

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

“All models are wrong—but some are useful.” (G.E.P. Box)
The Basic Idea
Traditionally, the design of novel drugs has essentially been a trial-and-error process despite the tremendous efforts devoted to it by pharmaceutical and academic research groups. It is estimated that only one in 5,000 compounds investigated in preclinical discovery research ever emerges as a clinical lead, and that about one in 10 drug candidates in development ever gets through the costly process of clinical trials. For each drug, the investment may be on the order of $600 million over 15 years from its first synthesis to FDA approval. In 2000, U.S. pharmaceutical companies spent more than $22 billion in research and development, which, after inflation adjustment, represents a four-fold increase from the corresponding figure some 20 years ago. In an attempt to counter these rapidly increasing costs associated with the discovery of new medicines, revolutionary advances in basic science and technology are reshaping the manner in which pharmaceutical research is conducted. For example, the use of DNA microarrays facilitates the identification of novel disease genes and also opens up other interesting opportunities in disease diagnosis, pharmacogenomics and toxicological research (toxicogenomics). The development of combinatorial chemistry and parallel synthesis methods has increased both the quantity and chemical diversity of potential leads against new targets. Our ability to discover useful leads has been greatly enhanced through astonishing advances in high-throughput screening (HTS) technologies. Through miniaturization and robotics, we now have the capacity to screen millions of compounds against therapeutic targets in very short period of time. Central to this new drug discovery paradigm is the rapid explosion of computational techniques that allow us to analyze vast amount of data, prioritize HTS hits and guide lead optimization. The advances and applications of computational methods in drug design are beginning to have a significant impact on the prosperity of the pharmaceutical industry.
Modern approaches to computer-aided molecular design fall into two general categories. The first includes structure-based methods which utilize the three-dimensional structure of the ligand-bound receptor. Many innovative algorithms have been developed and implemented to construct de novo ligands that fit the receptor binding-site in a complementary manner; some of these will be discussed in Chapter 5. The second approach includes ligand-based methods in which the physicochemical or structural properties of ligand molecules are characterized. A classic example of this concept is a quantitative structure-activity relationship (QSAR) model, which grants a theoretical ground for lead optimization.
For the past four decades the development of QSAR has had a momentous impact upon medicinal chemistry. Hansch pioneered the field by demonstrating that the biological activities of drug molecules can be correlated by a function of their physicochemical parameters:
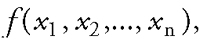
where f is a mathematical function and xi are the molecular descriptors providing information about the physicochemical or structural attributes of the molecules. The major challenges for QSAR practitioners are to find an appropriate set of molecular descriptors and a suitable function that can accurately elucidate the experimental data.
Development of QSAR Models
Ever since the seminal work of Hansch almost 40 years ago, QSAR research has evolved from the use of simple regression models with a few electronic or thermodynamics variables to an important discipline that is being applied to a wide range of problems.1–4 In the following Sections, we will outline the typical steps in the development of a QSAR model.
Descriptor Generation
The first step is the tabulation of experimental or computational physicochemical parameters which provide a description of similarities and differences of the compounds under investigation. The computation of descriptor values is generally straightforward because many commercial and academic computer-aided molecular design (CAMD) packages have been developed to handle this kind of calculation, often with great ease. However, it is more difficult to know a priori the type of descriptor which might be relevant to the biological activity of interest. In many cases, a standard set of descriptors chosen from experience may be used.5
Dimensionality of QSAR Descriptors
Molecular descriptors can vary greatly in their complexity. A simple example may be a structural key descriptor, which takes the form of a binary indicator variable that encodes the presence of certain substructure or functional features. Other descriptors, such as HOMO and LUMO energies, require semi-empirical or quantum mechanical calculations and are therefore more time-consuming to compute. Molecular descriptors are often categorized according to their dimensionality, which refers to the structural representation in which the descriptor values are derived.6 The 1D-descriptors are generally constitutive (e.g., molecular weight) descriptors. The 2D-descriptors include structural fragments fingerprints or molecular connectivity indices. It has been argued that structure key descriptors such as UNITY7 and Daylight8 implicitly account for many physicochemical parameters as well as atom-types and connectivity information.6 The molecular connectivity indices, which are based on graph theory concepts, can differentiate molecules according to their size, degree of branching, shape, and flexibility. Some of the most well-known topological descriptors are the Weiner index (W), the Zagreb index, Hosoya index (Z), Kier and Hall molecular connectivity index (χ), Kier's shape index (κ) , molecular flexibility index (), and Balaban indices (Jx and Jy). As implied by the name, 3D-descriptors are generated from a three-dimensional representation of molecules. Some examples include molecular volume, solvent-accessible surface area, molecular interaction fields, or spatial pharmacophores. With very few exceptions9–11 the descriptor values are computed from a static conformation, which is either a standard conformation with ideal geometries generated from programs such as CORINA12 or CONCORD,13 or a conformation that is fitted against a target X-ray structure or a pharmacophore.
Fragment-Based Physicochemical Descriptors
In addition to intrinsic dimensionality, molecular descriptors can be classified according to their physicochemical attributes. It is recognized that the dominant factors in receptor-drug binding are based on steric, electrostatic, and hydrophobic interactions. For many years medicinal chemists have attempted to model these principal forces of molecular recognition by using empirical physicochemical parameters, which ultimately led to the introduction of fragment constants in early QSAR studies. These descriptors are constants that account for the effect on a congeneric series of molecules of different substituents attached to the common core. The best-known electronic fragment constants are the Hammett σm and σp constants, denoting the electronic effect of parameters at meta and para positions (Note: the σ constant for ortho substituents is generally unreliable because of its steric interaction with the adjacent core group). Another common pair of electronic parameters is F and R, which are the inductive and the resonance components of the σp parameter, respectively. Perhaps the most widely used fragment constant in the field of QSAR is the hydrophobicity parameter, π. It measures the difference of the hydrophobic character of a given substituent relative to hydrogen. To represent the size of a substituent, molar refractivity (MR) is often used, though it has been shown that MR is not a pure bulk parameter since it also captures molecular polarizability of substituents.3
Novel QSAR Descriptors
Novel descriptors continue to appear in the literature; the more currently fashionable types encode combinations of steric, hydrophobic and electrostatic properties, not only for molecular fragments, but the whole molecule as well. For example, polar surface area contains information about both electronics and the size of a molecule and is commonly used in intestinal absorption modeling (see Chapter 4).9 Electrotopological state (E-state) indices capture both molecular connectivity and the electronic character of a molecule.14,15 The GRID16 and CoMFA17 programs take advantage of molecular interaction fields by using different probe types (steric, electrostatic or lipophilic) in a 3D lattice environment. Other variants of molecular field type, such as the molecular similarity-based CoMSIA,18 have also been reported in the literature. Most of the 3D-descriptors require a pre-aligned set of molecules. In cases where the exact molecular alignment is not obvious, one may consider the use of spatially invariant 3D descriptors (i.e., the descriptor values depends on conformation but not spatial orientation). A few innovative descriptors have been introduced for this purpose, including the use of autocorrelation vectors of molecular surface properties, including molecular surface-weighted holistic invariant molecular (MS-WHIM) descriptors MS-WHIM,19–22 and molecular quadrupolar moments.23 Another interesting descriptor, EVA,24,25 is based on normal coordinate frequency analysis and has been validated on a number of standard QSAR benchmark data sets recently.26–28 Burden's eigenvalue index29—originally developed for chemical indexing and later developed to become the BCUT metric30 for molecular diversity analysis—has also been found useful as a QSAR descriptor in emerging applications.31,32
Amino Acid Descriptors
Peptides are often used as probes of a binding site due to ease of synthesis and also to the prevalence of endogenous peptide ligands in nature. Consequently, significant effort has been expended on the development of robust parameters specifically designed to represent amino acids in QSAR applications. Examples are principal properties (z-scales), which are derived from a principal component analysis of 29 experimental and theoretical physicochemical parameters for the 20 naturally occurring amino acids (see Chapter 5),33–36 and the isotropic surface area (ISA) and electronic charge index.37 These latter indices are derived from 3D conformers of the side-chain units and are therefore more readily parameterized for unnatural amino acids. Both sets of parameters have been found to be useful for exploring peptide structure-activity relationships.
Feature Selection
Having generated a set of molecular descriptors, the next step is to apply a statistical or pattern recognition method to correlate these descriptors with the observed biological activities (see also the introduction to feature extraction given in Chapter 2). Partly due to the ease with which a great variety of theoretical descriptors may be generated, QSAR researchers are often confronted with high-dimensional data sets; i.e., the task in such a situation is to solve an underdetermined problem for which there are more variables (descriptors) than objects (compounds). The situation is even more complicated than it appears, because the underlying physicochemical attributes of the molecules that are correlated with their biological activities are often unknown, so that a priori feature selection is not feasible in most cases. Thus, the selection of the best variables for a QSAR model can be very challenging. To reduce the risk of chance correlation and overfitting of data, the entire data set is usually preprocessed using a filter to remove descriptors with either small variance or no unique information.38,39 A feature selection routine then operates on the reduced data set and identifies which of the descriptors have the most prominent influence on the activity to build a model. There are two major advantages of feature selection. First, it can help to define a model that can be interpreted. Second, the reduced model is often more predictive, partly because of the better signal-to-noise ratio which is a consequence of pruning the non-informative inputs.
In the past, variable or feature selection was made by a human expert who relied on experience and scientific intuition, or by a correlation analysis of the data set, or by application of statistical methods such as forward selection or backward elimination. However, when the dimensionality of the data is high and the interrelations between variables are convoluted, human judgment can be unreliable. Also, a simple forward or backward stepping algorithm fails to take into account information that involves the combined effect of several features, so that the optimal solution is not necessary obtained. This problem has been summarized by Kubinyi: “Selection of variables is time-consuming, difficult and, despite many different statistical criteria for the evaluation of the resulting models, a highly subjective and ambiguous procedure”.40 This suggests the need for a method which is applicable to complex multivariate data, easy to use and, of course, supplies a good solution to the problem.
Recent developments in computer science have allowed the creation of intelligent algorithms capable of finding the best, or nearly the best, solutions for such a combinatorial optimization problem (“complex adaptive systems”). A number of fully automated, objective feature selection methods have been introduced. In this section, we will review some of the most common selection strategies used in the current QSAR applications.
Forward Stepping and Backward Elimination
One of the simplest feature selection methods is a stepwise procedure. For forward stepping selection, new descriptors are added to the model one at a time until no more significant variables can be found. Most often, statistical significance is judged by the F-statistics. In backward elimination, the model begins with a full set of descriptors and less informative descriptors are pruned systematically. Both techniques are available as standard routines in many statistical or molecular modeling packages and are very fast to execute. The major shortcoming of this stepwise approach is that the algorithm fails to take into account information that involves any coupled (correlated, parallel) effect among multiple descriptors. Specifically, it is possible that a descriptor is eliminated in an earlier round because it may appear to be redundant at that stage; but later it could become the most relevant descriptor when others have been eliminated. This method is very sensitive to multiple local minima, and often finds a non-optimal solution—a problem which is related to the coupled descriptor effect just described.
Neural Network Pruning
The vast majority of ANN applications in QSAR concern the mapping of descriptor values to biological activities (i.e., parameter estimation of model). Wikel and Dow pioneered the (unconventional) use of ANN in variable selection for QSAR studies.41 This method is considered a “magnitude-based” method for which the inputs with the sensitivities of all variables are computed and the less sensitive variables (as reflected by smaller magnitude in their associated weights) are pruned. Other researchers have proposed “error-based” pruning methods for which the sensitivity of each input is probed by the corresponding change of the training error when its associated weights are eliminated.42
Simulated Annealing
Simulated annealing (SA) is a popular optimization method that is modeled from a physical annealing process.43 The principle of this method is similar to Monte Carlo simulations, except for the use of a cooling schedule for the temperature parameter. As the Boltzmann-type probability factor changes with the lowering of the temperature over the course of the simulation, solutions of lower quality are more readily accepted at the beginning of the optimization than during later stages. The result of SA often depends on the particular cooling schedule used during optimization. The most commonly used schedule is a geometric cooling scheme, where the temperature is decreased by a constant factor (typically in the range 0.90 to 0.99) at each annealing step. Other advanced cooling schedules have been proposed that help enhance the exploration of configuration space. They include the methods of geometric re-heating and adaptive cooling. The major advantages of SA are that the algorithm is easily understood, straightforward to implement, very robust, and generally applicable to almost all types of combinatorial optimization problems. In most cases, one can find good quality solutions to the problem at hand. However, as for all stochastic optimization methods, simulations that are initialized with different random seeds or cooling schedules can lead to very different outcomes. Compared to the forward stepping/backward elimination procedure, this algorithm requires substantially more computing resources.
Genetic Algorithm
The genetic algorithm (GA) idea mimics Nature to solve complex combinatorial optimization problems. GA is a stochastic optimization method that simulates evolution using a simplistic model of molecular genetics.44 The key aspect of a GA is that it investigates many solutions simultaneously, each of which explores different regions of configuration space. The details of this algorithm have been discussed in Chapter 1 and are not repeated here.
