

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Protection against cancer by p53 is due mainly to its activity as a transcription factor. The function of p53 in transactivation of target genes is analyzed here with emphasis on the dilemma between cell growth arrest and apoptosis pathways. The question as to which of these p53 functions is required for tumor suppression in vivo is revisited in the light of new studies that renew the focus on senescence and growth arrest mechanisms in protection against cancer. Global gene expression analysis by microarrays, employing either transcription, or conformation switches for p53 activation were utilized to distinguish primary (direct) p53 targets from secondary (indirect) ones and to probe pathways of inhibition of p53-induced apoptosis. The profile of gene expression indicates that p53 is a central node in the cellular network of growth control modulation and its activation results in altered expression of more than a thousand genes. Some of these are co-activators of p53 and may be involved in the decision making of the choice between p53 functions. The major conclusion is that the response to p53 activation is heterogeneous and is mainly dependent on the cellular context, which is evident from the pattern of p53-induced genes in different cell types and in various organs in response to irradiation. The analysis of gene expression profiles following activation or suppression of apoptosis by either chemotherapy or cytokines, respectively, may facilitate the identification of ways to bypass the loss of p53 activity and to design new modalities for cancer treatment.
Introduction
After the discovery of p53 in 1979, it became essential to clone the p53 cDNA in order to understand its cellular function. The first murine p53 cDNA was cloned in 1983 at the Weizmann Institute. In 1981 M. Oren came back from A. Levine lab in Stony Brook and joined the lab of D. Givol with the purpose of cloning p53. Initially a small fragment (~300 bp) from the 3' non-coding region was obtained, followed by a full-length clone that contained the entire cDNA sequence and the translated amino acids sequence.1 The mouse genome contains also a p53 pseudogene that showed a similar size to that of the cDNA and was used to characterize the isolated p53 gene by heteroduplex analysis. Later on the entire gene of p53 containing all the exons spanning 13 kbp was characterized and sequenced to determine the exon structure and intron borders.2 This gene was cloned into an expression vector and used to study the transforming activity of p53 in combination with Ras. Apparently this gene contained a point mutation at position 135 (Ala→Val) that rendered it transforming for NIH3T3 cells at 37°C.3 However, later on it was found that at 32°C this mutant p53 acquired the wild type (wt) conformation and exhibited inhibition of oncogene (e.g., Myc and Ras) -mediated focus formation.4-5 This construct or variants of it made up of a chimera composed from cDNA and genomic fragments was used since then as the temperature-sensitive (ts) p53, denoted p53val135, that can shift from transforming (mutant) to suppressing (wild type) p53 by a temperature shift from 37°C to 32°C.5
The human full-length p53 was cloned in 1984 by D. Givol from a cDNA library of SV-40 transformed fibroblasts6 and in hindsight could have been expected to be wild type p53 without mutations7 since the p53 in these cells was inactivated by the binding of SV-40 T antigen. Indeed this human clone did not transform NIH3T3 cells but this line of research was unfortunately abandoned because of lack of interest in non-oncogenic p53 at that time. An important advance was made when this human cDNA was used to map the chromosomal localization of p53 to chromosome 17p136,8 because this information linked genetics with cancer mutations. Couple of years later Vogelstein's lab determined the loss of heterozygosity (LOH) sites in colon cancer and showed that colon carcinoma have high incidence (75%) of LOH at 17p139 and they requested the human p53 cDNA to determine if p53 chromosomal location coincides with the LOH site on chromosome 17. Indeed they found that p53 was deleted from one allele and the second allele had a mutation.10 This is precisely what the Knudson's two-hit hypothesis predicted for tumor suppressor genes11-12 and was sufficient to rename and define p53 as a tumor suppressor. Similarly, the lab of J. Minna used this p53 clone to analyze tumors from the lung cancer NSCLC and found homozygous deletions, abnormal sized mRNA for p53 or loss of expression, along with a variety of mutations reinforcing the tumor suppressor nature of p53.13 Concomitant in vitro experiments showed that wild type p53 could inhibit the transformation of NIH3T3 cells by oncogenes.4,14 These studies merged genetic and clinical data with functional information to conclude that p53 is a tumor suppressor.
p53 as a Transcription Factor
The molecular function of p53 was defined to be that of a transcription factor15 by showing that it binds specifically to its target DNA fragments that had a symmetrical structure of two 10 bp palindromes, separated by a spacer of 0-14 bp as follows: PuPuPu(CA/TA/TG)PyPyPy (0-14N) PuPuPu(CA/TA/TG)PyPyPy. The three dimensional structure of p53 with its target DNA molecule demonstrated that the core domain of p53 involves in DNA binding16 and the amino acid residues of p53 that make contact with the DNA showed the highest mutation rate in cancer, whereas p53 that is mutated in these positions fail to bind the target DNA. These results defined the DNA sequence element with which p53 interacts in order to activate transcription of its target genes.15 This p53 target site was identified in regions upstream to the RNA start site of genes and also was identified and confirmed functionally in introns.
Recent developments in the human genome sequencing efforts made it possible to identify such p53 binding sites on a genomic scale leading to global identification of p53 target genes thereby throwing light on the cellular functional networks controlled by p53. An algorithm that enables the finding of such DNA sequence in the genome database was used to scan 2583 genes for p53 target sites.17 Using this algorithm, about 300 genes (out of 2583) received a score greater than the cut off value (a score of 93) and qualified as potential p53 targets. The directory of the p53 sites (http://linkage.rockefeller.edu/p53/) is a useful tool to search for new targets. Among the 2583 genes analyzed, 226 (8.7%) were found to have perfect match with the consensus p53 binding sequence, 304 had a similarity score above 93. The variability of the consensus sequence of the DNA target, based on a collection of 37 motifs, was qualified by assigning a proportional weight for each position in the target DNA sequence. The highest weight was given to the fourth C and the seventh G within the internal tetramer (CA/TA/TG) of the palindrome, which are conserved in most target sites whereas the rest of the sequence shows more variability.17 A different attempt to scan in-silico all p53 target sites on chromosome 21 and 22 was performed recently,18 and 48 high confident sites were found. Extrapolation of this analysis to the entire genome predicts 1600 p53 target sites whereas previous estimate was 200-400 sites.15 Surprisingly a significant number of these sites (36%) are located at the 3' of the genes, even more than those at the 5' of the gene, and several genes showed p53 target at both ends.18 This raises the possibility of simultaneous paired-transcription of coding and non-coding strands of the same gene that could contribute to a new level of regulation of gene expression, in the case of p53 or of other transcription factors.
