Bayesian learning techniques, or evolutionary algorithms performing some kind of adaptive random search.87,103 Most frequently applied are gradient techniques using the “back-propagation of errors” (bp) algorithm for weight update.100 Gradient descent follows the deepest descent on the error surface (Eq. 10):
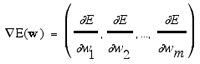
For network training each weight value is incrementally updated according to Equation 11:
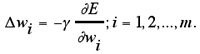
γ is a constant defining the step size of each iteration along the downhill gradient direction. If the weights of a two-layered network (Fig. 13a) must be updated we make these weight changes individually for each input pattern x in turn, and obtain the “delta rule” (also termed “Adaline rule” or “Widrow-Hoff rule”),100,131 as given in Equation 1.12:
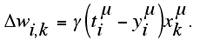
Using a continuously differentiable activation function for the hidden units of a multi-layered network, it is an easy step to calculate the update values (“deltas”) for hidden unit weights following this bp procedure. A thorough description of this algorithm can be found in many textbooks on neural networks. Several straightforward modifications of bp have been described.101 As with all optimization methods, gradient techniques can have problems with premature convergence due to local energy barriers or plateaus of the error surface.132 This will not be a problem if the network is small enough to establish a comparatively low-dimensional error surface lacking striking local optima. However, the classical bp algorithm might easily get trapped in a local optimum when large networks must be trained. For complicated networks especially adaptive evolutionary optimization can be useful.68,85,99 Besides considerations concerning network architecture, considerable effort must be spent in selecting an appropriate ANN training technique.
References
- 1.
- Kier LB. Book review: Neural Networks in QSAR and Drug Design. J Med Chem. 1997;40:2967.
- 2.
- Eglen R, Schneider G, Böhm HJ. High-throughput screening and virtual screening: Entry points to drug discovery In: Virtual Screening for Bioactive Molecules; Böhm HJ, Schneider G, eds. Weinheim, New York: Wiley-VCH, 20001–14.
- 3.
- Papadimitriou CH, Steiglitz K. Combinatorial Optimization-Algorithms and ComplexityMineola, New York: Dover Publications,1998.
- 4.
- Walters WP, Stahl MT, Murcko MA. Virtual screening-An overview. Drug Discovery Today. 1998;3:160–178.
- 5.
- Walters WP, Ajay, Murcko MA. Recognizing molecules with drug-like properties. Curr Opin Chem Biol. 1999;3:384–387. [PubMed: 10419858]
- 6.
- Bevan P, Ryder H, Shaw I. Identifying small-molecule lead compounds: The screening approach to drug discovery. Trends Biotechnol. 1995;13:115–121. [PubMed: 7766218]
- 7.
- Grepin C, Pernelle C. High-throughput screening. Drug Discovery Today. 2000;5:212–214. [PubMed: 10790267]
- 8.
- MORPACE Pharma Group Ltd. From data to drugs: Strategies for benefiting from the new drug discovery technologies. Scrip Reports
July 1999. - 9.
- Drews J. Rethinking the role of high-throughput screening in drug research. Decision Resources. Intern Biomedicine Management Partners Orbimed Advisors. 1999
- 10.
- Zhang JH, Chung TD, Oldenburg KR. Confirmation of primary active substances from high throughput screening of chemical and biological populations: A statistical approach and practical considerations. J Comb Chem. 2000;2:258–265. [PubMed: 10827934]
- 11.
- Bures MG, Martin YC. Computational methods in molecular diversity and combinatorial chemistry. Curr Opin Chem Biol. 1998;2:376–380. [PubMed: 9691077]
- 12.
- Böhm HJ, Schneider G. eds.Virtual Screening for Bioactive Molecules Weinheim, New York: Wiley-VCH, 2000.
- 13.
- Schneider G, Wrede P. Optimizing amino acid sequences by simulated molecular evolution In: Jesshope C, Jossifov V, Wilhelmi W, eds. Parallel Computing and Cellular Automata. Berlin; Akademie-Verlag; Mathem Res 199381335–346.
- 14.
- Karan C, Miller BL. Dynamic diversity in drug discovery: Putting small-molecule evolution to work. Drug Discovery Today. 2000;5:67–75. [PubMed: 10652457]
- 15.