Tabu Search
Tabu search is an iterative procedure for solving combinatorial optimization problems. Compared to either SA or GA, the use of Tabu search related to computational molecular design in the literature has been relatively sparse. The basic concept of Tabu search is to explore the search space of all feasible solutions by a sequence of intelligent moves through incorporation of adaptive memory.45 To ensure that new regions of parameter space will be investigated, new moves in Tabu search are not chosen randomly. Instead, some moves that have been previously visited are classified as forbidden (hence Tabu or Taboo), or are severely penalized to avoid cyclic embarking or becoming trapped in a local minimum. Central to this paradigm is Tabu list management that concerns the maintenance and update of moves that are considered forbidden within a particular iteration of moves. In the context of descriptor selection, the Tabu memory may contain a list of individual descriptors or combinations thereof, which have been shown to be uninteresting during the previous rounds of model building.46
Exhaustive Enumeration
Although GAs or Tabu searching explore many possible solutions simultaneously, there is no guarantee that the best solution will ever emerge from simulations. In the presence of complex non-linear system behavior, the exhaustive enumeration of all possible sets of descriptors is the only method which ensures the discovery of the globally optimal solution, although such brute-force approach is often practically impossible. This is due to an exponential increase of the number of descriptor combinations that can be formed from a given number of descriptors (Note: The number of all possible subsets is 2N _ 1, where N is the number of descriptors considered). For a data set that contains 50 descriptors, the number of possibilities is already greater than 1015.
Other Feature Selection Methods
Many feature selection methods have appeared in the literature and it is beyond the scope of this text to provide a comprehensive review. Here other interesting approaches are discussed briefly. GOLPE (Generating Optimal Linear PLS Estimations) is a variable selection procedure to obtain the best predictive PLS (vide infra) models. In this approach, combinations of variables derived from a fractional factorial design strategy are used to run several PLS analyses, and the variables that contribute significantly to prediction accuracy are selected, while all others are discarded.47,48 Livingstone and coworkers recently developed a novel method called Unsupervised Forward Selection (UFS) for eliminating redundant variables.39 UFS begins with the two least correlated descriptors in the data set and iteratively adds more descriptors based on an orthogonality criterion. Variables that have squared multiple correlation coefficients greater than a user-defined threshold with those already chosen will be rejected. The resulting selection of descriptors has low redundancy and multi-colinearity.
Model Construction
Having selected relevant features, the final stage of QSAR model building is executed by a feature mapping procedure—also referred to as the parameter estimation problem. The goal is to formulate a mathematical relationship and to estimate the model parameters. The quality of the parameter set is usually judged by comparing the result of the fitted data to observed data.49 Quite often, feature selection and parameter estimation are performed simultaneously to produce a QSAR model.
Linear Methods
Multiple linear regression (MLR), or ordinary least squares (OLS), was the traditional method for QSAR applications in the past. The major advantage of this method is its computational simplicity, offering the possibility to easily interpret the resulting equation. However, this method becomes inapplicable as soon as the number of input variables equals or exceeds the number of observed objects. As a rule of thumb, the ratio of objects and variables should be at least five for MLR analysis; otherwise there is a corresponding large risk in chance correlation.50 A common way to reduce the number of inputs to MLR without explicit feature selection is through feature extraction by means of principal component regression (PCR). In this procedure, the complete set of input descriptors is transformed to its orthogonal principal components, relatively few of which may suffice to capture the essential variance of the original data. The principal components are then used as the input to a regression analysis.
Another very powerful multivariate statistical method for application to an underdetermined data set is partial least squares (PLS).51 Briefly, PLS attempts to identify a few latent structures, or linear combinations of descriptors, that best correlate with the observables. Cross-validation is employed to avoid overfitting of data. Unlike MLR, there is no restriction in PLS on the ratio between data objects and variables, and the PLS method can analyze several response variables simultaneously. In addition, PLS can deal with strongly collinear input data and tolerates some missing data values.
Non-Linear Methods
Traditionally, non-linear correlation in the data are explicitly dealt with by a predetermined functional transformation before entering a MLR. Unfortunately, the introduction of non-linear or cross-product terms in a regression equation often requires knowledge which is not available a priori. Moreover, it adds to the complexity of the problem and often leads to insignificant improvement in the resulting QSAR. To overcome this deficiency of linear regression, there is an increasing interest in techniques that are intrinsically non-linear. Some of them are mapping methods that attempt to preserve the original Euclidean distance matrix when high-dimensional data are projected to lower (typically two or three) dimensions. Examples of such are non-linear mapping (NLM), self-organizing map (SOM),52 or a ReNDeR-type neural network53 (see discussion in Chapter 2). However, although such maps do offer visual cues to structure-activity relationships, they rarely provide quantitative relationship between structural descriptors and activity. At the present time, artificial neural networks (ANN) are probably the most widely used non-linear methods in chemometric and QSAR applications (see Chapter 1). ANN are computer-based simulations which contain some elements that exist in living nervous systems. What makes these networks powerful is their potential for performance improvement over time as they acquire knowledge about a problem, and their ability to handle fuzzy real world data. With the presence of hidden layers, neural networks are able to implicitly perform non-linear mapping of the physicochemical parameters to the biological activities of the compounds. During the training phase, a network builds an internal feature model from data patterns presented to its input. New similar patterns will then be recognized; the network has the ability for generalization and, more importantly, it is able to make quantitative predictions for queries of similar nature.
Another emerging non-linear method is genetic programming (GP), whose initial use was to evolve computer programs.54 Recently, Kell and coworkers have published an exciting adaptation of GP to analyze mass spectral data.55 It is noteworthy to point out that in GP, the evolutionary algorithm is responsible not only for the selection of descriptors, but also for parameter estimation; i.e., the discovery of the appropriate mathematical transformation relating the descriptors and the response function. The functional tree implementation suggested by Kell operates on simple mathematical expressions that are readily manipulated by genetic operators. Ultimately, these simple mathematical functions are combined leading to non-linear multivariate regression. An advantage of GP over ANN is that the trees evolved are more interpretable and therefore provide valuable insights in the logistics of the decision-making process.56 Another notable difference between GP and conventional ANN is that the GP tree can be evolved to arbitrary complexity in order to solve a problem, although the use of evolutionary neural networks does allow for adaptation of neural network architecture (such as number of hidden nodes) during training.57 In both cases, the QSAR practitioner should be aware of the risk of data overfitting.
Recently, Tropsha and coworkers published a novel non-linear QSAR approach adapted from the k-nearest-neighbor principle (kNN-QSAR).58,59 Briefly, the activity of an unknown compound is predicted as the average activity of the k most similar compounds as measured by their Euclidean distances in multidimensional descriptor space. A simulated annealing procedure may be applied to the selection of an optimal set of descriptors based on predictive statistical parameters. This method is extremely fast and is also generally applicable to large data sets that may contain many structurally diverse compounds.
Model Validation
Model validation is a critical but often neglected component of QSAR development. In a recent review,60 Kövesdi and coworkers state that “[..] In many respects, a proper validation process is more important than a proper training. It is all too easy to get a very small error on the training set, due to the enormous fitting ability of the neural network, and then one may erroneously conclude the network would perform excellently”. The first benchmark of a QSAR model is usually to determine the accuracy of the fit to the training data (“re-classification”), most commonly reported by residual root-mean-squares (rms) error or the Pearson correlation coefficient (see the next Section for definitions). However, because QSAR models are often used for activity prediction of compounds not yet synthesized, the more important statistical measures are those giving an indication of their prediction accuracy.
The most popular procedure for estimation of the prediction accuracy is cross-validation, which includes techniques such as jack-knife, leave-one-out (LOO), leave-group-out (LGO) and bootstrap analysis. The first group of methods is based on data splitting, where the original data set is randomly divided into two subsets. The first is a set of training compounds used for exploration and model building, and the second is the so-called “validation set” for prediction and model validation. The leave-one-out procedure systematically removes one data point at a time from the training set, and on the basis of this reduced data set constructs a model that is subsequently used to predict the removed sample. This procedure is repeated for all data points, so that a complete set of predicted values can be obtained. It has been argued that the LOO procedure tends to overestimate the model “predictivity” and that resulting QSAR models are “over-optimistic”.61 It is worth noting that LOO cross-validation is often confused with jack-knifing. Technically, jack-knifing is used to estimate the bias of a statistic. A typical application of jack-knifing is to compute the statistical parameters of interest for each subset of data, and to compare the average of these subset statistics with the one that is obtained from the entire sample in order to estimate the bias of the latter. In LOO, the main focus is on the estimation of the generalization error based on the prediction of the leave-out samples.62 As an alternative to LOO, a LGO procedure can be applied, which typically sets aside between 5% to 20% of the entire data set as a validation subset. In the literature, this procedure is also known as “k-fold cross-validation”, indicating that the entire data is divided into k groups of approximately equal size. An added bonus of a LGO procedure is a vast reduction in computing resource relative to a standard LOO cross-validation.
Bootstrapping represents another type of re-sampling method that is distinct from data-splitting. It is a statistical simulation method which generates sample distributions from the original data set.63 The concept of bootstrapping is founded on the premise that the sample represents an estimate of the entire population, and that statistical inference can be drawn from a large number of pseudo-samples to estimate the bias, standard error, and confidence intervals of the parameter of interest. The pseudo- (or bootstrap-) samples are created from the original data set by sampling with replacement, where some objects may appear in multiple instances. The usual point of contention about the bootstrap procedure concerns the minimal number of samplings required for computing reliable statistics. An empirical rule given by Davison and Hinkley suggests that the number of bootstrap-samples should be at least 40 times the number of sample objects.64
Another popular means of statistical validation is through a randomization test. In this procedure, the output values (typically biological responses) of the compounds are shuffled randomly, and the scrambled data set is re-examined by the QSAR method against real (unscrambled) input descriptors to determine the correlation and predictivity of the resulting “model”. The entire procedure is repeated multiple times (typically 20–50 models) on many differently scrambled data sets. If there remains a strong correlation between the selected descriptors and the randomized response variables, then the significance of the proposed QSAR model is regarded as suspect.
Finally, the most stringent validation of a QSAR model is through the prediction of new test compounds. It is important that the compounds in an external test set must not be used in any manner during the model building process (e.g., optimizing network parameters or determining a stopping point for neural network training). Otherwise the introduction of bias from the test set compromises the validation process.
Model Quality
A variety of statistical parameters have been reported in the QSAR literature to reflect the quality of the model. These measures give indications as to how well the model fits existing data, i.e., they measure the explained variance of the target parameter y in the biological data. Some of the most common measures are listed below. The first is the Pearson product-moment correlation coefficient (r), which measures the linearity between two variables (Eq. 2). If two variables are perfectly linear with positive slope, then r = 1. However, the Pearson correlation coefficient can be highly influenced by outliers or a skewed data distribution. Under such circumstances, the Spearman rank correlation coefficient rS (Eq. 3) is a robust alternative to r when normality is unreasonable or outliers are present. The Spearman rank correlation coefficient is calculated from the ranks of scores, not the scores themselves. In other words, it is a measure of the strength of the linear relationship between population ranks, and is therefore less sensitive to the presence of outliers in the data. Furthermore, Spearman rank correlation measures the monotony of two random variables; if two variables are perfectly monotonically increasing, then rs = 1. It is noteworthy that if rS is noticeably greater than r, a transformation of data might lead to a stronger linear relationship. For classification, Matthews' correlation coefficient (c) is a popular statistical parameter to denote the quality of fit (Eq. 18 of chapter 2). This accounts not only for correct predictions, i.e., true positives (P) and true negatives (N), but also incorrect predictions (false positives/overprediction (O) and false negatives/underpredictions (U)). Similar to the other types of correlation coefficients, the Matthews' correlation coefficient ranges from 1 (perfect correlation) to _1 (perfect anti-correlation).
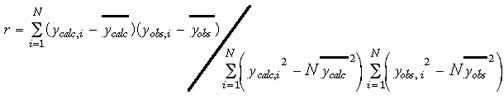
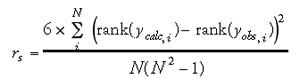
Other commonly used goodness-of-fit measures are the residual standard deviation (s) (Eq. 4) and the root-mean-square difference (rmsd) (Eq. 5) between the calculated versus observed values:
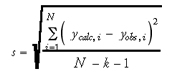
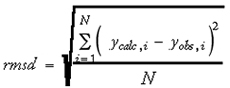
where N is the number of data objects and k is the number of terms used in the model. For these quantities, smaller values indicate a better fit of the data.
For cross-validation, PRESS (Eq. 6) and q2 (Eq. 7) have been suggested to provide good estimates of the real prediction error of a model:
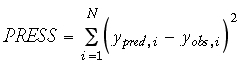
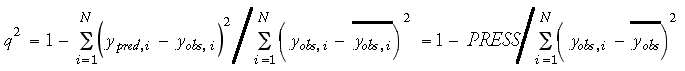
It should be noted that, contrary to its name, q2 can be a negative value. Generally speaking, a q2 value of 0.5-0.6 is regarded as the minimum acceptance criteria for a reliable QSAR model.
Generally, the following characteristics are regarded as good traits of a robust and high-quality QSAR model:
- All descriptors used in the model are significant, and specifically, none of descriptors should be found to account for single peculiarities (e.g., unique compound-descriptor association).
- There should be no leverage or outlier compounds in the training set, otherwise the statistical parameters reported by the model may not be meaningful.
- The cross-validation performance should be significantly better than that of randomized tests but not very different from that of the training set and external test predictions.
Application of Adaptive Methods in QSAR
Variable Selection
The crux of data reduction is to select a subset of features retaining the maximal information content of the data.65 Feature extraction, exemplified by principal component analysis and discriminant analysis, transforms a set of features into a reduced representation that captures the major variance of data. Feature selection, on the other hand, attempts to identify a small subset of features from all available features. The following is a brief account of how artificial intelligence methods have been applied to feature selection in QSAR applications.