The direct identification of gene targets by p53 used at the beginning subtractive hybridization and had to rely on identification of differentially induced genes by p53. Some of the early candidates detected by this method were not very meaningful considering the then known activity of p53. The first identified target of p53 that was in line with the growth suppression effect of p53 was p21waf. A cell line that expresses the aforementioned human p53 cDNA8 under steroid inducible promoter19 was used to clone the cDNA of the differentially enriched mRNA from the induced p53-containing cells and a major new cDNA clone identified, was that of p21waf.20 The differentially expressed cDNA clones showed overwhelming excess of p21 clones and the promoter of the cloned p21 gene contained two p53 target sites as far as 2.2 kb upstream to the RNA start. At the same time, using a different approach, p21 was isolated as the gene responsible for senescence21 and was also identified as a universal inhibitor of CDK which blocked the cell cycle at G1/S transition and inhibited cell growth.22-23 Interestingly, even before the discovery of p21 as a mediator of growth arrest, the apoptotic function of p53 was demonstrated,24 but there was no clue as to whether or how p21 can be involved in apoptosis. The first identified mediators of p53 induced apoptosis were Bax25 and Fas/APO1.26 Hence, the transcription activation by p53 was split into two functional avenues with separate sets of genes, one that involves cell cycle regulation leading to growth arrest and can be explained by transactivation of p21, and another one that must lead to apoptosis and involves genes that should also be turned on by p53. The choice between transcription activation of either of these pathways continues to be the main dilemma of p53 function in tumor suppression. This choice is tightly linked to the question of how the cell makes the decision whether to enter growth arrest or to undergo cell death upon p53 induction. It is also relevant to the mechanism of tumor suppression by p53 since the present dogma prefers apoptosis as the dominant way by which p53 protects against cancer. In this review we will discuss the systems that were selected for analysis of p53 targets that use inducible or infection vehicles, the methods used for analysis with special emphasis on microarrays, and the main results that expand our knowledge on the regulation of cell response to stress.
The p53 Network
Systems of Analysis
The analysis of p53 downstream targets needed appropriate systems that upon induced expression of p53 lead either to cell cycle arrest or to induction of apoptosis. Two different switches were employed to turn p53 on or off: a conformational switch based on the temperature-sensitive (ts) p53 that is independent of transcription, and a transcriptional switch based on induced p53 transcription by a variety of inducible promoters (Table 1). A number of studies were carried out where overexpression of wild type p53 resulted in cell cycle arrest thereby providing an attractive strategy to pan out genes that regulate cell cycle in a p53 dependent manner.27-35 To an equal extent, extensive analyses have been done on the apoptotic pathway as well that was mediated by p53.29,36-46
Table 1
System of p53 activation and cell lines utilised to determine p53 modulated gene expression profile.
A broad range of cell lines representing all major tumor types were used in an effort to maximize the identification of p53 regulated genes in all the tumor types (Table 1). Kannan et al, 2001,28 have used a temperature sensitive p53 (ts p53Val135) expressed in the human lung cancer cell line H1299 to analyze the p53 mediated transcriptional profile. This ts-p53 protein changes from a mutant to wild-type conformation by a temperature shift from 37°C (non-permissive) to 32°C (permissive temperature) and induces either cell cycle arrest27 or apoptosis29,37 depending on the cell line that is being used. As this conformational switch does not require protein synthesis, it allowed us to distinguish the primary targets that are directly induced by p53 from the secondary indirectly activated genes, by using cycloheximide to inhibit protein synthesis where the levels of p53 remained unchanged.28 In later studies the opposite approach was taken by screening genes during inhibition of the p53 induced apoptosis by cytokines and other factors.37 Similar conformational switch was used in other cell types as well.31,42
Different from the aforementioned conformational switch, overexpression of wild-type p53 was achieved by using transcriptional switches, employing recombinant viral methodologies, chemotherapeutic drugs or irradiation (Table 1). Cells which lack wild type p53 were infected with adenoviruses encoding wild type p53 under a CMV promoter (Ad5CMV-p53) and RNA was harvested at different time points.33-34,38-39,41,46 On the other hand, studies that were aimed at identifying the genes involved in p53 mediated apoptosis used mainly chemotherapeutic agents viz., Etoposide, a p53 activating topoisomerase II inhibitor,30,45 Doxorubicin44 and Camptothecin, a DNA damaging agent that functions through p53-dependent mechanism.43 Zhao et al, 2000,36 have used both UV and Gamma irradiation to induce p53 expression and were able to detect distinct sets and subsets of gene expression patterns specific to each mode of activation. Kannan et al,27 have employed a muristerone inducible transcriptional switch to over-express p53 and p21. Similarly, a tet-off system was used to study the identification and classification of p53 regulated genes32 and Zn induced metallothionein promoter was used by Zhao et al.36
Heterogeneity of the Microarray Based p53 Expression Profile Data
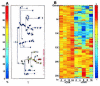
Microarray based transcription profiling data indicated that genes induced by gamma radiation, UV radiation and Zinc induced p53 resulted in distinct sets of genes. Also, not surprisingly, the gene expression pattern thus observed was cell-type or cell-line specific36 and showed different expression kinetics as determined by clustering and expression profiles (fig. 1). For example, cluster 2 contained the early genes that as early as 2 hours after induction showed expression and include p21 and gadd45 and cluster 4 contained late genes that were expressed only after 8 hours. Similar clusters were also observed for the down regulated genes (fig. 1). This and other results led to the conclusion that the p53 dependent gene expression pattern depends on the levels of p53 protein in the cell, the type of induction of p53 and more importantly the cell type examined.36 Similar genomic approach by SAGE analysis revealed that every gene induced by p53 through retroviral infection of DLD-1 cells was also induced by another transcriptional switch (e.g., the rTA system) used, indicating that the cell type rather than mode of p53 induction determines the downstream p53 target genes. Interestingly, they were able to identify only six genes upregulated by Adriamycin and 5-FU, though identical levels of p53 were found after the treatment, alluding to the fact that in addition to cell type, the nature of the induction signal is also important.32 This observation on the cell-type specificity is correlated with the study on p53-induced genes in vivo in response to irradiation. By comparing expressed genes in p53+/+ or p53-/- mice, striking tissue specificity with distinct regulation of p53-induced genes in different tissue compartments was observed.47 For example in the liver the major up regulated gene was p21 whereas in the spleen PUMA was induced in the white pulp, but Noxa and Bid were upregulated in the red pulp and all three were induced in the intestine. This selectivity in transactivation by p53 following DNA damage in vivo correlates with either growth arrest or a variable pro-apoptotic response to γ-irradiation and it is extremely important for cancer therapy. This analysis indicates that to a certain extent p53 behaves like a tissue specific gene when activated by various stress conditions.