- Frank SA. The design of natural and artificial adaptive systems In: Rose MR, Lauder GV, eds. Adaptation San Diego: Academic Press, 1996451–505.
- 16.
- Koza JR. The genetic programming paradigm: Genetically breeding populations of computer programs to solve problems In: Soucek B and the IRIS Group, eds. Dynamic, Genetic, and Chaotic Programming New York: John Wiley and Sons, 1992203–321.
- 17.
- Schneider G, Schuchhardt J, Wrede P. Simulated molecular evolution and artificial neural networks for sequence-oriented protein design: an evaluation. Endocytobiosis Cell Res. 1995;11:1–18.
- 18.
- Schneider G. Evolutionary molecular design in virtual fitness landscapes In Böhm HJ, Schneider G, eds: Virtual Screening for Bioactive Molecules Weinheim, New York: Wiley-VCH, 2000161–186.
- 19.
- Schneider G, Lee ML, Stahl M, Schneider P. De novo design of molecular architectures by evolutionary assembly of drug-derived building blocks. J Comput Aided Mol Des. 2000;14:487–494. [PubMed: 10896320]
- 20.
- Devillers J. ed. Genetic Algorithms in Molecular Modeling London: Academic Press, 1996.
- 21.
- Narendra KS, Annaswamy AM. Stable Adaptive Systems London: Prentice Hall, 1989.
- 22.
- Dracopoulos DC. Evolutionary Learning Algorithms for Neural Adaptive Control London: Springer, 1997.
- 23.
- Luenberger DG. Introduction to Dynamic Systems New York: John Wiley and Sons, 1979.
- 24.
- High-Throughput Screening Drug Discovery Today 20005(12)Suppl.
- 25.
- Bavarian B. Introduction to neural networks for intelligent control. IEEE Control Systems Magazine. 1988 April:3–7.
- 26.
- Narenda KS, Mukhopadhyay S. Intelligent control using neural networks. IEEE Control Systems Magazine. 1992 April:11–18.
- 27.
- Kubinyi H. A general view on similarity and QSAR studies In: van de Waterbeemd H, Testa B, Folkers G, eds. Computer-Assisted Lead Finding and Optimization-Current Tools for Medicinal Chemistry Weinheim, New York: Wiley-VCH, 19979–28.
- 28.
- Leach AR. Molecular Modeling-Principles and Applications Harlow: Pearson Education, 1996.
- 29.
- So SS, Karplus M. Evolutionary optimization in quantitative structure-activity relationships: An application of genetic neural networks. J Med Chem. 1996;39:1521–1530. [PubMed: 8691483]
- 30.
- Schneider G, Neidhart W, Adam G. Integrating virtual screening to the quest for novel membrane protein ligands. CNSA. 2001;1:99–112.
- 31.
- Rosenbusch JP. The critical role of detergents in the crystallization of membrane proteins. J Struct Biol. 1990;104:134–138. [PubMed: 2088442]
- 32.
- Sakai H, Tsukihara T. Structures of membrane proteins determined at atomic resolution. J Biochem. 1998;124:1051–1059. [PubMed: 9832606]
- 33.
- Heberle J, Fitter J, Sass HJ, Buldt G. Bacteriorhodopsin: The functional details of a molecular machine are being resolved. Biophys Chem. 2000;85:229–248. [PubMed: 10961509]
- 34.
- Holland JH. Adaptation in Natural and Artificial Systems Ann Arbor: University of Michigan Press 1975.
- 35.
- Rechenberg I. Optimierung technischer Systeme nach Prinzipien der biologischen Evolution 2nd ed. Stuttgart: Frommann-Holzboog 1994.
- 36.
- Schwefel HP. Numerical Optimization of Computer Models Chichester: Wiley, 1981.
- 37.
- Bäck T, Schwefel HP. An overview of evolutionary algorithms for parameter optimization. Evol Comput. 1993;1:1–23.
- 38.
- Goldberg DE. Genetic Algorithms in Search, Optimization, and Machine Learning Reading: Addison-Wesley, 1989.
- 39.
- Davis L. ed. Handbook of Genetic Algorithms New York: Van Nostrand Reinhold, 1991.
- 40.
- Rawlins G. ed. Foundations of Genetic Algorithms Los Altos: Morgan Kauffmann, 1991.