Artificial Neural Networks
The earliest application of ANN for variable selection in a QSAR application was reported by Wikel and Dow.41 After training a neural network by using all descriptors, those inputs having large weights between the input and the hidden nodes were selected for further analysis. The “magnitude-based” algorithm was tested on the widely-studied Selwood data set,66 which is comprised of a series of 31 antifilarial antimycin analogs, each parameterized by 53 physicochemical descriptors (Note: Livingstone, a co-author of the Selwood paper, has recently expressed the opinion that this particular data set should not be regarded as a “standard” but rather a “difficult” data set due to poor data distribution)39. In their pioneering work, Wikel and Dow employed a color map to indicate the magnitude of the weight values. This led to the identification of a set of three relevant descriptors. Used in a multiple linear regression they produced to a QSAR model that was marginally better (r = 0.77, rcv = 0.68) than the three-descriptor model originally published by Selwood (r = 0.74, rcv = 0.67).66 Despite this encouraging result, it was argued that this descriptor selection scheme seemed somewhat subjective. Specifically, an overtrained network (vide infra), which is characterized by large weight values of nearly all of the used descriptors, can result in poor discrimination between the relevant and irrelevant descriptors. Thus, it is important to adopt a robust early stopping criteria during neural network training in order to achieve correct pruning of unnecessary input descriptors.41,42
Recently, Tetko and coworkers benchmarked five neural network-based descriptor pruning methods in a series of three studies.42,67,68 Two magnitude- and three error-based methods were examined. The magnitude-based methods are similar in principle to the Wikel and Dow method, in which the pruning follows direct analysis of the magnitudes of the outgoing weights. The error-based method, on the other hand, detects sensitivity of the input by monitoring the change of the network error due to the elimination of some neuron weights associated with some inputs. Unlike the magnitude-based sensitivity method, the error-based pruning method assumes that the training error is at a minimum, so if an early stopping criteria is applied then the assumption will no longer be justified.42 Overall, the authors concluded that no significant advantage of one method over the others was evident from their analyzed data sets. In general, error-based sensitivity methods are computationally more demanding, particularly those which require higher derivatives of the error functions (e.g., optimal brain damage,69 or optimal brain surgeon algorithms70). All algorithms give similar results for both linear and non-linear simulated sets of data (artificial structured data sets) and are capable of identifying the least sensitive input descriptors. In all cases, predictivity of the ANN can be improved by the removal of redundant input descriptors. The five pruning algorithms were also tested with three real QSAR data sets, with the conclusion that the behavior of the different pruning methods seems to deviate more significantly in real QSAR modeling. However, the authors stated that it is very difficult to determine which pruning method would be universally applicable since the efficacy of each method is most likely data-dependent.
Genetic Algorithm
The earliest application of GAs in the role of descriptor-selection chemometric and QSAR applications was reported by Leardi and coworkers in 1992.71 Their initial test was performed on an artificial data set containing 16 objects and 11 descriptors. The simplicity of this test set allowed them to compare the result of a GA solution with that of an exhaustive enumeration of all possible subset selections. They observed that, on average, the globally optimal solution was found by the GA within one-quarter of the time required by the exhaustive search. This was in contrast with the result obtained by the stepwise regression method, which found a solution that was ranked only sixth overall in the list generated by the exhaustive search. After the initial validation, the GA-MLR method was applied to a real chemometric data set consisting of 41 samples and 69 descriptors. For this example, the stepwise regression approach yielded a 12-descriptor model with a cross-validated variance of 83%. The top models from five independent GA simulations yielded models with cross-validated variances of 89%, 85%, 81%, 91%, and 84%, demonstrating the stochastic nature of GA optimization. It is appropriate to perform multiple runs on the same input data when a GA is employed for feature selection. Besides, it is quite possible that the simplistic implementation of GA used in this exercise failed to escape from local optima. More advanced evolutionary algorithms, such as the “ring” or “island” models of parallel GAs, which partition the population into sub-populations and allow for a “migration” operator between sub-populations, may lead to improved convergence.56,72
Rogers and Hopfinger proposed a new GA-based method, termed genetic function approximation (GFA), for descriptor selection. A conventional GA, which contains crossover and mutation operators, is coupled with a MLR for parameter estimation. There are two principal enhancements in the GFA approach. The first is the introduction of a few non-linear basis functions such as splines and quadratic functions. The second is the incorporation of the lack-of-fit (LOF) error measure, which penalizes the use of large numbers of descriptors as a fitness criterion to safeguard against overfitting. With their GFA algorithm, Rogers and Hopfinger discovered a number of linear QSAR models for the Selwood antimycin data set which were significantly better than those obtained by Selwood and by Wikel and Dow.41,66 Interestingly, there seems to be only little overlap, other than the use of clogP—the sole descriptor encoding hydrophobicity—among the three studies. Similar to the finding by Leardi et al, the top 20 GFA models have a range of cross-validated r values from 0.85 to 0.81, again supporting the notion that many independent QSAR models can provide useful activity correlation on the same data. The use of multiple statistical models in the context of consensus scoring was also suggested by the authors, who observed that averaging the predictions of many top-rated models can lead to better predictions compared to any individual model.
At about the same time, two research groups published another GA variant also using the Selwood data set as a benchmark. The algorithm investigated termed MUSEUM (Mutation and Selection Uncover Models) by Kubinyi or Evolutionary Programming (EP) by Luke.40,73 The major difference of the algorithm compared to GFA is the absence of a crossover operator and its reliance solely on point mutation to generate new solutions. Independently, both groups discovered other excellent three-descriptor combinations that might have been found by GFA but were probably destroyed during the evolution because GFA did not employ elitism to preserve the best solutions for the next generation.
Very recently, Waller and Bradley proposed a novel variable selection technique called FRED (Fast Random Elimination of Descriptors) which contains elements from both evolutionary programming and Tabu search.46 In contrast to the other common genetic and evolutionary algorithms, the complete solutions (i.e., the descriptor combinations) are not propagated to the next generation, but rather, only those descriptors are retained which contribute positively to the genetic makeup of the fittest solutions. Descriptors that appear to be less useful are kept in a Tabu list, and are subsequently eliminated if they are not found to be beneficial during later iterations. Application of the FRED algorithm to the Selwood data yielded a final population of three-descriptor combinations that can be represented by 13 different input variables. This analysis was consistent with the results of the previously published methods. In particular, the selection of descriptors shared much similarity with the sets of descriptors that are chosen in the top GFA, MUSEUM, or EP models. The authors argued that it would be more difficult for poorer descriptors to be masked by some exceptionally good combination of descriptors and subsequently proliferate to the next generations, because only potentially good (single) descriptors are being passed to subsequent generations. It should be emphasized that the result of the FRED algorithm is to prune a potential list of descriptors by eliminating the less relevant descriptors; at the end of the calculation the best solutions are not necessarily guaranteed by the algorithm. Accordingly, one interesting utility of the FRED algorithm would be to treat it as a pre-filter for redundant descriptors so that an exhaustive enumeration could be applied.
All of the above variants of GAs are used in conjunction with MLR. A natural extension is to replace MLR with PLS, which is often regarded as a modern alternative and has also played a critical role in the development of the CoMFA methodology. Interestingly, relatively few researchers have investigated methods of variable selection for PLS analysis in the past (other than filtering out the variables with insignificant variance). One explanation might be that PLS has a high tolerance towards noisy data, and any number of input variables may be used. This attitude has changed somewhat over the past few years, as more people have begun to recognize the benefits of feature selection, and the use of hybrid approaches such as GA-PLS or GOLPE-PLS has become increasingly popular. Some examples of the application of GA-PLS include the QSAR studies performed by Funatsu and coworkers.74–78 In a QSAR study of 35 dihydropyridine derivatives,74 these researchers discovered that the cross-validation statistics (q2 = 0.69) of the GA-PLS model based on only six descriptors is superior to the full PLS model using all 12 descriptors (q2 = 0.62). Furthermore, elimination of the less relevant descriptors makes the QSAR model more interpretable, and the selected descriptors were then consistent with an earlier analysis of Gaudio et al, who had performed an extensive investigation on the same set of compounds.79 The usefulness of variable selection in PLS analysis was further demonstrated in a subsequent QSAR study of 57 benzodiazepines.75 Two GA-PLS models—based on 10 and 13 descriptors—yielded the essentially identical q2 value (0.84), and were again significantly better than the model derived from a PLS analysis using all 42 descriptors (q2 = 0.71). The apparent improvement in predictivity was verified by an external validation. Using D-optimal design, the data set was partitioned into a training set of 42 compounds and a test set of 15 compounds. The r2 values of the test predictions for the two GAPLS models were 0.70 and 0.74, respectively, which compares favorably to the solution of the full PLS model (r2 = 0.59). Overall, the results from this and other research groups underscore the value of descriptor selection in the context of QSAR modeling.80
Parameter Estimation
Artificial Neural Networks
The key strength of a neural network is its ability to allow for flexible mapping of the selected features by implicitly manipulating their functional dependence. Unlike multiple linear regression analysis, ANN handle both linear and non-linear relationships without adding complexity to the model. This capability partly offsets the longer computing time required by a neural network simulation because it avoids the need for separate examination of each possible non-linearity.81 In addition, it has been suggested that neural networks are parsimonious approximators of mathematical functions;82 that is to say, an ANN tends to fit the required function with fewer parameters than other methods, and is particularly effective if non-linearity is evident in the data set. Thus, this type of approach seems to be exceptionally well suited for the study of quantitative structure activity relationships.
The first applications of neural networks in the area of QSAR were published by Aoyama, Suzuki, and Ichikawa in 1990 with the promise that “the effective application of such neural networks may bring forth a breakthrough in the current state of QSAR analysis”.83,84 In their initial applications neural networks were used to perform tasks that were previously accomplished by multiple linear regression analysis. Three data sets were examined: a set of mitomycin analogues with anticarcinogenic activity; a series of antihypertensive arylacryloylpiperazines, and a large series of benzodiazepines used as tranquilizers. In these studies, substituent fragment descriptors, together with a few structural indicator variables, were used to encode molecular structures. In all cases the neural networks were able to deduce QSAR models that were superior to MLR fits. However, the use of an excessive number of connecting weights (in one example, 420 weights were used to fit 60 compounds) seemed questionable,85 partly because this contradicted a previously established guideline for MLR: the ratio of compounds versus parameters should be at least five.50 In addition, the authors included both linear and squared terms of molecular descriptors in the analysis, which seems unnecessary since an ANN ought to be able to uncover the appropriate functional transform of each descriptor.
This initial promise—as well as some obvious limitations—of the first ANN applications to the field of QSAR motivated many subsequent investigations aiming to gain a better understanding of this novel tool.85 These were exemplified by the outstanding work of Andrea and Kalayeh, who performed a comprehensive investigation of QSAR of a large data set of dihydrofolate reductase (DHFR) inhibitors.86 This data set had been previously analyzed by Silipo and Hansch using MLR,87 and contains 256 compounds that were characterized using seven substituents descriptors augmented by 6 indicator variables (Table 1). It is noteworthy that the indicator variables had been introduced by Silipo and Hansch to capture certain commonalities and structural features that could not be easily explained using standard fragment descriptors. It should be recognized that the net effect of some combinations of indicator variables and substituent parameters is to encode non-linear effects. For example, one of the terms in a regression equation may be a fragment-based descriptor (e.g., MR) that reflects how activity generally increases with the size of a given substituent. But at the same time, there may be an indicator variable present in the equation that penalizes the presence of an excessively large group at the same position (i.e., the binding pocket has a finite size). Thus, the net effect is that the relationship between substituent size and bioactivity is non-linear. With the exception of I1, which accounts for possible differences in DHFR active sites or assay conditions, all indicator variables were related to substituent positions already encoded by other fragment parameters. In the published MLR model, the indicator variables explained a significant amount of variance, as well as many outliers in the data set. For this data set, it was found that the r2 value of MLR decreased from 0.77 to 0.49, and the number of outliers (defined by the authors as those compounds with an absolute prediction error greater than 0.8) in the model increased from 20 to 61 when indicator variables (I2 to I6) were excluded from the analysis. However, because indicator variables provide little or no insight into the physicochemical factors that govern biological activity, their utility in de novo design of new analogues is limited. In this regard, it is encouraging to observe that it was not necessary to utilize these indicator variables in the ANN model. In fact, using only seven substituent descriptors and (the non-structural) I1, the ANN model yielded a r2 value of 0.85 and only 12 outliers. Thus, the neural network seemed to circumvent the need for indicator variables and was able to extract relevant information directly from the various hydrophobic, steric, and electronic parameters. In addition, Andrea and Kalayeh conducted a cross-validation experiment on a subset of 132 compounds (DHFR from Walker 256 leukemia tumors, i.e., compounds with I1 = 1) and obtained a r2 cv value of 0.79 for ANN. This result once again compared very favorably with the corresponding statistics from MLR, which yielded r2cv values of only 0.64 and 0.30 for models with and without the use of indictor variables, respectively. In addition, in contrast to the first ANN applications of Aoyama and coworkers, Andrea and Kalayeh clearly demonstrated that a neural network implicitly handles higher-order effects and also showed that it was not necessary to include non-linear transformation of the descriptors as inputs to the network.
Table 1
Descriptors used by Andrea and Kalayeh.
Andrea and Kalayeh also presented the first example of ANN overfitting in the area of QSAR. By demonstrating that the training error typically decreases with the number of hidden nodes while the test set error initially decreases, but will later increase, when an excessive number of hidden nodes is deployed. Furthermore, they considered test set statistics as a criteria to select an optimal neural network architecture (Note: For this reason their test set should not be regarded as an external set in the true sense because it was involved during model building). They also proposed a parameter, r, which is the ratio of the number of data points to the number of network weights, to help to define optimal network architecture. Though it was later shown that r by itself may not be sufficient to minimize the risk of overfitting,88 the general principles that were elucidated in this work are still valid and have probably saved many researchers from the perils of flawed QSAR models; i.e., a QSAR model may yield outstanding performance for the training set but no predictivity for new compounds.