Primary and Secondary Targets
The tumor suppressor function of p53 mainly stems from its ability to act as a transcription factor that could sense and integrate growth, oncogenic and stress signals in a concerted manner to maintain cell growth. It is very well documented that activation of p53 leads to modulation in the expression of downstream p53 target genes that in turn control either cell cycle arrest or apoptosis. Since p53 is a transcription factor, a first hand knowledge about its primary targets is an important step in understanding the p53 network. Use of cycloheximide (CHX, a protein synthesis inhibitor) and ts-p53 along with microarray technology on a cell culture system helped us to study this problem since the p53 was stable during the time course of the experiment and RNA synthesis was not affected by the inhibitor. We were able to identify 38 and 24 primary targets genes that were up and down regulated respectively by p53 directly (Figure 2). The results revealed altered expression of a lot of novel genes that controlled other aspects of cell function including adhesion, DNA repair and cell signaling28 in addition to well known p53 targets like p21, Fos, Bax and Bak (Table 2). The results of this experiment also indicated that upon p53 activation at least 10 fold more genes than the number of primary targets genes are activated in the cell, placing p53 at the center of a gene expression network of cellular response (fig. 3). Similar approaches to find out the primary targets in the in vivo context were carried out by treating mice (p53 null or wild type p53) with cycloheximide and irradiation resulted in the identification of marked differences between the gene expression profile in intestine, spleen and thymus (Margalit O and Rechavi G, unpublished).
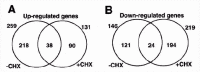
Table 2
Partial list of genes identified as trans-activated by p53 through microarray analyses.
Major p53 Targets and Alternate Pathways Identified by Microarrays: Who Makes the Decisions?
Upon activation, the ability of the ts-p53 (val135) to induce cell cycle arrest but not apoptosis in the lung cancer H1299 cells and vice versa in the myeloid LTR6 cells provided us with an opportunity to dissect the distinct pathways that are key to the tumor suppressor function of p53. This further supports the notion that the p53 transcription program is dependent on the cell type more than the mode of activation. Comparison of p53 regulated gene expression profile in LTR6 cells to that of H1299 cells revealed that only 15% of the genes are common to both systems, leading us to believe in the presence of two distinct transcriptional programs in response to p53 signaling, one leading to growth arrest and the other to apoptosis depending on the cellular context. The proapoptotic genes induced only in LTR6 cells like Apaf-1, Sumo1 and gelsolin may suggest a possible explanation for apoptosis in LTR6 cells.29 Genetic and cellular data indicated previously that Apaf-1 deficiency (e.g., by promoter methylation) abrogates the apoptotic effect of p53 and substitutes for p53 loss in promoting tumor formation, particularly in melanoma.48 Apaf-1 associates with Cytochrome C and caspase 9 to form the apoptosome, a crucial intermediate in the mitochondrial pathway of apoptosis and activation of caspase 3. The identification of a p53 target site in the promoter sequence of Apaf-129,49 added to our understanding of the role of p53 in this pathway. In addition, we found that p53 trans-activation of Apaf-1 was enhanced by Zac-1, a transcription factor that has previously been shown to inhibit cell proliferation. It appears that Zac-1 functions like a co-activator of p53. Furthermore, it was demonstrated that Zac-1 itself is a possible direct target of p53 since the sequence upstream to the first coding exon of Zac-1 contains a p53 recognition site and the luciferase construct containing this region was activated by p53. These results suggest the existence of a tightly controlled self-amplifying mechanism of transcriptional activation leading to apoptosis by p53.50
This is a part of a more general emerging picture that suggests that p53 is not working alone in transactivation but rather it is collaborating with a wide group of co-activators or co-repressors in its effect on gene transcription. Important examples are a group of acetyl transferase enzymes like p300 and CBP that were shown to associate with p53 as co-activators. The deacetylating enzyme HDAC may be a co-repressor for p53. A very recent study demonstrated that p300 is the dominant factor in determining p53 dependent apoptosis.51 Absence of p300 in colorectal cancer cells increases apoptosis in response to DNA damage, (e.g., UV, etoposide, doxorubicin) and this is accompanied by increase in PUMA and decrease in p21waf expression. On the other hand the presence of p300 reverses the situation, increases p21 expression and drives the cells to growth arrest. The expression ratio of PUMA over p21waf is determined by p300. The results put a question mark on who makes the decision in the dilemma between growth arrest and apoptosis: Is it p53 itself or its associate activators? Obviously this will open new directions for research on candidate chemotherapeutic agents against cancer.