- 41.
- Forrest S. Genetic Algorithms: Principles of natural selection applied to computation. Science. 1993;261:872–878. [PubMed: 8346439]
- 42.
- Reeves CR. Modern Heuristic Problems for Combinatorial Problems Oxford: Blackwell Scientific, 1993.
- 43.
- Kirkpatrick S, Gelatt C D Jr., Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–680. [PubMed: 17813860]
- 44.
- Ackley DH, Hinton GE, Sejnowski TJ. A learning algorithm for Boltzmann machines. Cognitive Science. 1985;9:147–169.
- 45.
- Glover FW, Laguna M. Tabu search Boston: Kluwer Academic Publishers, 1997.
- 46.
- Baxter CA, Murray CW, Clark DE, Westhead DR, Eldrige MD. Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins: Struct Funct Genet. 1998;33:367–382. [PubMed: 9829696]
- 47.
- Globus A, Lawton J, Wipke T. Automatic molecular design using evolutionary techniques. Nanotechnology. 1999;10:290–299.
- 48.
- Newell A. Heuristic programming: Ill-structured problems In: Arnofsky J, ed. Progress in Operations Research New York: Wiley, 1969363–414.
- 49.
- Davis L. A genetic algorithms tutorial In: Davis L, ed. Handbook of Genetic Algorithms New York: Van Nostrand Reinhold,19911–101.
- 50.
- De Jong KA. Introduction to the second special issue on genetic algorithms. Machine Learning. 1990;5(4):351–353.
- 51.
- Forrest S, Mitchell M. What makes a problem hartford genetic algorithm? Some anomalous results in the explanation. Machine Learning. 1993;13:285–319.
- 52.
- Kauffmann SA. The Origins of Order-Self-Organization and Selection in Evolution New York: Oxford University Press, 1993.
- 53.
- Giuliani A, Benigni R. Modeling without boundary conditions: An issue in QSAR validation In: van de Waterbeemd H, Testa B, Folkers G, eds. Computer-Assisted Lead Finding and Optimization-Current Tools for Medicinal Chemistry Weinheim, New York: Wiley-VCH, 199751–63.
- 54.
- Mathews JH. Numerical Methods for Mathematics, Science, and Engineering Englewood Cliffs: Prentice-Hall International, 1992.
- 55.
- Levitan B, Kauffman S. Adaptive walks with noisy fitness measurements. Mol Divers. 1995;1:53–68. [PubMed: 9237194]
- 56.
- Schulz AS, Shmoys DB, Williamson DP. Approximation algorithms. Proc Natl Acad Sci USA. 1997;94:12734–12735. [PMC free article: PMC34165] [PubMed: 9370525]
- 57.
- Clark DE, Westhead DR. Evolutionary algorithms in computer-aided molecular design. J Comput Aided Mol Des. 1996;10:337–358. [PubMed: 8877705]
- 58.
- Desjarlais JR, Clarke ND. Computer search algorithms in protein modification and design. Curr Opin Struct Biol. 1998;8:471–475. [PubMed: 9729739]
- 59.
- Koza JR. Genetic Programming Cambridge: The MIT Press, 1992.
- 60.
- Clark DE. ed. Evolutionary Algorithms in Molecular Design Weinheim, New York: Wiley-VCH, 2000. URL: http://panizzi.shef.ac.uk/cisrg/links/ea_bib.html .
- 61.
- Rechenberg I. Cybernetic solution path of an experimental problemRoyal Aircraft Establishment, Library Translation 1122. Farnborough.
- 62.
- Bremermann HJ. Optimization through evolution and recombination In: Yovits MC, Jacobi GT, Goldstine GD, eds. Self-organizing Systems Washington: Spartan Books, 196293–106.
- 63.
- Davidor Y, Schwefel HP. An introduction to adaptive optimization algorithms based on principles of natural evolution In: Soucek B and the IRIS Group, eds. Dynamic, Genetic, and Chaotic Programming New York: John Wiley and Sons,1992183–202.
- 64.
- Dunham B, Fridshal D, Fridshal R, North JH. Design by natural selection IBM Res Dept RC-476,
June 20, 1961. - 65.
- Johnson MA, Maggiora GM. eds. Concepts and Applications of Molecular Similarity New York: John Wiley, 1990.