Following the publications of Aoyama and co-workers83,84 and Andrea and Kalayeh86 in the early 1990s, the use of ANN in the area of structure-activity relationships has flourished. Figure 1 shows a histogram of the number of publications related to the application of ANN in QSAR analysis according to a bibliographic search.89 These reports include many correlative structure-activity studies using standard descriptors,88,90–114 or some more novel descriptors such as topological indices,88,115,116 molecular similarity matrices,117–121 quantum chemistry descriptors,122–125 autocorrelation vectors of surface properties,19,126 hydrogen bonding and lipophilicity properties,127,128 and thermodynamic properties based on ligand-receptor interactions.110 More recently, a number of novel ANN applications have reached beyond the premise of structure correlation with in vitro or in vivo potency and have ventured to solve some more challenging problems such as the prediction of pharmacokinetic properties,129–132 bioavailability,133 toxicity,106,125,134–141 carcinogenicity,142,143 prediction of mechanism of action,144 or even the formulation of a discrimination function to distinguish drug-like versus nondrug-like compounds145–147 (see Chapter 4). In addition to these quantitative studies, ANN has also been employed as a visualization tool to reveal qualitative trends in SAR analyses.53,117,118,144,148 The widespread use of ANN in the area of molecular design has stimulated the continuous influx of novel neural network technologies to the field, including the introduction of Bayesian neural networks,145,149–151 cascade-correlation learning architecture,67,152 evolutionary neural networks,57 polynomial neural network153 and intercommunication architecture.154 This cross-fertilization between artificial intelligence and pharmaceutical research is likely to continue as more robust ANN toolkits become commercially or freely available. Many excellent technical reviews have been written on the application of ANN in QSAR, and interested readers are encouraged to refer to them.60,81,82,85,155–164
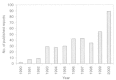
Figure 1
Number of published reports on application of neural networks in the field of QSAR.
It is possible to summarize with a set of general guidelines for effective use of neural networks in QSAR modeling (and statistical modeling in general). First, the law of parsimony calls for the use of a small neural network, if possible. The number of adjustable parameters should be small with respect to the number of data points used for model construction, otherwise poor predictive behavior may result due to data overfitting. Neural network modeling can also benefit from the use of a large data set, which can facilitate location of a generalized solution from the underlying correlation in the data. In addition, the training patterns must be representative of the data to be predicted. This will lead to a more realistic predictive performance on the external test set with respect to the training result. It is advantageous to make use of efficient training algorithms; for example, those that make use of second derivatives of the error function for the weight update, which can give better convergence characteristics. Finally the input descriptors must obviously be relevant for the data modeling process. The golden rule of “garbage in, garbage out” can never be over-emphasized.165
Hybrid Methods
GA-NN
The natural evolution of the next generation of QSAR methods is to apply artificial intelligence methods in both descriptor selection and parameter estimation. An example of such hybrid approach was proposed by So and Karplus, who have combined GA with ANN for QSAR analysis.119,120,166,167 This method, called genetic neural network (GNN), was first applied to an examination of the Selwood data set.166 The major aim of this work was to use a GA to select a suitable set of descriptor for use in the development of a QSAR. The effectiveness of the GA was demonstrated by the ability to select an optimal set of descriptors, as compared to exhaustive enumeration, in the GNN models. It appears that the improvement of the GNN QSAR over other published models (Table 2) is due to the selection of non-linear descriptors which the ANN is able to assimilate.
Table 2
Comparison of linear regression and neural network QSAR models of the Selwood data set. All models are based on three molecular descriptors.
In their next study,167 an improvement to the core GNN simulator was made by replacing the problematic steepest descent training algorithm by a more robust scaled conjugate gradient optimizer,168 leading to substantial performance gains in both convergence and the speed of computation. To provide an extended test of the enhanced GNN simulator, it was applied to a set of 57 benzodiazepines which had been previously studied by Maddalena and Johnston using a backward elimination descriptor selection strategy and neural network training.99 It was found that the GNN protocol discovered a number of 6-descriptor QSAR models that are superior to the best (and arguably more complex) models reported by Maddalena and Johnston.
After appreciable success with standard fragment-based 2D descriptors, So and Karplus extended the use of GNN to the analysis of a molecular similarity matrix to derive 3D QSAR models.119,120 Molecular similarity is a measure based on the similarity between the physical or structural attributes of a set of molecules.169 This type of descriptor differs from conventional substituent parameters (e.g., p, s, and MR) in the sense that it does not encode physicochemical properties which are specific for molecular recognition. The similarity index is derived from numerical integration and normalization of the field values, and represents a global measure of the resemblance between a pair of molecules based on their spatial and/or electrostatic attributes. Thus, instead of a correlation between substituent properties and activities, a similarity-based QSAR method establishes an association between global properties and activity variation among a series of lead molecules. The implicit assumption is that globally similar compounds have similar activities.170 Figure 2 is a schematic diagram showing the different stages in the construction of a SMGNN (Similarity Matrix Genetic Neural Network) QSAR model. The initial validation was performed on a corticosteroid-binding globulin steroid data set,119 which had been extensively studied in the past by many novel 3D-QSAR methods.17,19,20,22,117,119,171–176 The first SMGNN application focused mainly on method validation, in particular the sensitivity and effect of parameters related to: (a) electrostatic potential calculations (type of atomic charges; truncation scheme for electrostatic potential, and dielectric constant); (b) similarity index (Carbó,177 Hodgkin,178 linear and exponential formulae179); (c) grid parameters (spacing, size, and location); and (d) number of similarity descriptors in QSAR model. The results of the sensitivity studies demonstrated that the SMGNN QSAR obtained was very robust with respect to variation in most of the user-defined electrostatic parameters. The fact that the various similarity indices are highly correlated also means that the choice of an index had negligible effect in determining the quality of the QSAR. The grid-related settings also had relatively little impact on the overall result. The key parameter seems to be the number of descriptors used in the model; it is important to have enough descriptors to characterize the data set but not so many that overfitting can arise. Overall, the SMGNN model is superior to those obtained from PLS and GA-MLR method; and also compares favorably with the results from other established 3D-QSAR methods. This approach was further validated using eight different data sets, with impressive results.120 The biological activities and physicochemical properties of a broad range of chemical classes were successfully correlated. One of the shortcomings of the SMGNN method is that interpretation of the QSAR model is difficult because the similarity index is not related to physicochemical attributes of the molecules. However, it is remarkable that the SMGNN QSAR model was consistent with all known SAR for CBG-steroid binding and, therefore, seems to handle the physical attributes leading to optimal binding in an implicit manner.
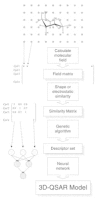
Figure 2
Schematic diagram for the construction of SMGNN 3D-QSAR models. Reprinted with permission from: So S-S, Karplus M. J Med Chem 1997; 40:4347–4359. ©1997 American Chemical Society.
Recently, Borowski and coworkers implemented and extended the SMGNN methodology to evaluate a set of 5-HT2A receptor antagonists in a 3D QSAR study.121 The data set included 26 2- and 4-(4-methylpiperazino)pyrimidines, as well as clozapine, which was used as a reference compound. Due to molecular symmetry the pyrimidines can have multiple mappings to the clozapine reference structure. Five alternative alignment schemes were suggested by the authors, and the q2 values of the models from each alignment were compared with the values derived from 30 randomly chosen alignment sets, which served as a baseline to test statistical significance. The alignment set with a particularly high predictivity was assumed to contain the correct superimposition of the bioactive conformations of these molecules. An interesting finding was that, although it was recognized that the piperazine nitrogen ought to be protonated upon binding to the 5-HT2A receptor, setting an explicit positive charge on the ligand was detrimental to the performance of the SMGNN QSAR model. This is because the charge has a pronounced effect in the electrostatic calculation, rendering the similarity indices discriminating. One suggestion from the authors is to consider only the neutral, deprotonated form during the similarity calculation. The best steric and electrostatic SMGNN models both contain five descriptors, and yield q2 values of 0.96 and 0.93, respectively. Both models are significantly better than random models with scrambled output values, which return q2 values of 0.28 ± 0.16 and 0.29 ± 0.14, respectively. In summary, the results of this independent study strongly support the use of the SMGNN methodology in 3D QSAR studies.
The research group of Peter Jurs is also very active in the development and application of GA-NN type hybrid methods in QSAR and QSPR studies.138 In their procedure, the full data set is usually divided into three parts. The majority (70–80%) of the compounds belongs to the training set (tset), and the reminder of compounds are usually evenly divided to give a cross-validation set (cvset) to guide model development, and an external prediction test (pset) to validate the newly developed QSAR models. Jurs has defined three types of statistical models derived from their multivariate data analysis:
- Type I model is a linear regression model whose selection of descriptors is based on a stochastic combinatorial optimization routine (e.g., SA or GA);
- Type II model is a non-linear ANN model that directly adopts the descriptors used in the Type I model;
- Type III model is a fully non-linear ANN model developed in conjugation with a SA or GA for descriptor selection.
The quality of a model is based on the following fitness (or cost) function (Eq. 8):38
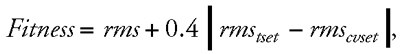
where the coefficient of 0.4 was determined empirically to yield models with enhanced external predictivity.
In a recent application this GA-NN method was used to study the QSAR of 157 compounds with inhibitory activity against acyl-CoA:cholesterol O-acyltransferase (ACAT), a biological target implicated in the reduction of triglyceride and cholesterol levels.38 Twenty-seven compounds were removed from the initial data set due to high experimental uncertainty of their IC50 values, and the remaining compounds were partitioned to obtain tset, cvset and pset with 106, 11, and 13 compounds, respectively. A large number of descriptors were generated using their in-house automated data analysis and pattern recognition toolkit (ADAPT) software package, and were pruned according to a minimal redundancy and variance criteria. The best Type I model is a nine-descriptor MLR that has a rmstset of 0.42 and rmspset of 0.43 log units. Using the same set of descriptors, they generated a Type II ANN-based model that has significantly lower rms errors of 0.36 and 0.34 log units for the tset and pset. Finally, to take full advantage of the non-linear modeling capability of ANN, they conducted a more comprehensive search using a combined GA-NN simulation. The top Type III model employed eight descriptors and yielded an rms error of 0.27 for both tset and pset. Four of the eight descriptors used in the Type III model are identical to those selected by the linear Type I model. It is suggested that unique descriptors in the Type III model provide relevant information and are also non-linear in nature.
The general applicability of the GA-NN approach in QSAR was further verified in another study where a large set of sodium ion-proton antiporter inhibitors were investigated.180 Following the established procedure, Kauffman and Jurs divided the 113 benzoylguandine derivatives into a 91-member tset, an 11-member cvset, and an 11-member pset. Using an SA feature selection algorithm, they searched for predictive models containing from 3 to 10 descriptors. The optimal Type I linear regression model used 5 descriptors and yielded a rms error of 0.47 and r2 = 0.46 for the tset. The predictive performance of the pset was, however, rather poor (rms = 0.55; r2 = 0.01), indicating a general deficiency of this linear model. The replacement of MLR by ANN in functional mapping led to moderate improvement. The corresponding Type II model reported a rms error of 0.36 and r2 of 0.68 for the tset, and significantly lower prediction errors for the pset compounds (rmspset = 0.42 and r2pset = 0.44). The greatest increase of accuracy was seen in the construction of the Type III model, where the rms error of the tset dropped to 0.28 and r2 increased to 0.81. The corresponding pset statistics were 0.38 and 0.44, respectively.
The authors also explored the consensus scoring concept proposed by Rogers and Hopfinger,181 and examined the effect of prediction averaging using a committee of five ANNs. They confirmed that the composite predictions were more reliable than those from individual predictors, largely because they make better use of the available information. The rms error of the prediction set for the consensus model was 0.30 compared to an error of 0.38 ± 0.09 from the five separate trials. This result is also consistent with an earlier GNN study on the Selwood data set, stating that averaging of the outputs of the top-ranking GNN models led to marginally better cross-validation statistics compared to the individual models.166
One major drawback of the QSAR model derived from the ADAPT descriptors concerns the ability to design novel analogues with desirable bioactivity. For example, the five descriptors selected by the GA in the non-linear Type III model were MDE-14, which is a topological descriptor encoding the distance (edges) between all primary and quaternary carbon atom pairs; GEOM-2, the second major geometric moment of the molecule; DIPO-0, the dipole moment; PNSA-3, a combined descriptor with atomic charge weighted partial negative surface area; and RNCS-1, the negatively charged surface area. Because these are whole-molecule descriptors, even a seemingly small substituent modification (e.g., changing methyl to hydroxy) can sometimes lead to significant changes of the entire set of descriptor values. Thus, even when an optimal set of descriptor values is known, it can still be a challenging task to engineer a molecule that fulfils the necessary conditions. One brute-force solution is to enumerate a massive virtual library and deploy the QSAR model as a filtering tool. Another possibility is to perform iterative structure optimization using the predicted activity as the cost function. The latter approach is the basis of many de novo design programs. For example, the EAinventor package provides an interface between a structure optimizer, with some embedded synthetic intelligence, and an user-supplied scoring function. This is a powerful combination which creates a synergy between synthetic consideration and targeted potency (see Chapter 5).
In the conclusion of a recent review article on neural networks,85 Manallack and Livingstone wrote:
“We feel that the combination of GAs and neural networks is the future for the [QSAR] method, which may also mean that these methods are not limited to simple structure-property relationships, but can extend to database searching, pharmacokinetic prediction, toxicity prediction, etc.”
Novel applications utilizing hybrid GA-NN approaches are beginning to appear in the literature, and will be discussed in detail in Chapter 4.