Induction of neuronal cell death by camptothecin, a DNA damaging agent that functions through a p53 dependent mechanism resulted in increased Apaf-1 expression. Isogenic cell lines that undergo p53-mediated apoptosis after treatment with ionizing radiation or Doxorubicin were also found to have Apaf-1 up regulation through microarray analysis.44 Results from these studies indicate that Apaf-1 can be a key mediator of p53-induced apoptosis. Some of the previously known and unknown key p53 target genes that were identified in microarrays were listed on Table 2. Another candidate gene that was up-regulated in these studies was the Bcl2-binding component 3 (bbc3)29 that later on was cloned, renamed as PUMA and analyzed in detail to be a major factor in p53 induced apoptosis52-53 (fig. 4). The global analysis of gene expression by the SAGE (serial analysis of gene expression) screening method identified a group of genes, denoted PIGs (p53 Induced Genes) predicted to encode proteins that could generate or respond to oxidative stress. Perhaps the formation of reactive oxygen species; and the oxidative degradation of mitochondrial components, culminating in cell death are related to the p53 pathway of apoptosis.54 The microarray experiments always detected some if not all of the PIG genes, like PIG327 and PIG8.29
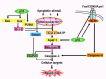
Figure 4
A scheme that integrates the genes in the mitochondrial and cell surface receptor apoptotic pathways that are the targets of activation by p53, as depicted by microarrays.
Inhibition of p53 Induced Apoptosis without Affecting p53 Transactivation
The study of p53-regulated apoptosis is important for cancer research because it may help design strategies for cancer therapy. The genes or factors involved in inhibition of p53-induced apoptosis could be informative for the treatment of cancer since in ~50% of cancer the p53 is in the wild type form but nevertheless p53 activated apoptosis is inhibited. It was shown previously55 that apoptosis induction by p53 in the myeloid leukemia cells M1, could be inhibited by various compounds including cytokines such as interleukin-6 (IL-6) and calcium-mobilizing compounds such as the Ca2+ATPase inhibitor thapsigargin (TG). This inhibition can be due to either inhibition of p53 transcription function, or to p53 degradation or to other means and a microarray analysis was carried out to study this effect37 (fig. 5). Clustering analysis of 1,786 genes, the expression level of which changed after activation of wild-type p53 in either the absence or presence of IL-6 or TG, showed that neither IL-6, nor TG cause a general inhibition of the ability of p53 to up-regulate or down-regulate gene expression. IL-6 or TG also did not affect expression of various p53 targets implicated as mediators of p53-induced apoptosis; nevertheless apoptosis was inhibited effectively. These compounds thus can bypass the effect of wild-type p53 on gene expression and inhibit apoptosis. Hence IL-6 and TG activated different p53-independent pathways of gene expression that include up-regulation of antiapoptotic genes, thereby facilitating tumor development as well as tumor resistance to radiation and chemotherapy in cells that retain wild-type p53.37 This is important for designing therapy that can maximize the anti-tumor effect of p53-induced apoptosis by inhibiting the effect of such cytokines. Cytokines decrease apoptosis in normal cells and therefore inhibition of cytokine activity may improve cancer therapy by enhancing apoptosis in cancer cells. Anti-cytokine therapy against cytokines that inhibit apoptosis can increase the susceptibility of cancer cells to cytotoxic agents such as radiation and chemotherapy. Extending the same line of thinking, there could also be inhibitors that can improve cancer therapy by inhibiting the antiapoptotic function of other compounds.37

New p53 Regulated Pathways Identified by Microarrays
Gene Suppression
Equally important to the identification of up-regulated genes by p53 is the discovery of pattern of downregulated genes upon p53 induction (Table 3). Kannan et al, 2001,28 reported, 24 genes that were downregulated by p53 including BRCA1 and cyclin E. There were several genes reported by various groups to be downregulated by p53 that control cell cycle, DNA replication and signal transduction.27-28,36 Recently a genome-wide study by Kho et al, 2004,56 have identified 189 genes repressed compared to 41 genes activated upon p53 induction during 5-FU induced apoptosis. They have reported that, similar to upregulated primary target genes, there were primary repressed genes like PTTG1 and CHEK1 that had p53 target site and secondary repressed genes like PLK that may be repressed by indirect mechanisms. These results also mirror the repression pattern observed by us.28 3 summarizes a list of p53 trans-repressed genes that involve in cell cycle arrest and apoptosis mediated by p53.
Table 3
Partial list of genes identified as trans-represented by p53 through microarray analyses.
All these results, taken together, suggest that p53 regulates concerted opposing signals (e.g., activation of pro-apoptotic and suppression of anti-apoptotic genes) and exerts its effect through a network of transcriptional changes, which collectively alter the cell phenotype in response to stress. The power of microarray analysis has yielded us an opportunity to look at alternate pathways that were previously not known to involve p53. A cursory look at the p53 mediated transcription profile resulted in the identification of alteration in genes involved in the cell cycle control circuitry along with other functions like cell adhesion, signaling, transcription, neuronal growth, DNA repair, oxidative stress, ECM, cell migration and angiogenesis27-29,36 (Tables 2 and 3). The new p53 regulated pathways identified by microarrays include metabolic enzymes, DNA repair, signal transduction and secreted factors. We will describe several examples to illustrate the relevance of these new pathways for p53 function.