- 66.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. [PubMed: 11259830]
- 67.
- Clark DE, Pickett SD. Computational methods for the prediction of ‘drug-likeness’ Drug Discovery Today. 2000;5:49–58. [PubMed: 10652455]
- 68.
- Schneider G, Schuchhardt J, Wrede P. Artificial neural networks and simulated molecular evolution are potential tools for sequence-oriented protein design. Comput Appl Biosci. 1994;10:635–645. [PubMed: 7704662]
- 69.
- Saravanan N, Fogel DB, Nelson KM. A comparison of methods for self-adaptation in evolutionary algorithms. Biosystems. 1995;36:157–166. [PubMed: 8573696]
- 70.
- Conrad M, Ebeling W, Volkenstein MV. Evolutionary thinking and the structure of fitness landscapes. Biosystems. 1992;27:125–128. [PubMed: 1467458]
- 71.
- Schneider G, Schuchhardt J, Wrede P. Peptide design in machina: Development of artificial mitochondrial protein precursor cleavage-sites by simulated molecular evolution. Biophys J. 1995;68:434–447. [PMC free article: PMC1281708] [PubMed: 7696497]
- 72.
- Davis L. Adapting operator probabilities in genetic algorithms In: Schaffer JD, ed. Proceedings of the Third International Conference on Genetic Algorithms and Their Applications San Mateo: Morgan Kaufmann,198961–69.
- 73.
- Bäck T, Hoffmeister F, Schwefel HP. A survey of evolution strategies In: Whitley D, ed. Proceedings of the Fourth International Conference on Genetic Algorithms San Mateo: Morgan Kaufmann,19912–9.
- 74.
- Julstrom BA. What have you done for me lately? In: Eshelman LJ, ed. Proceedings of the Sixth International Conference on Genetic Algorithms San Mateo: Morgan Kaufmann,199581–87.
- 75.
- Tuson A, Ross P. Adapting operator settings in genetic algorithms. Evol Comput. 1998;6:161–184. [PubMed: 10021745]
- 76.
- Kitano H. Designing neural networks using genetic algorithms with graph generation systems. Complex Systems. 1990;4:461–476.
- 77.
- Mitchell M. An Introduction to Genetic Algorithms Cambridge: The MIT Press, 1998.
- 78.
- Tuson A, Clark DE. New techniques and future directives In: Clark DE, ed. Evolutionary Algorithms in Molecular Design Weinheim, New York: Wiley-VCH,2000241–264.
- 79.
- Weber L. Practical approaches to evolutionary design In: Böhm HJ, Schneider G, eds. Virtual Screening for Bioactive Molecules Weinheim, New York: Wiley-VCH,2000187–205.
- 80.
- Heitkoetter J, Beasley D. The hitch-hiker's guide to evolutionary computation: A list of frequently-asked questions (FAQ). USENET: comp.ai.genetic. Available via anonymus FTP from: rtfm.mit.edu/pub/usenet/news.answers/ai-faq/genetic .
- 81.
- So SS. Quantitative structure-activity relationships In: Clark DE, ed. Evolutionary Algorithms in Molecular Design New York, Weinheim: Wiley-VCH,200071–97.
- 82.
- Sheridan RP, SanFeliciano SG, Kearsley SK. Designing targeted libraries with genetic algorithms. J Mol Graph Model. 2000;18:320–334. [PubMed: 11143552]
- 83.
- Goldberg DE, Korb B, Deb K. Messy genetic algorithms: Motivation, analysis, and first results. Complex Syst. 1989;3:493–530.
- 84.
- Parrill AL. Introduction to evolutionary algorithms In: Clark DE, ed. Evolutionary Algorithms in Molecular Design New York, Weinheim: Wiley-VCH,20001–13.
- 85.
- Schneider G, Schuchhardt J, Wrede P. Evolutionary optimization in multimodal search space. Biol Cybernetics. 1996;74:203–207.
- 86.
- Raudys S. How good are support vector machines? Neural Netw. 2000;13:17–19. [PubMed: 10935455]
- 87.
- Ripley BD. Pattern Recognition and Neural Networks Cambridge: Cambridge university Press, 1996.
- 88.