Comparison to Classical QSAR Methods
Chance Correlation, Overfitting, and Overtraining
In the examples presented in the previous Section, we have discussed the utility of GA for the selection of descriptors to be used in combination with multivariate statistical methods in QSAR applications. Although variable selection is appropriate for the typical size of a data set in conventional QSAR studies (i.e., 20–200 descriptors)—particularly if the initial pool of descriptors exceeds the number of data objects—selection may still carry a great risk of chance correlation.40 The ratio of the number of descriptors to the number of objects used in model building can be a useful parameter indicating the likelihood of chance correlation. As a general guideline, it has been suggested that a ratio of greater than five suggests that GA-optimized descriptor selection may produce unreliable statistical models.80 Obviously, other factors that are related to signal-to-noise, redundancies, and collinearity in the data can also be critical. To further reduce this risk, it is also recommended that randomization tests should be performed as an integral part of standard validation procedures in any application that involves descriptor selection. In addition, it may be beneficial to implement an early stopping point during GA evolution in order to prevent overfitting of data. Based on empirical observation, the fitness of the population usually increases very rapidly during the early phase, and then the improvement slowly levels off. The reason for this behavior is that the modeling of useful information in the data is usually made quite rapidly during the initial stage. Later, the GA begins to fit the noise or idiosyncrasies of the data to the model, sometimes using additional parameters. To determine an optimal stopping point for GA optimization, Leardi suggested a criterion that is based on the difference in the statistical fit between the real and the randomized data set.80 In this scheme, the evolution cycle that corresponds to the maximum difference between the two sets of statistics is considered an optimal termination point. Related to this concept, another intriguing idea is to combine the statistics gathered in both real and randomized training, yielding a composite cost function that may be used to evaluate individual solutions during the course of GA optimization (Dr. Andrew Smellie, personal communication). It is also known that the use of GA can sometimes produce solutions containing non-essential descriptors hidden within a subset of useful descriptors.46 A useful means to eliminate these irrelevant descriptors from the GA selection is through a hybridization operator, which periodically examines the entire population and discards the non-contributing descriptors using a backward elimination procedure.80 This idea originated from the observation that forward selection in the GA-selected subset can greatly reduce the number of irrelevant inputs.182
It has been demonstrated that ANN often produces superior QSAR models compared to models derived by the more traditional approach of multiple linear regression. The key strength of the neural network is that, with the presence of hidden layers, neural networks are able to perform non-linear mapping of the molecular descriptors to the biological activities of the compounds. The quality of the fit to the training data can be particularly impressive for networks that have many adjustable weights. Under such circumstances, the neural network simply memorizes the entire data set and behaves effectively as a look-up table. Thus, it is doubtful that the network would be able to extract a relevant correlation of the input patterns and give a meaningful assessment of other unknown examples. This phenomenon is known as overfitting of data, where a neural network may reproduce the training data set almost perfectly but erroneous predictions will be realized on other unseen test objects. It is fair to point out that the purpose of QSAR is to understand the forces governing the activity of a particular class of compounds, and to assist drug design, and that a look-up table will therefore not aid medicinal chemists in the design of new drugs. What is needed is a system that is able to provide reasonable predictions for the compounds which are previously unknown. So, the use of ANN with an excessive number of network parameters should be avoided. There are two advantages of adopting networks with relatively few processing nodes: First, the efficiency of each node increases and, consequently, the time of the computer simulation is reduced significantly. Second, and probably more importantly, the network can generalize the input patterns better, and this often results in superior predictive power. However, caution is again needed to ensure that the network is not overconstrained. Since a neural network with too few free parameters may not be able to learn the relevant relationships in the data. Such an analysis will collapse during training and again no reliable predictions can be sought. Thus, it is important to find an optimal network topology to deliver a balance between these two extreme situations.
While the numbers of nodes in the input and output layers in a neural network are typically pre-determined by the nature of the data set, the users can control the number of hidden units—and subsequently the number of adjustable weights—in the network. It has been suggested that a parameter, r, can help to determine an optimal setting for the number of hidden units.86 The definition of r is the ratio of the number of data points in the training set to the number of adjustable network parameters. The number of network variables is simply the sum of the number of connections and the number of biases in the network. A three-layered back-propagation network with I input units, H hidden units and O output units will have H × (I + O) connections and H + O biases. The total number of adjustable parameters is therefore H × (I + O) + H + O. The range 1.8 < r < 2.2 has been suggested by Andrea and Kalayeh as a guideline for acceptable r values.86 It is claimed that for r << 1, the network will simply memorize the training patterns; for r>> 3, the network will have difficulty generalizing from the data. The concept of the r ratio has made a significant impact upon the design of neural network architecture in many subsequent QSAR studies.85 It is now possible to make a reasonable initial choice for the number of hidden nodes. Nevertheless, the suggested range of 1.8 < r < 2.2 is perhaps empirical, and is also expected to be case-dependent. For example, some redundancies may already exist in the training patterns, so that the effective number of data points is in fact smaller than anticipated. On a related note, there is another rough guideline that allows the user to choose the number of hidden nodes independently from the number of data points. It is the so-called geometric pyramid rule, which states that the number of nodes in each layer follows a pyramid shape, decreasing progressively from input layer to output layer in a geometric ratio.60 That is to say, a good starting estimate of the number of hidden nodes will be the geometric mean of the numbers of input and output nodes in the network.
Overtraining of a neural network is related to the problem of overfitting of data. While overfitting is often regarded as a problem with excessive neural network parameters, overtraining refers to a prolonged training process. Both can significantly influence the quality of a model. Interestingly, the profile of training error as a function of the epoch cycle is very similar to the situation with the GA-optimization previously discussed. During the initial phase of ANN training, the neural network will quickly establish some crude rules that lead to a rapid minimization of the error. In the mid-phase of the training process, the neural network will begin to learn fine structure and other peculiarities which may in part be due to simple memorization.85 Correspondingly, the rate of decrease of the training error will slow down significantly. An obvious solution to this problem is to stop training before the final convergence of rms error so that the neural network has optimal predictive capability (forced stop).183 This can be achieved through the use of a disjoint validation (or control) data set to monitor the course of neural network training process. The training is halted when the predictive performance of the control set begins to deteriorate.
Functional Dependencies
One of the major criticisms of the application of ANN in QSAR research is that neural networks lack transparency, and are often very difficult to interpret. This contrasts with a MLR equation, where the influence of each molecular descriptor on the biological activity is immediately obvious from the coefficients. To improve the interpretability of the ANN model, a technique known as “functional dependence monitoring” has been introduced (see also Chapter 5, in particular Figure 5.6 and the corresponding text passages). Usually, all but one of the input parameters was kept constant, and the one remaining input descriptor is varied between the minimum and the maximum of its known range (but sometimes extrapolation is allowed). Prediction of biological activity is made for this particular set of descriptor values, and the resulting plot provides the functional dependence of the biological descriptor. This procedure is repeated for all input descriptors. It is hoped that the identification of the functional dependence will assist medicinal chemists in the design of more useful analogues.
To demonstrate the potency of the approach, this monitoring scheme was applied to a set of dihydrofolate reductase (DHFR) inhibitors which were extensively studied by Hansch and coworkers.184 In Hansch's analysis of a set of 68 2,4-diamino-(5-substituted benzyl)pyrimidine analogues, a correlation equation was formulated (Eq. 9):
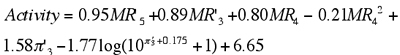
The corresponding functional dependence plots were made after successful training of a neural network on the same set of compounds using MR'5, MR'3, MR4, and p'3 as inputs (Figure 3). In this calculation, the non-variable input descriptors were pegged at a quarter of their maximum ranges. In Figure 3 the corresponding plots from the regression model are shown. It is evident that the neural network result is consistent with the regression equation. Both neural network and the regression analysis suggest that the biological activity is linearly dependent on MR'5 and MR'3, and a parabolic dependence is found for MR4. It is clear from Equation 9 that the functional dependence on π'3 is highly non-linear; remarkably, the neural network came up with a very similar plot using this monitoring scheme.
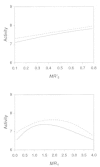
Figure 3A
Biological activity as a function of the individual physicochemical parameter for a neural network model (solid line) and multivariate regression model of Equation 9 (dashed line).
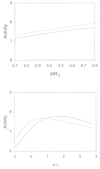
In summary, building a regression equation as complex as Equation 9 cannot be inspired by a flash of brilliance; it requires a laborious development phase. In regression analysis, the inclusion of higher-order and cross-product terms is often made on a trial-and-error basis. However, the staggering diversity of such terms often makes this task very difficult. Despite this shortcoming, it is fair to point out that with a careful design and the inclusion of appropriate non-linear transformations of descriptors, a multivariate regression model can achieve results that are comparable in quality to a well-trained ANN. To date, the most successful studies in this area are reported by Lucic and coworkers, who have demonstrated that the use of non-linear multivariate regressions can sometimes outperform ANN models for a number of benchmark QSAR and QSPR data sets.185,186 This success was achieved through a very efficient descriptor selection routine, using an enlarged descriptor set containing squared and cross-product terms. In neural networks non-linearity is implicitly handled. The descriptor monitoring scheme permits a crude analysis of the functional form and highlights those molecular descriptors that possibly play an important role for biological activity. Furthermore this technique can be used to identify whether and how non-linear terms contribute to the QSAR analysis.
The Inverse QSAR Problem
Many QSAR studies using neural networks have been published; some of them reporting resounding success. Such ANN applications often conclude with the discovery of a statistically significant correlation, or sometimes with validation using an external test set. From a practical standpoint, the more important aspect concerns molecular design, and it would therefore be interesting to use these models to predict the required structural features leading to the generation of novel bioactive analogs.
The reason that the so-called Inverse QSAR Problem appears to be tricky that the functional form connecting the input descriptors with biological activity is not a simple mathematical relationship. This contrasts with regression analysis, where a linear equation is well-defined and optimal values for certain molecular properties can be readily identified. For example, optimal values of MR4 and π3' in Equation 9 are known to be 1.85 and 0.73. One remedy for a neural network model is to monitor the functional dependence of individual descriptors and guess what range of values would be optimal. So and Karplus attempted to identify more potent benzodiazepine (BZ) ligands based on this approach.167 They applied the genetic neural network (GNN) methodology to a series of 57 benzodiazepines that had been studied by Maddalena and Johnston.99 The core structure of BZ is shown in Figure 4, together with the conventional numbering scheme for its substituents. These BZs contain six variable substituent positions, though the data were extensive for only positions R7, R1, and R2'. Each substituent was parameterized by seven physicochemical descriptors, including dipole moment (μ), lipophilicity (π), molar refractivity (MR), polar constant (F), resonance constant (R), Hammett meta constant (σm) and para (σp) constant. Using the GNN methodology, three top-ranking 6-descriptor neural network models were developed: Model I used π7, F7, MR1, σm2, π6, and MR8; Model II used MR1, π7, σm7, σm2', MR6', and σm8; and Model III used MR1, π7, σm7, σm2', σp2', and σp6'. These QSAR models yielded r2 values of 0.91 and q2 values in the range 0.87–0.86. In this study the authors also attempted to predict new compounds by a minimum perturbation approach in which one substitution position was focused on at a time. In theory, an exhaustive screening of all six physicochemical parameter spaces—or a more elaborated multivariate experimental design method—can be applied to searching for improved BZ analogues, but such procedures were not attempted. Instead, the most active compound in the training set served as the template which was then subjected to small structural modifications. To minimize the likelihood of generating compounds that had poor steric fit with the receptor, new substituents at a given position were restricted to those that were not significantly greater in bulk than the largest known substituent from a compound of at least moderate activity. The influence of each of the six parameters on the biological activity was monitored by a modified functional dependence monitoring procedure. In this implementation, all but one of the inputs were fixed at the parameter values that corresponded to the substituents of the template, and the one remaining input descriptor was varied between the minimum and the maximum of its known range. The resulting functional dependence plots are shown in Figure 5. Based on these plots, it was concluded that the most potent compounds would have a value of MR1 within the range of 1 to 3, thus limiting the substitution option to hydrogen. The relatively flat dependence of the MR8 and sm8 curves indicated that modification at this substituent position was unlikely to lead to significant change in potency and, therefore, additional synthetic effort concerning regiospecific substitution at this position might not be justified. The plot for the 2'-position suggested that increasing the σm and σp parameters would enhance the predicted receptor affinity. Thus, the template, which has a Cl atom in the 2'-position, was replaced with a substituent that had greater σm and σp values than those of Cl. Three relatively simple substituents, CN, NO2 and SO2F, were identified. Examination of Figure 5 also suggests that the activity might be improved by decreasing the descriptor values (π6', MR6', and σp6') relative to hydrogen at the 6'-position. To this end, substituents such as NH2 and F were considered. Due to the symmetry of the 2'- and 6'-position, the three new 2'-substituents (CN, NO2, and SO2F) which were considered earlier were probed for the 6'-position as well. Finally, the dependence plot for position 7 suggested that an increase in both lipophilicity and polar effect would also increase activity. However, finding suitable molecular fragments with such characteristics is non-trivial, since the two properties are naturally anti-correlated, although CH2CF3, SO2F, and OCF3 are feasible candidates. On the basis of this analysis, a number of compounds containing these favorable combinations were suggested, most of them possessing predicted activities considerably higher than that of the template compound. This design exercise shows that through careful analysis it is possible to utilize neural network QSAR models to design compounds that are predicted to be more potent.
Figure 4
Core structure of benzodiazepines and the substituent numbering scheme.