Secreted Factors
Two different studies, one aimed at identifying p53 mediated growth arrest responsive genes27,29 and another aimed at identifying p53 mediated apoptosis responsive genes upon Etoposide treatment,45 has identified MIC1 gene, a member of the TGF-β family as a novel p53 target gene. Later the promoter region of this gene was found to contain the putative p53 target site.30 The MIC1 product was found to be secreted and the conditioned medium from cells expressing this protein can suppress the growth of certain TGF-β receptor and smad-4 expressing tumor cells. This implies a connection between p53 and the growth inhibitory TGF-β family genes and underscores a potentially important novel paracrine mechanism of growth suppression by p53. An extended study on such p53 targets using irradiation and cDNA arrays revealed that in addition to its activity on targets of intracellular growth arrest mediators, p53 induces several secreted growth inhibitory factors with anti-cancer effect on the near-by cells that also can potentiate the cytotoxic effect of chemotherapeutic drugs on tumor cells.57 Another type of secreted factors like thrombospondin 1, inhibit vascularisation and angiogenesis and therefore cut off blood supply to the tumor cells thus preventing tumor growth and metastases.58
Metabolic Pathways
Microarray analysis of differential gene expression in p53 mediated apoptosis resistant and apoptosis sensitive tumor cell line identified several metabolic enzymes to be involved in p53 mediated growth arrest and apoptosis and one example is proline oxidase that is a transcription target for p53.39 A proline oxidase antisense vector repressed the p53-induced up-regulation of proline oxidase, release of cytochrome c from mitochondria, and the apoptosis in renal carcinoma cells. The localization of proline oxidase to mitochondria, suggests that it can directly influence the apoptotic pathway and thus tumorigenesis.59 In another study, an orphan adaptor protein TRAF4, that mediate cellular signaling by binding to members of TNFR superfamily and IL-1/Toll like receptor superfamily is found to be overexpressed and involved in p53 mediated proapoptotic signaling.42 Other metabolic enzymes and processes like ribosomal protein synthesis and histone modifications are also indicated by the analysis of p53 induced genes.
Signal Transduction
Transcription profiling analysis of a glioblastoma cell line showed up-regulation by p53 induction of the expression of hCDC4b, one of the four subunits of SCF (ubiquitin Ligase) complex responsible for degradation of cyclin E.33 This indicates that a previously unrecognized transcriptional target of p53 was being employed to negatively regulate cyclin E, which might stop the cell-cycle progression at G1-S. This would represent a novel p53 dependent mechanism to control cell division in addition to the well-known p21 pathway. Using the same cell line, another study has identified upregulation of the phosphatase DUSP5 gene by p53 induction that resulted in dephosphorylation of Erk1/2 followed growth suppression of human cancer cells. This represents a novel mechanism by which p53 might negatively regulate cell cycle progression by down regulating mitogen or stress activated protein kinases.34 In another study, expression array analysis identified heparin-binding epidermal growth factor-like growth factor (HB-EGF) as being markedly up-regulated by p53. In response to DNA damage, HB-EGF was induced in wild-type, but not in mutant p53-containing cells. This in turn activated the MAPK cascade through pathways involving Ras and Raf. This expression of HB-EGF protects cells from H2O2 induced apoptosis through MAPK activation. Additionally, the PI3K/Akt pathway was activated in response to HB-EGF induction, whereas inhibition of MAPK and Akt activation after DNA damage decreased cell survival in wild type p53-containing cells. These findings point to a novel aspect of p53 function; p53-induced growth factors such as HB-EGF, which activate MAPK and Akt signaling, may be involved in a compensatory mechanism to alleviate adverse effects of cellular stresses. Hence p53 induction counteracts p53 growth suppression through activation of MAPK and PI3K/Akt signaling cascades.60 Perhaps, also in many other cases the outcome of p53 induction is the balance between effects of pro and anti apoptotic genes and this balance can be modulated by small changes due to factors like cytokines.
Tumor Suppression Function of p53 by Growth Arrest: Reassessment
Although in vitro studies and knock-out mice (mainly p21 deficient mice) point to the pro-apoptotic function of p53 as the dominant factor in tumor suppression, it is still possible that the growth arrest function is important in vivo. The major theme of this review was that the dilemma of p53 transactivation in mediating tumor suppression is biased towards preference of apoptosis as the mechanism of cancer prevention and therefore effort should be made to discover the p53- induced apoptotic genes. This is partly due to the fact that very few systems can separate the growth arrest function from the apoptotic one but as was shown recently the p53 mutant R172P (R175P in human) probably does it. This mutant lost the capacity to activate apoptosis but retained the growth arrest function and up-regulates p21 like wild type p53.61 Recent experiments renew the interest in the growth inhibitory function of p53 in vivo as an important factor in suppressing cancer. Lozano and colleagues62 prepared a knocked-in mouse containing this mutant (R172P) and compared it with p53-/- mouse for the spontaneous appearance of tumors. Remarkably at 7 month of age 85% of the p53R172P mice remained alive and tumor free whereas from the p53 deficient mice 90% died, mainly from lymphoma.62 Comparison of the MEFs from the two mice strains showed that both were resistant to apoptosis but only in those derived from the R172P mutant mice, p21 level was elevated. These results suggest that the growth arrest function of p53 is an important part of its tumor suppression and the current dogma about the dominance of apoptosis in tumor suppression should be reconsidered.63 This raises the question as to what may be the mechanism for this anti tumor effect of this mutant p53. One possibility is that it results from the generation of replicative senescence as, for example, was shown by p53 effect in H1299 cells64-65 and other cells66 and perhaps may be important in cancer prevention in various tissues, in particular in the intestine.67-68 An additional unexpected observation was that the tumors derived from the p53-/- mice showed aneuploid karyotypes where the few tumors that developed late in the R172P mutant showed a diploid or sometimes tetraploid but not aneuploid karyotypes. This was due to extra number of centrosomes in the tumors from the p53-/- mice that cause unequal segregation of chromosomes and genome instability.62 It is suggested that p21waf regulates the involvement of cyclin E in centrosome duplication and prevents the generation of more than two centrosomes per cell. Since most cancer cells show chromosomal aberration and aneuploidy, it is possible that p53 gained its title as “guardian of the genome” also because of its role in preventing aneuploidy, perhaps through the activity of p21. In view of the marked heterogeneity in gene expression profile in various organs as a result of p53 activation more information of tissue specific microarray analysis after in vivo p53 induction is needed.