- Katz WT, Snell JW, Merickel MB. Artificial neural networks. Methods Enzymol. 1992;210:610–636. [PubMed: 1584053]
- 89.
- Soucek B. The IRIS Group, eds. Dynamic, Genetic, and Chaotic Programming New York: John Wiley and Sons, 1992.
- 90.
- Hofstadter DR. Gödel, Escher, Bach: An Eternal Golden Braid New York: Basic Books, 1979.
- 91.
- Agatonovic-Kustrin S, Beresford R. Basic concepts of artificial neural network (ANN) modeling and its application in pharmaceutical research. J Pharm Biomed Anal. 2000;22:717–727. [PubMed: 10815714]
- 92.
- Milne G W A. Mathematics as a basis for chemistry. J Chem Inf Comput Sci. 1997;37:639–644. [PubMed: 9254910]
- 93.
- Hinton GE. How neural networks learn from experience. Sci Am. 1992;267:144–151. [PubMed: 1502516]
- 94.
- Hampson S. Generalization and specialization in artificial neural networks. Prog Neurobiol. 1991;37:383–431. [PubMed: 1754687]
- 95.
- Hornik K, Stinchcombe M, White H. Multilayer feed-forward networks are universal approximators. Neural Networks. 1989;2:359–366.
- 96.
- Kosko B. Neural Networks and Fuzzy Systems-A Dynamical Systems Approach to Machine Intelligence Englewood Cliffs: Prentice Hall International, 1992.
- 97.
- Zupan J, Gasteiger J. Neural Networks for Chemists Weinheim: VCH, 1993.
- 98.
- Schneider G, Wrede P. Artificial neural networks for computer-based molecular design. Prog Biophys Mol Biol. 1998;70:175–222. [PubMed: 9830312]
- 99.
- Schneider G, Schuchhardt J, Wrede P. Development of simple fitness landscapes for peptides by artificial neural filter systems. Biol Cybernetics. 1995;73:245–254. [PubMed: 7548312]
- 100.
- Rumelhart DE, McClelland JL. The PDB Research Group. Parallel Distributed Processing Cambridge: MIT Press,1986.
- 101.
- Hertz J, Krogh A, Palmer RG. Introduction to the Theory of Neural Computation Redwood City: Addison-Wesley, 1991.
- 102.
- Amari SI. Mathematical methods of neurocomputing In: Barndorff-Nielsen OE, Jensen JL, Kendall WS, eds.Networks and Chaos-Statistical and Probabilistic Aspects London: Chapman & Hall,19931–39.
- 103.
- Bishop CM. Neural networks for pattern recognition Oxford: Clarendon Press, 1995.
- 104.
- Mitchie D, Spiegelhalter DJ, Taylor CC. eds. Machine Learning: Neural and Statistical Classification New York: Ellis Horwood, 1994.
- 105.
- Bratko I, Muggleton S. Applications of inductive logic programming. Commun Assoc Comput Machinery. 1995;38:65–70.
- 106.
- Langley P, Simon HA. Applications of machine learning and rule induction. Commun Assoc Comput Machinery. 1996;38:54–64.
- 107.
- Weigend AS, Gershenfeld NA. eds. Time Series Prediction: Forecasting the Future and Understanding the Past Reading: Addison-Wesley, 1993.
- 108.
- Carpenter GA, Grossberg S. eds. Pattern Recognition by Self-Organizing Neural Networks Cambridge: The MIT Press, 1991.
- 109.
- Anderson JA, Rosenfeld E. eds. Neurocomputing: Foundations of Research Cambridge: MIT Press, 1988.
- 110.
- Devillers J. ed. Neural Networks in QSAR and Drug Design London: Academic Press, 1996.
- 111.
- Downs GM, Willett P. Clustering of chemical structure databases for compound selection In: van de Waterbeemd H, ed. Advanced Computer-Assisted Techniques in Drug Discovery Weinheim: VCH,1995111–130.
- 112.
- Willett P. ed. Methods for the analysis of molecular diversity In: Perspectives in Drug Discovery and Design Vols 7/81997.
- 113.
- Agrafiotis DK, Lobanov VS, Rassokhin DN, Izrailev S. The measurement of molecular diversity In: Böhm HJ, Schneider G, eds. Virtual Screening for Bioactive Molecules Weinheim, New York: Wiley-VCH,2000265–300.