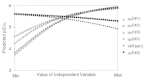

Although it is relatively easy to implement the minimum perturbation method outlined above, it should be recognized that this stepwise approach is unlikely to yield compounds that correspond to the optimal combination of input descriptor values. This is because descriptors from each position are treated independently so that the effect of inter-descriptor coupling amongst substituents is not taken into account. To overcome this limitation, ANN may, for example, serve as a fitness function in evolutionary molecular design. This approach was pioneered by Schneider and Wrede for the example of peptide de novo design, and later extended to arbitrary small molecules.187 For further details, see Chapter 5. An alternative design philosophy is to apply a combinatorial optimization technique to perform an extensive search on descriptor space and determine optimal combinations of parameter values, and then map these values to appropriate sets of functional groups. This idea was explored by Burden and coworkers, who applied a GA to search for novel DHFR inhibitors that have maximal predicted potency according to an ANN QSAR model.103 Using a data set of 256 diaminodihydrotriazines, they established a 5–8–1 neural network model that accurately predicted the pIC50 values against two different tumor cell lines. The five descriptors used were π3, π4, MR3 and MR4, the hydrophobicity substituent parameters and molar refractivities at the 3- and 4-positions, respectively, and Ss3,4, the sum of the Hammett parameter at these two positions. Upon completion of neural network training, they utilized a commercial GA package to probe the activity surface. Three different strategies were suggested to conduct these searches. The first was to constrain the search strictly within descriptor ranges defined by the training set, and the second allowed for a +/−10% extrapolation for each descriptor. Both can be regarded as conservative measures. In the third search, the parameter range explored by the GA was bound by substituent values that are chemically reasonable (i.e., −1 < σ < 1.5, −2 < π < 6, 0 < MR < 100). The results obtained for the L1210 cell lines are shown in Table 3. The optimal values determined by the GA search were π3 = 5.34, MR3 = 32.2, π4 = −1.88, MR4 = 15.3, and Σs3,4 = −0.91. The chemical groups that have parameter values closest to the optimums were identified, based on the tabulated values of substituent parameters,188 and a few novel analogues were proposed. As expected, these compounds were predicted by the model to have high potency (pIC50 = 9.35 − 8.89), which is considerably greater than the most potent compound in the training set (pIC50 = 8.37). Although the four hydrophobicity and molar refractivity parameters are within the scope of the training compounds, it should be noted that the descriptor values of Ss3,4 were outside the range of the training set, so that the increase in pIC50 value should be treated with appropriate caution.
Table 3
Optimal descriptor values and substituents identified by a GA search.
It is fair to say that reports showing real practical applications of GA and ANN in pharmaceutical design are still relatively sparse. However, several practical applications are discussed in Chapter 5. One outstanding recent example is reported by Schaper and coworkers, who have developed neural network systems that can distinguish the affinity for 5-HT1A and α1-adrenergic receptors using a data set of 32 aryl-piperazines.114 The pKi values of the training set range from 5.3 − 8.7 and 4.8 − 8.3 for 5-HT1A and α1-adrenergic receptors, respectively. However, no significant selectivity is observed in any compound in the training set. Using SAR information derived from the neural network systems, three new analogs were specifically designed to validate the predictions. These compounds were synthesized and their experimental and predicted pKi values against the two receptors are listed in Table 4. The discovery of a potent and highly selective 5-HT1A ligand is evidently a great triumph in QSAR research and computer-aided molecular design in general. There is great optimism that the use of adaptive systems in lead optimization will continue to flourish in the future.
Table 4
Experimental and predicted pKi values against 5-HT1A and α1-adrenergic receptors for the three designed compounds according to ANN QSAR models.
Outlook and Concluding Remarks
Because QSAR methods, in principle, require only ligand information, they will remain the best computational probes for such cases where there is little or no information available about the 3D structure of the therapeutic target. In our opinion, the next major advance in QSAR research will come from innovative techniques that can deal with the desperately needed increase in data handling capacity, the fact that just a decade ago, scientists routinely screened only tens of compounds each year—the typical size of a “classical” QSAR data set. Now, however, the use of new technologies, including combinatorial chemistry, robotic high-throughput screening and miniaturization give the ability to screen tens of millions of compounds a year. Such a vast volume of experimental data demands a corresponding expansion of the capacity of computational analysis. Obtaining correlations from data sets of this order of magnitude is not a trivial task, although the problem becomes more tractable when practical issues are taken into consideration. Due to the sometimes fuzzy nature of HTS data, new analysis methods must cope with a higher degree of noise. In particular, precise numerical modeling or prediction of biological activity—a characteristic of traditional QSAR approaches—may no longer be necessary or even appropriate. It is to be expected that exciting new developments in QSAR methods will continue to emerge. At the same time, one must realize that there is no guarantee of success even with the most spectacular technological advances at hand. Ultimately, the impact of any computational tool will depend critically on its implementation and integration in the drug discovery process, and on the readiness of medicinal chemists to consider “computer-generated designs” for their work. It is our conviction that medicinal and computational chemists both have responsibility to optimize the balance between the exploration and exploitation of a hypothesis. On this note, we would like to conclude this Chapter with a comment by Hugo Kubinyi:48
“There is a fundamental controversy between statisticians and research scientists, i.e., between theoreticians and practitioners, as to how statistical methods should be applied with or without all the necessary precautions to avoid chance correlations and other pitfalls. Statisticians insist that only good practice guarantees a significance of the results. However, this is most often not the primary goal of a QSAR worker. Proper series design is sometimes impossible owing to the complexity of synthesis or to a lack of availability of appropriate starting materials, problems that are often underestimated by theoreticians. A medicinal chemist is interested in a quick and automated generation of different hypotheses from the available data, to continue his research with the least effort and maximum information, even if it is fuzzy and seemingly worthless information. Predictions can be made from different regression models and the next experiments can be planned on a more rational basis to differentiate between alternative hypotheses.”
References
- 1.
- Hansch C, Fujita T. ρ-σ-π analysis. A method for the correlation of biological activity and chemical structure. J Am Chem Soc. 1964;86:1616–1626.
- 2.
- Kubinyi H. ed.QSAR: Hansch Analysis and related approaches In: Mannhold R, Krogsgaard-Larsen P, Timmerman H, eds. Methods and Principles in Medicinal Chemistry Weinheim: VCH,1993.
- 3.
- van de Waterbeemd H. ed. Chemometric methods in molecular design In: Mannhold R, Krogsgaard-Larsen P, Timmerman H, eds. Methods and Principles in Medicinal Chemistry Weinheim: VCH,1995.
- 4.
- van de Waterbeemd H. ed. Advanced computer-assisted techniques in drug discovery In: Mannhold R, Krogsgaard-Larsen P, Timmerman H, eds. Methods and Principles in Medicinal Chemistry Weinheim: VCH,1995.
- 5.
- Labute P. A widely applicable set of descriptors. J Mol Graph Model. 2000;18:464–477. [PubMed: 11143563]
- 6.
- Bajorath J. Selected concepts and investigations in compound classification, molecular descriptor analysis, and virtual screening. J Chem Inf Comput Sci. 2001;41:233–245. [PubMed: 11277704]
- 7.
- UNITY Tripos, Inc., St. Louis, MO.
- 8.
- FINGERPRINT Daylight Chemical Information Systems, Inc., Mission Viejo, CA.
- 9.
- Palm K, Kuthman K, Ungell AL. et al. Evaluation of dynamic polar molecular surface area as predictor of drug absorption: Comparison with other computational and experimental predictors. J Med Chem. 1998;41:5382–5392. [PubMed: 9876108]
- 10.
- Kelder J, Grootenhuis PD, Bayada DM. et al. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm Res. 1999;16:1514–1519. [PubMed: 10554091]
- 11.
- Santos-Filho OA, Hopfinger AJ. A search for sources of drug resistance by the 4D-QSAR analysis of a set of antimalarial dihydrofolate reductase inhibitors. J Comput-Aided Mol Design. 2001;15:1–12. [PubMed: 11217916]
- 12.
- Sadowski J, Gasteiger J. From atoms and bonds to 3-dimensional atomic coordinates—Automatic model builders. Chem Rev. 1993;7:2567–2581.
- 13.
- CONCORD University of Texas, Austin, TX.
- 14.
- Kier LB, Hall LH. An electrotopological state index for atoms in molecules. Pharm Res. 1990;7:801–807. [PubMed: 2235877]
- 15.
- Hall LH, Kier LB. Electrotopological state indices for atom types: a novel combination of electronic, topological, and valence shell information. J Chem Inf Comput Sci. 1995;35:1039–1045.
- 16.
- Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important molecules. J Med Chem. 1985;28:849–857. [PubMed: 3892003]
- 17.
- Cramer RD, Patterson DE, Bunce JD. Comparative Molecular Field Analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–5967. [PubMed: 22148765]
- 18.
- Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem. 1994;37:4130–4146. [PubMed: 7990113]
- 19.
- Wagener M, Sadowski J, Gasteiger J. Autocorrelation of molecular surface properties for modeling corticosteroid binding globulin and cytosolic Ah receptor activity by neural networks. J Am Chem Soc. 1995;117:7769–7775.
- 20.
- Pastor M, Cruciani G, McLay I. et al. GRid-INdependent descriptors (GRIND): A novel class of alignment-independent three-dimensional molecular descriptors. J Med Chem. 2000;43:3233–3243. [PubMed: 10966742]
- 21.
- Gancia E, Bravi G, Mascagni P. et al. Global 3D-QSAR methods: MS-WHIM and autocorrelation. J Comput Aided Mol Des. 2000;14:293–306. [PubMed: 10756483]
- 22.
- Bravi G, Gancia E, Mascagni P. et al. MS-WHIM, new 3D theoretical descriptors derived from molecular surface properties: a comparative 3D QSAR study in a series of steroids. J Comput Aided Mol Des. 1997;11:79–92. [PubMed: 9139115]
- 23.
- Silverman BD, Platt DE. Comparative molecular moment analysis (CoMMA): 3D-QSAR without molecular superposition. J Med Chem. 1996;39:2129–2140. [PubMed: 8667357]
- 24.
- Ferguson AM, Heritage T, Jonathon P. et al. EVA: A new theoretically based molecular descriptor for use in QSAR/QSPR analysis. J Comput Aided Mol Des. 1997;11:143–152. [PubMed: 9089432]
- 25.
- Turner DB, Willett P. The EVA spectral descriptor. Eur J Med Chem. 2000;35:367–375. [PubMed: 10858598]
- 26.
- Turner DB, Willet P, Ferguson AM. et al. Evaluation of a novel infrared range vibration-based descriptor (EVA) for QSAR studies. 1. General application. J Comput Aided Mol Des. 1997;11:409–422. [PubMed: 9334906]
- 27.
- Turner DB, Willett P, Ferguson AM. et al. Evaluation of a novel molecular vibration-based descriptor (EVA) for QSAR studies. 2. Model validation using a benchmark steroid dataset. J Comput Aided Mol Des. 1999;13:271–296. [PubMed: 10216834]
- 28.
- Turner DB, Willett P. Evaluation of the EVA descriptor for QSAR studies: 3. The use of a genetic algorithm to search for models with enhanced predictive properties (EVA_GA). J Comput Aided Mol Des. 2000;14:1–21. [PubMed: 10702922]
- 29.
- Burden FR. Molecular identification number for substructure searches. J Chem Inf Comput Sci. 1989;29:225–227.
- 30.
- Pearlman RS, Smith KM. Metric validation and the receptor-relevant subspace concept. J Chem Inf Comput Sci. 1999;39:28–35.
- 31.
- Pirard B, Pickett SD. Classification of kinase inhibitors using BCUT descriptors. J Chem Inf Comput Sci. 2000;40:1431–1440. [PubMed: 11128102]
- 32.
- Gao H. Application of bcut metrics and genetic algorithm in binary QSAR analysis. J Chem Inf Comput Sci. 2001;41:402–407. [PubMed: 11277729]
- 33.
- Hellberg S, Sjostrom M, Skagerberg B. et al. Peptide quantitative structure-activity relationships, a multivariate approach. J Med Chem. 1987;30:1126–1135. [PubMed: 3599020]
- 34.
- Sjostrom M, Eriksson L, Hellberg S. et al. Peptide QSARS: PLS modelling and design in principal properties. Prog Clin Biol Res. 1989;291:313–317. [PubMed: 2726864]
- 35.
- Eriksson L, Jonsson J, Hellberg S. et al. Peptide QSAR on substance P analogues, enkephalins and bradykinins containing L- and D-amino acids. Acta Chem Scand. 1990;44:50–55. [PubMed: 1698414]
- 36.
- Hellberg S, Eriksson L, Jonsson J. et al. Minimum analogue peptide sets (MAPS) for quantitative structure-activity relationships. Int J Pept Protein Res. 1991;37:414–424. [PubMed: 1917297]
- 37.
- Collantes ER, Dunn W J I. Amino acid side chain descriptors for quantitative-structure activity relationship studies of peptide analogues. J Med Chem. 1995;38:2705–2713. [PubMed: 7629809]
- 38.
- Patankar SJ, Jurs PC. Prediction of IC50 values for ACAT inhibitors from molecular structure. J Chem Inf Comput Sci. 2000;40:706–723. [PubMed: 10850775]
- 39.
- Whitley DC, Ford MG, Livingstone DJ. Unsupervised forward selection: A method for eliminating redundant variables. J Chem Inf Comput Sci. 2000;40:1160–1168. [PubMed: 11045809]
- 40.
- Kubinyi H. Variable selection in QSAR studies. I. An evolutionary algorithm. Quant Struct-Act Relat. 1994;13:285–294.
- 41.
- Wikel JH, Dow ER. The use of neural networks for variable selection in QSAR. Bioorg Med Chem Lett. 1993;3:645–651.
- 42.
- Tetko IV, Villa A E P, Livingstone DJ. Neural network studies. 2. Variable selection. J Chem Inf Comput Sci. 1996;36:794–803. [PubMed: 8768768]
- 43.