Retrospection and Future Directions
The analysis of p53-induced genes had begun with methods of subtractive hybridization and differential display and isolation of individual relevant genes in various systems.20 Many important genes were identified this way and their relevance was verified by Northern blots and by functional analysis using transfections. The main functions of such genes were cell cycle inhibition, apoptosis and genome stability. The introduction of microarray analysis to the p53 field in 1999 allowed the screen of the equivalent of thousands of Northern blots at once and depicting a gene expression profile that became meaningful in the context of the p53 system. Obviously the major genes that were previously identified were picked up by the microarray methodology as well, and new key genes in the pathway of p53 activation were also discovered (Tables 2 and 3). In addition the microarray method extended the pattern of p53 gene activation to many other cell functions previously unrecognized as related to p53. These include: gene suppression, upregulation of secreted factors, inhibitors of angiogenesis, genes for DNA repair enzymes and nucleotide metabolism, genes of ribosomal and protein synthesis activity, genes related to cytoskeletal functions and cell division, and the realization of the presence of a second envelop of co-activators or co-repressors that assist in the transactivation by p53. Hence p53 is one of the most highly connected nodes in the cellular network and an attack on p53 (by mutation) will disrupt basic cellular functions, particularly the responses to DNA damage and tumor-predisposing stresses.69 On the other hand this also indicates the possibility of correcting defects in p53 functions by employing by-passes at various points of the network in order to overcome tumor growth.
It should however be noted that the microarray gene expression profile is like a snapshot of global gene expression and lacks the functional dimension. It will be desirable to have an analytical system that enables global detection of gene expression combined with functional selection of the relevant genes. A recent study points to such a complementary approach by using libraries of siRNA to select for genes that are functionally involved in the p53 effect on cells. Berns et al, 200470 used RNA interference (RNAi) to perform loss-of-function genetic screens of cellular signaling pathways in mammalian cells. They developed expression vectors to direct the synthesis of short hairpin RNAs (shRNAs) that act as short interfering RNA (siRNA)-like molecules to stably suppress gene expression. In their recent report they constructed a set of retroviral vectors encoding 23,742 distinct shRNAs, which target 7,914 different human genes for suppression and used this RNAi library in human cells to identify new modulators of p53-dependent proliferation arrest. The cells express a temperature sensitive SV40 T antigen that will lose its inhibition of p53 at 39°C. The ts T antigen binds to p53 at 32°C and therefore cell proliferation is not inhibited by p53, but at 39°C (where T antigen is inactive) proliferation is inhibited by p53 and the cells will develop colonies only if p53 growth inhibition is bypassed or blocked by the gene knockdown due to the siRNA. This knockdown confers resistance to p53- dependent growth arrest and allows for the isolation of the genes involved from the selected colonies. In their experiment they identified five new targets that confer resistance to p53 dependent growth arrest. These are RPS6KA6 (ribosomal S6 kinase 4, RSK4), HTATIP (histone acetyl transferase TIP60), HDAC4 (histone deacetylase 4), KIAA0828 (a putative S-adenosyl-L-homocysteine hydrolase, SAH3) and CCT2 (T-complex protein 1, β-subunit). These new tools are similar to global gene expression profile detected by microarrays combined with a built-in functional output, and therefore will greatly facilitate a large-scale loss-of-function genetic screen in mammalian cells. In view of the many dilemma facing p53 function as well as variability and cell specificity pointed out in this review, the new gene expression knock-down/ cell selection methodology is a promising addition to the field and may facilitate the discovery of new p53 targets with functional implications.
In addition, methods that combine microarrays results with promoter identification of co-regulated genes and proteomics promise to decipher cellular networks and to shed more light on the p53 network.
Acknowledgment
We thank Prof. Eytan Domany for helpful discussion.
References
- 1.
- Zakut-Houri R, Oren M, Bienz BSL. et al. A single gene and a pseudogene for the cellular tumour antigen p53. Nature. 1983;306:594–597. [PubMed: 6646235]
- 2.
- Bienz B, Zakut-Houri R, Givol DSL. et al. Analysis of the gene coding for the murine cellular tumour antigen p53. EMBO J. 3:2179–2183. [PMC free article: PMC557662] [PubMed: 6092064]
- 3.
- Eliyahu D, Raz A, Gruss PSL. et al. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature. 1984;312:646–649. [PubMed: 6095116]
- 4.
- Eliyahu D, Michalovitz D, Eliyahu SSL. et al. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci. 1989;86:8763–8767. [PMC free article: PMC298370] [PubMed: 2530586]
- 5.
- Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62:671–680. [PubMed: 2143698]
- 6.
- McBride OW, Merry DE, Oren M. et al. Human p53 cellular antigen is on chromosome 17p13. Cytogenet Cell Genet. 1985;40:694.
- 7.
- Zakut-Houri R, Bienz-Tadmor BSL. et al. Human p53 cellular tumor antigen : cDNA sequence and expression in COS cells. EMBO J. 1985;4:1251–1255. [PMC free article: PMC554332] [PubMed: 4006916]
- 8.
- McBride OW, Merry D, Givol D. The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17p13). Proc Natl Acad Sci. 1986;83:130–134. [PMC free article: PMC322805] [PubMed: 3001719]
- 9.
- Vogelstein B, Fearon ER, Hamilton SRSL. et al. Genetic alterations during colorectal-tumor development. New Engl J Med. 1988;319:525–532. [PubMed: 2841597]
- 10.
- Baker SJ, Fearon ER, Nigro JM. et al. ,Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas Science 1989244217–221. [PubMed: 2649981]
- 11.
- Knudson AGJr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. [PMC free article: PMC389051] [PubMed: 5279523]
- 12.
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. [PubMed: 11905807]
- 13.
- Takahashi T, Nau MM, Chiba ISL. et al. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989;246:491–494. [PubMed: 2554494]
- 14.
- Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–1093. [PubMed: 2525423]
- 15.
- El-Deiry WS, Kern SE, Pietenpol JASL. et al. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. [PubMed: 1301998]
- 16.