- 114.
- Good AC, Peterson SJ, Richards WG. QSAR from similarity matrices. Technique validation and application in the comparison of different similarity evaluation methods. J Med Chem. 1993;36:2929–2937. [PubMed: 8411009]
- 115.
- Good AC, So SS, Richards WG. Structure-activity relationships from molecular similarity matrices. J Med Chem. 1993;36:433–438. [PubMed: 8474098]
- 116.
- Dean PM. ed. Molecular Similarity in Drug Design London: Blackie Academic & Professional, 1995.
- 117.
- Sen K. ed. Molecular Similarities I Topics in Current Chemistry Vol 173 Berlin: Springer, 1995.
- 118.
- Sen K. ed. Molecular Similarities II Topics in Current Chemistry Vol 174 Berlin: Springer, 1995.
- 119.
- Barnard JM, Downs GM, Willett P. Descriptor-based similarity measures for screening chemical databases In: Böhm HJ, Schneider G, eds. Virtual Screening for Bioactive Molecules Weinheim, New York: Wiley-VCH,200059–80.
- 120.
- So SS, Karplus M. Three-dimensional quantitative structure-activity relationships from molecular similarity matrices and genetic neural networks. 1. Method and validations. J Med Chem. 1997;40:4347–4359. [PubMed: 9435904]
- 121.
- So SS, Karplus M. Three-dimensional quantitative structure-activity relationships from molecular similarity matrices and genetic neural networks. 2. Applications. J Med Chem. 1997;40:4360–4371. [PubMed: 9435905]
- 122.
- Loew GH, Villar HO, Alkorta I. Strategies for indirect computer-aided drug design. Pharm Res. 1993;10:475–486. [PubMed: 8483829]
- 123.
- Marrone TJ, Briggs JM, McCammon JA. Structure-based drug design: computational advances. Annu Rev Pharmacol Toxicol. 1997;37:71–90. [PubMed: 9131247]
- 124.
- Drews J, Ryser S. eds. Human Disease-From Genetic Causes to Biochemical Effects Berlin: Blackwell Science, 1997.
- 125.
- Bongard M. Patterns Recognition Rochelle Park: Spartan Books, 1970.
- 126.
- Sumpter BG, Getino C, Noid DW. Theory and applications of neural computing in chemical science. Annu Rev Phys Chem. 1994;45:439–481.
- 127.
- Kolmogorov AN. On the representation of continuous functions of several variables by superposition of continuous functions of one variable and addition. Dokl Akad Nauk SSSR. 1957;114:953–956.
- 128.
- Cybenko G. Approximations by superpositions of a sigmoidal function. Mathematics of Control, Signals, and Systems. 1989;2:303–314.
- 129.
- Cybenko G. Continuous valued neural networks with two hidden layers are sufficient Technical Report, Department of Computer Science, Tufts University, Medford, MA, 1988.
- 130.
- Baum EB, Haussler D. What size net gives valid generalization? Neural Computation. 1989;1:151–160.
- 131.
- Widrow B, Hoff ME. Adaptive switching circuits In: 1960 IRE WESCON Convention Record, Part 4. New York: IRE, 196096–104.
- 132.
- McInerny JM, Haines KG, Biafore S, Hecht-Nielsen R. Back propagation error surfaces can have local minima In: International Joint Conference on Neural Networks 2. Washington, 1989. New York: IEEE Press:627.
- A Conceptual Framework - Madame Curie Bioscience DatabaseA Conceptual Framework - Madame Curie Bioscience Database
- Biocompatibility of Polyurethanes - Madame Curie Bioscience DatabaseBiocompatibility of Polyurethanes - Madame Curie Bioscience Database
- Ceramide in the Regulation of Neuronal Development: Two Faces of a Lipid - Madam...Ceramide in the Regulation of Neuronal Development: Two Faces of a Lipid - Madame Curie Bioscience Database
- Learning from Deficiency: Gene Targeting of Caspases - Madame Curie Bioscience D...Learning from Deficiency: Gene Targeting of Caspases - Madame Curie Bioscience Database
- Neuronal Survival and Cell Death Signaling Pathways - Madame Curie Bioscience Da...Neuronal Survival and Cell Death Signaling Pathways - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...