- Kirkpatrick S, Gelatt C D J, Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–680. [PubMed: 17813860]
- 44.
- Holland JH. Adaption in Natural and Artificial Systems. Ann Arbor: The University of Michigan Press. 1975
- 45.
- Glover F. Future paths for integer programming and links to artificial intelligence. Comp Oper Res. 1986;5:533–549.
- 46.
- Waller CL, Bradley MP. Development and validation of a novel variable selection technique with application to multidimensional quantitative structure-activity relationship studies. J Chem Inf Comput Sci. 1999;39:345–355.
- 47.
- Clementi S, Wold S. How to choose the proper statistical method In: van de Waterbeemd H, ed. Chemometric Methods in Molecular Design New York: VCH Publishers, Inc.,1995319–338.
- 48.
- Kubinyi H. Evolutionary variable selection in regression and PLS analyses. J Chemometrics. 1996;10:119–133.
- 49.
- Wehrens R, Buydens L M C. Chemometrics In: Clark DE, ed. Evolutionary Algorithms in Molecular Design Weinheim: Wiley-VCH,200099–114.
- 50.
- Topliss JG, Edwards RP. Chance factors in studies of quantitative structure-activity relationships. J Med Chem. 1979;22:1238–1244. [PubMed: 513071]
- 51.
- Wold S, Eriksson L. Statistical validation of QSAR results In: van de Waterbeemd H, ed. Chemometric Methods in Molecular Design New York: VCH Publishers, Inc.,1995309–318.
- 52.
- Saarinen J, Kohonen T. Self-organized formation of colour maps in a model cortex. Perception. 1985;14:711–719. [PubMed: 3837872]
- 53.
- Livingstone DJ, Hesketh G, Clayworth D. Novel method for the display of multivariate data using neural networks. J Mol Graph. 1991;9:115–118. [PubMed: 1768641]
- 54.
- Koza JR. Genetic Programming: On the Programming of Computers by Means of Natural Selection. Cambridge: MIT Press. 1992
- 55.
- Gilbert RJ, Goodacre R, Woodward AM. et al. Genetic programming: A novel method for the quantitative analysis of pyrolysis mass spectral data. Anal Chem. 1997;69:4381–4389. [PubMed: 21639171]
- 56.
- Tuson A, Clark DE. New techniques and future directions In: Clark DE, ed. Evolutionary Algorithms in Molecular Design Weinheim: Wiley-VCH, 2000.
- 57.
- Kyngäs J, Valjakka J. Evolutionary neural networks in quantitative structure-activity relationships of dihydrofolate reductase inhibitors. Quant Struct-Act Relat. 1996;15:296–301.
- 58.
- Hoffman B, Cho SJ, Zheng W. et al. Quantitative structure-activity relationship modeling of dopamine D1 antagonists using comparative molecular field analysis, genetic algorithms-partial least-squares, and k nearest neighbor methods. J Med Chem. 1999;42:3217–3226. [PubMed: 10464009]
- 59.
- Zheng W, Tropsha A. Novel variable selection quantitative structure-activity relationship approach based on the k-nearest neighbor principle. J Chem Inf Comput Sci. 2000;40:185–194. [PubMed: 10661566]
- 60.
- Kövesdi I, Dominguez-Rodriguez MF, Ôrfi L. et al. Application of neural networks in structure-activity relationships. Med Res Rev. 1999;19:249–269. [PubMed: 10232652]
- 61.
- Shao J. Linear-model selection by cross-validation. J Am Stat Assoc. 1993;88:486–494.
- 62.
- http://www.faqs.org/faqs/ai-faq/neural-nets/part3/section-12.html.
- 63.
- Wehrens R, Putter H, Buydens L M C. The bootstrap: A tutorial. Chemo Intell Lab Sys. 2000;54:35–52.
- 64.
- Davison AC, Hinkley DV. Bootstrap Methods and Their Applications Cambridge: Cambridge University Press, 1997 .
- 65.
- Castellano G, Fanelli AM. Variable selection using neural-network models. Neurocomputing. 2000;31:1–13.
- 66.
- Selwood DL, Livingstone DJ, Comley JC. et al. Structure-activity relationships of antifilarial antimycin analogues: A multivariate pattern recognition study. J Med Chem. 1990;33:136–142. [PubMed: 2296013]
- 67.
- Kovalishyn VV, Tetko IV, Luik AI. et al. Neural network studies. 3. Variable selection in the cascade-correlation learning architecture. J Chem Inf Comput Sci. 1998;38:651–659.
- 68.
- Tetko IV, Villa AP, Aksenova TI. et al. Application of a pruning algorithm to optimize artificial neural networks for pharmaceutical fingerprinting. J Chem Inf Comput Sci. 1998;38:660–668. [PubMed: 9691475]
- 69.
- LeChun Y, Denker JS, Solla SA. Optimal brain damage In: Touretzky DS, ed. Advances in Neural Processing Systems 2 (NIPS*2) San Mateo:Morgan-Kaufmann Publishers,1990598–605.
- 70.
- Hassibi B, Stork D. Second order derivatives for network pruning: Optimal brain surgeon In: Hanson S, Cowan J, Giles C, eds. Advances in Neural Processing Systems 5 (NIPS*5) San Mateo:Morgan-Kaufmann Publishers,1993164–171.
- 71.
- Leardi R, Boggia R, Terrile M. Genetic algorithms for a strategy for feature selection. J Chemometrics. 1992;6:267–281.
- 72.
- Cantu-Paz E. A summary of research on parallel genetic algorithm Illinois Genetic Algorithm Laboratory, University of Illinois at Urbana-Champaign, 1997 .
- 73.
- Luke BT. Evolutionary programming applied to the development of quantitative structure-activity relationships and quantitative structure-property relationships. J Chem Inf Comput Sci. 1994;34:1279–1287.
- 74.
- Hasegawa K, Miyashita Y, Funatsu K. GA strategy for variable selection in QSAR studies: GA based PLS analysis of calcium channel antagonists. J Chem Inf Comput Sci. 1997;37:306–310. [PubMed: 9157101]
- 75.
- Hasegawa K, Funatsu K. GA strategy for variable selection in QSAR studies: GAPLS and D-optimal designs for predictive QSAR model. J Mol Struct (Theochem). 1998;425:255–262.
- 76.
- Hasegawa K, Kimura T, Funatsu K. GA strategy for variable selection in QSAR studies: Enhancement of comparative molecular binding energy analysis by GA-based PLS method. Quant Struct-Act Relat. 1999;18:262–272.
- 77.
- Hasegawa K, Kimura T, Funatsu K. GA strategy for variable selection in QSAR studies: Application of GA-based region selection to a 3D-QSAR study of acetylcholinesterase inhibitors. J Chem Inf Comput Sci. 1999;39:112–120. [PubMed: 10094610]
- 78.
- Kimura T, Hasegawa K, Funatsu K. GA strategy for variable selection in QSAR studies: GA-based region selection for CoMFA modeling. J Chem Inf Comput Sci. 1998;38:276–282.
- 79.
- Gaudio AC, Korolkovas A, Takahata Y. Quantitative structure-activity relationships for 1,4-dihydropyridine calcium channel antagonists (nifedipine analogues): A quantum chemical/classical approach. J Pharm Sci. 1994;83:1110–1115. [PubMed: 7983594]
- 80.
- Leardi R, González AL. Genetic algorithms applied to feature selection in PLS regression: How and when to use them. Chemo Intell Lab Sys. 1998;41:195–207.
- 81.
- Ajay A unified framework for using neural networks to build QSARs. J Med Chem. 1993;36:3565–3571. [PubMed: 8246224]
- 82.
- Duprat AF, Huynh T, Dreyfus G. Toward a principled methodology for neural network design and performance evaluation in QSAR. Application to the prediction of logP. J Chem Inf Comput Sci. 1998;38:586–594. [PubMed: 9691473]
- 83.
- Aoyama T, Suzuki Y, Ichikawa H. Neural networks applied to structure-activity relationships. J Med Chem. 1990;33:905–908. [PubMed: 2308139]
- 84.
- Aoyama T, Suzuki Y, Ichikawa H. Neural networks applied to quantitative structure-activity relationship analysis. J Med Chem. 1990;33:2583–2590. [PubMed: 2202830]
- 85.
- Manallack DT, Livingstone DJ. Neural networks in drug discovery: Have they lived up to their promise? Eur J Med Chem. 1999;34:195–208.
- 86.
- Andrea TA, Kalayeh H. Applications of neural networks in quantitative structure-activity relationships of dihydrofolate reductase inhibitors. J Med Chem. 1991;34:2824–2836. [PubMed: 1895302]
- 87.
- Silipo C, Hansch C. Correlation analysis. Its application to the structure-activity relationship of triazines inhibiting dihydrofolate reductase. J Am Chem Soc. 1975;97:6849–6861. [PubMed: 1237510]
- 88.
- Winkler DA, Burden FR, Watkins A J R. Atomistic topological indices applied to benzodiazepines using various regression methods. Quant Struct-Act Relat. 1998;17:14–19.
- 89.
- The bibliographic search was made on SciFinder (version 2000, the American Chemical Society) using the keyword neural network and QSAR.
- 90.
- Aoyama T, Ichikawa H. Neural networks applied to pharmaceutical problems. 4. Basic operating characteristics of neural networks when applied to structure activity studies. Chem Pharm Bull Tokyo. 1991;39:372–378.
- 91.
- So S -S, Richards WG. Application of neural networks: Quantitative structure-activity relationships of the derivatives of 2,4-diamino-5-(substituted-benzyl)pyrimidines as DHFR inhibitors. J Med Chem. 1992;35:3201–3207. [PubMed: 1507206]
- 92.
- Salt DW, Yildiz N, Livingston DJ. et al. The use of artificial neural networks in QSAR. Pestic Sci. 1992;36:161–170.
- 93.
- Tetko IV, Luik AI, Poda GI. Applications of neural networks in structure-activity relationships of a small number of molecules. J Med Chem. 1993;36:811–814. [PubMed: 8464034]
- 94.
- Wiese M, Schaper KJ. Application of neural networks in the QSAR analysis of percent effect biological data: comparison with adaptive least squares and nonlinear regression analysis. SAR QSAR Environ Res. 1993;1:137–152. [PubMed: 8790630]
- 95.
- Song XH, Yu RQ. Artificial neural networks applied to the quantitative structure-activity relationship study of dihydropteridine reductase inhibitors. Chemo Intell Lab Sys. 1993;19:101–109.
- 96.
- Ghoshal N, Mukhopadhyay SN, Ghoshal TK. et al. Quantitative structure-activity relationship studies of aromatic and heteroaromatic nitro-compounds using neural network. Bioorg Med Chem Lett. 1993;3:329–332.
- 97.
- Manallack DT, Livingstone DJ. Limitations of functional-link nets as applied to QSAR data analysis. Quant Struct-Act Relat. 1994;13:18–21.
- 98.
- Hirst JD, King RD, Sternberg MJ. Quantitative structure-activity relationships by neural networks and inductive logic programming. II. The inhibition of dihydrofolate reductase by triazines. J Comput-Aided Mol Des. 1994;8:421–432. [PubMed: 7815093]
- 99.
- Maddalena DJ, Johnston G A R. Prediction of receptor properties and binding affinity of ligands to benzodiazepine/GABAA receptors using artificial neural networks. J Med Chem. 1995;38:715–724. [PubMed: 7861419]
- 100.
- Hasegawa K, Deushi T, Yaegashi O. et al. Artificial neural network studies in quantitative structure-activity relationships of antifungal azoxy compounds. Eur J Med Chem. 1995;30:569–574.
- 101.
- van Helden SP, Hamersma H, van Geerestein VJ. Prediction of progesterone receptor binding of steroids using a combination of genetic algorithms and neural networks In: Devillers J, ed Genetic Algorithm in Molecular Modeling. London: Academic Press 1996 .
- 102.
- Burden FR. Using artificial neural networks to predict biological activity from simple molecular structural considerations. Quant Struct-Act Relat. 1996;15:7–11.
- 103.
- Burden FR, Rosewarne BS, Winkler DA. Predicting maximum bioactivity by effective inversion of neural network using genetic algorithm. Chemo Intell Lab Sys. 1997;38:127–137.
- 104.
- Novic M, Nikolovska-Coleska Z, Solmajer T. Quantitative structure-activity relationship of flavonoid p56lck protein tyrosine kinase inhibitors. A neural network approach. J Chem Inf Comput Sci. 1997;37:990–998.
- 105.
- Hosseini M, Maddalena DJ, Spence I. Using artificial neural networks to classify the activity of capsaicin and its analogues. J Chem Inf Comput Sci. 1997;37:1129–1137. [PubMed: 9392859]
- 106.
- Tang Y, Chen K -X, Jiang H -L. et al. QSAR/QSTR of fluoroquinolones: An example of simultaneous analysis of multiple biological activities using neural network method. Eur J Med Chem. 1998;33:647–658.
- 107.
- López-Rodríguez ML, Morcillo J, Fernández E. et al. Design and synthesis of 2-[4-[4-(m-(ethylsulfonamido)-phenyl) piperazin-1-yl]butyl]-1,3-dioxoperhydropyrrolo[1,2-c]imidazole (EF-7412) using neural networks. A selective derivative with mixed 5-HT1A/D2 antagonist properties. Bioorg Med Chem Lett. 1999;9:1679–1682. [PubMed: 10397500]
- 108.
- Schaper K -J. Free-Wilson-type analysis of non-additive substituent effects on THPB dopamine receptor affinity using artificial neural networks. Quant Struct-Act Relat. 1999;18:354–360.
- 109.