- Cho Y, Gorina S, Jeffrey SL. et al. Crystal structure of a p53 tumor suppressor-DNA complex : understanding tumorigenic mutations. Science. 1994;265:346–355. [PubMed: 8023157]
- 17.
- Hoh J, Jin S, Parrado TSL. et al. the p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci. 2002;99:8467–8472. [PMC free article: PMC124275] [PubMed: 12077306]
- 18.
- Cawley S, Bekiranov S, Ng HHSL. et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. [PubMed: 14980218]
- 19.
- Mercer WE, Shields MT, Amin MSL. et al. Negative growth regulation in a glioblastoma tumor cell line that conditionally expresses human wild-type p53. Proc Natl Acad Sci USA. 1990;87:6166–6170. [PMC free article: PMC54493] [PubMed: 2143581]
- 20.
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. [PubMed: 8242752]
- 21.
- Noda A, Ning Y, Venable SFSL. et al. Cloning of senescent cell derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. [PubMed: 8125163]
- 22.
- Harper JW, Adami GR, Wei NSL. et al. The p21 cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin dependent kinases. Cell. 1993;75:805–816. [PubMed: 8242751]
- 23.
- Xiong Y, Hannon GJ, Zhang HSL. et al. p21 is a universal inhibitor of cyclin kinases. Nature. 1983;366:634–635. [PubMed: 8259214]
- 24.
- Yonish-Rouach E, Resnitzky D, Lotem JSL. et al. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–347. [PubMed: 1852210]
- 25.
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. [PubMed: 7834749]
- 26.
- Owen-Schaub LB, Zhang W, Cusack JC. et al. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol. 1995;15:3032–3040. [PMC free article: PMC230534] [PubMed: 7539102]
- 27.
- Kannan K, Amariglio N, Rechavi GSL. et al. Profile of gene expression regulated by induced p53: connection to the TGF-B family. FEBS Lett. 2000;470:77–82. [PubMed: 10722849]
- 28.
- Kannan K, Amariglio N, Rechavi GSL. et al. DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene. 2001;20:2225–2234. [PubMed: 11402317]
- 29.
- Kannan K, Kaminski N, Rechavi GSL. et al. DNA microarray analysis of genes involved in p53 mediated apoptosis activation of apaf-1. Oncogene. 2001;20:3449–3455. [PubMed: 11423996]
- 30.
- Tan M, Wang Y, Guan KSL. et al. PTGF-B, a type B transforming growth factor (TGF-B) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-B signaling pathway. Proc Natl Acad Sci USA. 2000;97:109–114. [PMC free article: PMC26624] [PubMed: 10618379]
- 31.
- Li C, Shridhar K, Liu J. Molecular characterization of oncostatin M-induced growth arrest of MCF-7 cells expressing a temperature sensitive mutant of p53. Breast Cancer Res Treat. 2003;80:23–37. [PubMed: 12889596]
- 32.
- Yu J, Zhang L, Hwang PM, Rago C, Kinzler KW, Vogelstein B. Identification and classification of p53-regulated genes. Proc Natl Acad Sci USA. 1999;96:14517–14522. [PMC free article: PMC24468] [PubMed: 10588737]
- 33.
- Kimura T, Gotoh M, Nakamura YSL. et al. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer Sci. 2003;94:431–436. [PubMed: 12824889]
- 34.
- Ueda K, Arakawa H, Nakamura Y. Dual-specificity phosphatase 5 (DUSP5) as a direct transcriptional target of tumor suppressor p53. Oncogene. 2003;22:5586–5591. [PubMed: 12944906]
- 35.
- Ongusaha PP, Ouchi T, Kim KSL. et al. BRCA1 shifts p53-mediated cellular outcomes towards irreversible growth arrest. Oncogene. 20032;22:3749–3758. [PubMed: 12802282]
- 36.
- Zhao R, Gish K, Murphy MSL. et al. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–993. [PMC free article: PMC316542] [PubMed: 10783169]
- 37.
- Lotem J, Gal H, Kama RSL. et al. Inhibition of p53-induced apoptosis without affecting expression of p53 regulated genes. Proc Natl Acad Sci USA. 2003;100:6718–6723. [PMC free article: PMC164513] [PubMed: 12743373]
- 38.
- Maxwell SA, Davis GE. Differential gene expression in p53-mediated apoptosis-resistant vs. apoptosis-sensitive tumor cell lines. Proc Natl Acad Sci USA. 2000;97:13009–13014. [PMC free article: PMC27169] [PubMed: 11069295]
- 39.
- Maxwell SA, Davis GE. Biological and molecular characterization of an ECV-304-derived cell line resistant to p53 mediated apoptosis. Apoptosis. 2000;5:277–290. [PubMed: 11225849]
- 40.
- Stankovic T, Hubank M, Cronin DSL. et al. Microarray analysis reveals that TP53- and ATM-mutant B-CLLs share a defect in activating proapoptotic responses after DNA damage but are distinguished by major differences in activating prosurvival responses. Blood. 2004;103:291–300. [PubMed: 12958068]
- 41.
- Mirza A, Wu Q, Wang LSL. et al. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene. 2003;22:3645–3654. [PubMed: 12789273]
- 42.
- Sax JK, El-Deiry WS. Identification and characterization of the cytoplasmic protein TRAF4 as a p53-regulated proapoptotic gene. J Biol Chem. 2003;278:36435–36444. [PubMed: 12788948]
- 43.
- Fortin A, Cregan SP, MacLaurin JG. et al. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol. 2001;155:207–216. [PMC free article: PMC2198828] [PubMed: 11591730]
- 44.
- Robles AI, Bemmels NA, Foraker AB. et al. APAF-1 is a transcriptional target of p53 in DNA damage-induced apoptosis. Cancer Res. 2001;61:6660–6664. [PubMed: 11559530]
- 45.
- Wang Y, Rea T, Bian JSL. et al. Identification of the genes responsive to etoposide-induced apoptosis:application of DNA chip technology. FEBS Lett. 1999;445:269–273. [PubMed: 10094470]
- 46.
- Wu Q, Kirschmeier P, Hockenberry TSL. et al. Transcriptional regulation during p21waf1/cip1 induced apoptosis in human ovarian cancer cells. J Biol Chem. 2002;277:36329–36337. [PubMed: 12138103]
- 47.
- Fei P, Bernhard EJ, El-Deiry WS. Tissue-specific induction of p53 targets in vivo. Cancer Res. 2002;7316-7327 ;62
- 48.
- Soengas MS, Alarcon RM, Yoshida HSL. et al. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. [PubMed: 10102818]
- 49.
- Moroni MC, Hickman ES, Denchi ELSL. et al. Apaf-1 is a transcriptional target for E2f and p53. Nat Cell Biol. 2001;3:552–558. [PubMed: 11389439]
- 50.
- Rozenfeld-Granot G, Krishnamurthy JSL. et al. A positive feedback mechanism in the transcriptional activation of Apaf-1 by p53 and the coactivator Zac-1. Oncogene. 2002;21:1469–1476. [PubMed: 11896574]
- 51.
- Iyer NG, Chin S, Ozdag H, Daigo Y, Hu D, Cariati M, Brindle K, Aparicio S, Caldas C. p300 regulates p53 dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci. 2004;101:7386–7391. [PMC free article: PMC409928] [PubMed: 15123817]
- 52.
- Yu J, Wang Z, Kinzler KWSL. et al. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA. 2003;100:1931–1936. [PMC free article: PMC149936] [PubMed: 12574499]
- 53.
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. [PubMed: 11463392]
- 54.
- Polyak K, Xia Y, Zweier JLSL. et al. A model for p53 induced apoptosis. Nature. 1997;389:300–305. [PubMed: 9305847]
- 55.
- Lotem J, Sachs L. Different mechanisms for suppression of apoptosis by cytokines and calcium mobilizing compounds. Proc Natl Acad Sci USA. 1998;95:4601–4606. [PMC free article: PMC22536] [PubMed: 9539784]
- 56.
- Kho PS, Wang Z, Zhuang LSL. et al. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem. 279:21183–21192. [PubMed: 15016801]
- 57.
- Komarova EA, Diatchenko L, Rokhlin OWSL. et al. Stress-induced secretion of growth inhibitors: a novel tumor suppressor function of p53. Oncogene. 1998;17:1089–1096. [PubMed: 9764819]
- 58.
- Dameron KM, Volpert OV, Tainsky MASL. et al. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–1584. [PubMed: 7521539]
- 59.
- Maxwell SA, Rivera A. Proline oxidase induces apoptosis in tumor cells, and its expression is frequently absent or reduced in renal carcinomas. J Biol Chem. 2003;278:9784–9789. [PubMed: 12514185]
- 60.
- Fang L, Li G, Liu GSL. et al. P53 induction of heparin-binding EGF-like growth factor counteracts p53 growth suppression through activation of MAPK and PI3K/Akt signaling cascades. EMBO J. 2001;20:1931–1939. [PMC free article: PMC125417] [PubMed: 11296226]
- 61.
- Rowan S, Ludwig RL, Haupt YSL. et al. Specific loss of apoptotic but not cell cycle arrest function in a human tumor derived p53 mutant. EMBO J. 1996;15:827–838. [PMC free article: PMC450281] [PubMed: 8631304]
- 62.
- Liu G, Parant JM, Lang GSL. et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet. 2003;36:63–68. [PubMed: 14702042]
- 63.
- Attardi LD, de VriesA, Jacks T. Activation of the p53 dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene. 2004;23:973–980. [PubMed: 14749764]
- 64.
- Wang Y, Blandino G, Oren MSL. et al. Induced p53 expression in lung cancer cell line promotes cell senescence and differentially modifies the cytotoxicity of anti-cancer drugs. Oncogene. 1998;17:1923–1930. [PubMed: 9788435]
- 65.
- Wang Y, Blandino G, Givol D. Induced p21waf expression in H1299 cell line promotes cell senescence and protects against cytotoxic effect of radiation and doxorubicin. Oncogene. 1999;18:2643–2649. [PubMed: 10353608]
- 66.
- Sugrue MM, Shin DY, Lee SWSL. et al. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci USA. 1997;94:9648–9653. [PMC free article: PMC23243] [PubMed: 9275177]
- 67.
- Fazeli A, Steen RG, Dickinson SL. et al. Effects of p53 mutations on apoptosis in mouse intestinal and human colonic adenomas. Proc Natl Acad Sci USA. 1997;94:10199–10204. [PMC free article: PMC23339] [PubMed: 9294187]
- 68.
- Campisi J. Cellular senescence as a tumor suppressor mechanism. Trends Cell Biol. 2001;11:S27–S31. [PubMed: 11684439]
- 69.
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. [PubMed: 11099028]
- 70.
- Berns K, Hijmans EM, Mullenders J. et al. A large scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–7. [PubMed: 15042092]
- p53's Dilemma in Transcription: Analysis by Microarrays - Madame Curie Bioscienc...p53's Dilemma in Transcription: Analysis by Microarrays - Madame Curie Bioscience Database
- Genetics, Mutations, and Polymorphisms - Madame Curie Bioscience DatabaseGenetics, Mutations, and Polymorphisms - Madame Curie Bioscience Database
- Tumor-Initiating Cells, Cancer Metastasis and Therapeutic Implications - Madame ...Tumor-Initiating Cells, Cancer Metastasis and Therapeutic Implications - Madame Curie Bioscience Database
- Dynamic Kinetic Modelling of Mitochondrial Energy Metabolism - Madame Curie Bios...Dynamic Kinetic Modelling of Mitochondrial Energy Metabolism - Madame Curie Bioscience Database
- Calcium Channel Block and Inactivation: Insights from Structure-Activity Studies...Calcium Channel Block and Inactivation: Insights from Structure-Activity Studies - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...