- Chen Y, Chen D, He C. et al. Quantitative structure-activity relationships study of herbicides using neural networks and different statistical methods. Chemo Intell Lab Sys. 1999;45:257–276.
- 110.
- So S -S, Karplus M. A comparative study of ligand-receptor complex binding affinity prediction methods based on glycogen phosphorylase inhibitors. J Comput Aided Mol Des. 1999;13:243–258. [PubMed: 10216832]
- 111.
- Jalali-Heravi M, Parastar F. Use of artificial neural networks in a QSAR study of anti-HIV activity for a large group of HEPT derivatives. J Chem Inf Comput Sci. 2000;40:147–154. [PubMed: 10661561]
- 112.
- So S -S, van Helden SP, van Geerestein VJ. et al. Quantitative structure-activity relationship studies of progesterone receptor binding steroids. J Chem Inf Comput Sci. 2000;40:762–772. [PubMed: 10850780]
- 113.
- Moon T, Chi MH, Kim D -H. et al. Quantitative structure-activity relationships (QSAR) study of flavonoid derivatives for inhibition of cytochrome P450 1A2. Quant Struct-Act Relat. 2000;19:257–263.
- 114.
- López-Rodríguez ML, Morcillo J, Fernández E. et al. Synthesis and structure-activity relationships of a new model of arylpiperazines. 6. Study of the 5-HT1A/a1-adrenergic receptor affinity by classical Hansch analysis, artificial neural networks, and computational simulation of ligand recognition. J Med Chem. 2001;44:198–207. [PubMed: 11170629]
- 115.
- Huuskonen JJ, Livingstone DJ, Tetko IV. Neural network modeling for estimation of partition coefficient based on atom-type electrotopological state indices. J Chem Inf Comput Sci. 2000;40:947–955. [PubMed: 10955523]
- 116.
- Jaén-Oltra J, Salabert-Salvador MT, García-March FJ. et al. Artificial neural network applied to prediction of fluoroquinolone antibacterial activity by topological methods. J Med Chem. 2000;43:1143–1148. [PubMed: 10737746]
- 117.
- Good AC, So S -S, Richards WG. Structure-activity relationships from molecular similarity matrices. J Med Chem. 1993;36:433–438. [PubMed: 8474098]
- 118.
- Good AC, Peterson SJ, Richards WG. QSAR's from similarity matrices. Technique validation and application in the comparison of different similarity evaluation methods. J Med Chem. 1993;36:2929–2937. [PubMed: 8411009]
- 119.
- So S -S, Karplus M. Three-dimensional quantitative structure-activity relationships from molecular similarity matrices and genetic neural networks: I.Method and Validations. J Med Chem. 1997;40:4347–4359. [PubMed: 9435904]
- 120.
- So S -S, Karplus M. Three-dimensional quantitative structure-activity relationships from molecular similarity matrices and genetic neural networks: II. Applications. J Med Chem. 1997;40:4360–4371. [PubMed: 9435905]
- 121.
- Borowski T, Król M, Broclawik E. et al. Application of similarity matrices and genetic neural networks in quantitative structure-activity relationships of 2- or 4-(4-methylpiperazino)pyrimdines: 5–HT2A receptor antagonists. J Med Chem. 2000;43:1901–1909. [PubMed: 10821703]
- 122.
- Breindl A, Beck B, Clark T. Prediction of the n-octanol/water partitiion coefficient, logP, using a combination of semiempirical MO-calculations and a neural network. J Mol Model. 1997;3:142–155.
- 123.
- Vracko M. A study of structure-carcinogenic potency relationship with artificial neural networks. The using of descriptors related to geometrical and electronic structures. J Chem Inf Comput Sci. 1997;37:1037–1043.
- 124.
- Braunheim BB, Miles RW, Schramm VL. et al. Prediction of inhibitor binding free energies by quantum neural network. Nucleoside analogues binding to trypanosomal nucleoside hydrolase. Biochemistry. 1999;38:16076–16083. [PubMed: 10587430]
- 125.
- Vendrame R, Braga RS, Takahata Y. et al. Structure-activity relationship studies of carcinogenic activity of polycyclic aromatic hydrocarbons using calculated molecular descriptors with principal component analysis and neural network. J Chem Inf Comput Sci. 1999;39:1094–1104. [PubMed: 10614026]
- 126.
- Polanski J, Gasteiger J, Wagener M. et al. The comparison of molecular surfaces by neural networks and its applications to quantitative structure activity studies. Quant Struct-Act Relat. 1998;17:27–36.
- 127.
- Devillers J, Flatin J. A general QSAR model for predicting the acute toxicity of pesticides to Oncorhynchus mykiss. SAR QSAR Environ Res. 2000;11:25–43. [PubMed: 10768404]
- 128.
- Devillers J. A general QSAR model for predicting the acute toxicity of pesticides to Lepomis macrochirus. SAR QSAR Environ Res. 2001;11:397–417. [PubMed: 11328712]
- 129.
- Ritschel WA, Akileswaran R, Hussain AS. Application of neural networks for the prediction of human pharmacokinetic parameters. Methods Find Exp Clin Pharmacol. 1995;17:629–643. [PubMed: 8786678]
- 130.
- Opara J, Primozic S, Cvelbar P. Prediction of pharmacokinetic parameters and the assessment of their variability in bioequivalence studies by artificial neural networks. Pharm Res. 1999;16:944–948. [PubMed: 10397618]
- 131.
- Schneider G, Coassolo P, Lavé T. Combining in vitro and in vivo pharmacokinetic data for prediction of hepatic drug clearance in humans by artificial neural networks and multivariate statistical techniques. J Med Chem. 1999;42:5072–5076. [PubMed: 10602692]
- 132.
- Quiñones C, Caceres J, Stud M. et al. Prediction of drug half-life values of antihistamines based on the CODES/neural network model. Quant Struct-Act Relat. 2000;19:448–454.
- 133.
- Wessel MD, Jurs PC, Tolan JW. et al. Prediction of human intestinal absorption of drug compounds from molecular structure. J Chem Inf Comput Sci. 1998;38:726–735. [PubMed: 9691477]
- 134.
- Benigni R, Giuliani A. Quantitative structure-activity relationship (QSAR) studies in genetic toxicology: mathematical models and the “biological activity” term of the relationship. Mutat Res. 1994;306:181–186. [PubMed: 7512217]
- 135.
- Xu L, Ball JW, Dixon SL. et al. Quantitative structure-activity relationships for toxicity of phenols using regression analysis and computational neural networks. Environmental Toxicol Chem. 1994;13:841–851.
- 136.
- Hatrik S, Zahradnik E. Neural network approach to the prediction of the toxicity of benzothiazolium salts frommolecular structure. J Chem Inf Comput Sci. 1996;36:992–995. [PubMed: 8831139]
- 137.
- Zakarya D, Larfaoui EM, Boulaamail A. et al. Analysis of structure-toxicity relationships for a series of amide herbicides using statistical methods and neural network. SAR QSAR Environ Res. 1996;5:269–279. [PubMed: 9104783]
- 138.
- Eldred DV, Jurs PC. Prediction of acute mammalian toxicity of organophosphorus pesticide compounds from molecular structure. SAR QSAR Environ Res. 1999;10:75–99. [PubMed: 10491847]
- 139.
- Eldred DV, Weikel CL, Jurs PC. et al. Prediction of fathead minnow acute toxicity of organic compounds from molecular structure. Chem Res Toxicol. 1999;12:670–678. [PubMed: 10409408]
- 140.
- Niculescu SP, Kaiser K L E, Schultz TW. Modeling the toxicity of chemicals to Tetrahymena pyriformis using molecular fragment descriptors and probabilistic neural networks. Arch Environ Contam Toxicol. 2000;39:289–298. [PubMed: 10948278]
- 141.
- Basak SC, Grunwald GD, Gute BD. et al. Use of statistical and neural net approaches in predicting toxicity of chemicals. J Chem Inf Comput Sci. 2000;40:885–890. [PubMed: 10955514]
- 142.
- Gini G, Lorenzini M, Benfenati E. et al. Predictive carcinogenicity: A model for aromatic compounds, with nitrogen-containing substituents, based on molecular descriptors using an artificial neural network. J Chem Inf Comput Sci. 1999;39:1076–1080. [PubMed: 10614025]
- 143.
- Bahler D, Stone B, Wellington C. et al. Symbolic, neural, and bayesian machine learning models for predicting carcinogenicity of chemical compounds. J Chem Inf Comput Sci. 2000;40:906–914. [PubMed: 10955517]
- 144.
- Weinstein JN, Kohn KW, Grever MR. et al. Neural computing in cancer drug development. Predicting mechanism of action. Science. 1992;258:447–451. [PubMed: 1411538]
- 145.
- Ajay , Walters P, Murcko MA. Can we learn to distinguish between “drug-like” and “nondrug-like” molecules? J Med Chem. 1998;41:3314–3324. [PubMed: 9719583]
- 146.
- Sadowski J, Kubinyi H. A scoring scheme for discriminating between drugs and nondrugs. J Med Chem. 1998;41:3325–3329. [PubMed: 9719584]
- 147.
- Frimurer TM, Bywater R, Nærum L. et al. Improving the odds in discriminating “drug-like” from “non drug-like” compounds. J Chem Inf Comput Sci. 2000;40:1315–1324. [PubMed: 11128089]
- 148.
- Kirew DB, Chretien JR, Bernard P. et al. Application of Kohonen neural networks in classification of biological active compounds. SAR QSAR Environ Res. 1998;8:93–107. [PubMed: 9517011]
- 149.
- Burden FR, Winkler DA. Robust QSAR models using bayesian regularized neural networks. J Med Chem. 1999;42:3183–3187. [PubMed: 10447964]
- 150.
- Burden FR, Ford MG, Whitley DC. et al. Use of automatic relevance determination in QSAR studies using bayesian neural networks. J Chem Inf Comput Sci. 2000;40:1423–1430. [PubMed: 11128101]
- 151.
- Burden FR, Winkler DA. A quantitative structure-activity relationships model for the acute toxicity of substituted benzenes to Tetrahymena pyriformis using Bayesian regularized neural networks. Chem Res Toxicol. 2000;13:436–440. [PubMed: 10858316]
- 152.
- Micheli A, Sperduti A, Starita A. et al. Analysis of the internal representations developed by neural networks for structures to quantitative structure-activity relationship studies of benzodiazepines. J Chem Inf Comput Sci. 2001;41:202–218. [PubMed: 11206375]
- 153.
- Tetko IV, Aksenova TI, Volkovich VV. et al. Polynomial neural network for linear and non-linear model selection in quantitative structure-activity relationship studies on the internet. SAR QSAR Environ Res. 2000;11:263–280. [PubMed: 10969875]
- 154.
- Devillers J. Designing molecules with specific properties from intercommunicating hybrid systems. J Chem Inf Comput Sci. 1996;36:1061–1066. [PubMed: 8941992]
- 155.
- Maggiora GM, Elrod DW. Computational neural networks as model-free mapping devices. J Chem Inf Comput Sci. 1992;32:732–741.
- 156.
- Ichikawa H, Aoyama T. How to see characteristics of structural parameters in QSAR analysis: Descriptor mapping using neural networks. SAR QSAR Environ Res. 1993;1:115–130. [PubMed: 8790628]
- 157.
- Gasteiger J, Zupan J. Neural networks in chemistry. Angew Chem Int Ed Engl. 1993;105:503–527.
- 158.
- Livingstone DJ, Manallack DT. Statistics using neural networks: Chance effects. J Med Chem. 1993;36:1295–1297. [PubMed: 8487267]
- 159.
- Manallack DT, Ellis DD, Livingstone DJ. Analysis of linear and nonlinear QSAR data using neural networks. J Med Chem. 1994;37:3758–3767. [PubMed: 7966135]
- 160.
- Ajay On better generalization by combining 2 or more models—A quantitative structure-activity relationship example using neural networks. Chemo Intell Lab Sys. 1994;24:19–30.
- 161.
- Schuurmann G, Muller E. Backpropagation neural networks—Recognition vs prediction capability. Environ Toxicol Chem. 1994;13:743–747.
- 162.
- Devillers J. ed.Neural Networks in QSAR and Drug Design London: Academic Press, 1996. [PMC free article: PMC172907]
- 163.
- Kocjancic R, Zupan J. Application of a feed-forward artificial neural network as a mapping device. J Chem Inf Comput Sci. 1997;37:985–989.
- 164.
- Agatonovic-Kustrin S, Beresford R. Basic concepts of artificial neural network (ANN) modeling and its applications to pharmaceutical research. J Pharm Biomed Anal. 2000;22:717–727. [PubMed: 10815714]
- 165.
- Zupan J, Gasteiger J. Neural Networks for Chemists: An Introduction New York: VCH Publishers, 1993. [PMC free article: PMC158788]
- 166.
- So S -S, Karplus M. Evolutionary optimization in quantitative structure-activity relationship: An application of genetic neural network. J Med Chem. 1996;39:1521–1530. [PubMed: 8691483]
- 167.
- So S -S, Karplus M. Genetic neural networks for quantitative structure-activity relationships: improvements and application of benzodiazepine affinity for benzodiazepine/GABAA receptors. J Med Chem. 1996;39:5246–5256. [PubMed: 8978853]
- 168.
- Møller MF. A scaled conjugate gradient algorithm for fast supervised learning. Neural Networks. 1993;6:525–533.
- 169.
- Richards WG. Molecular similarity and dissimilarity In: Pullman A, Jortner J, Pullman B, eds. Modelling of Biomolecular Structures and Mechanisms Dordrecht: Kluwer Academic Publishers,1995365–369.
- 170.
- Kubinyi H.ed. 3D QSAR in Drug Design: Theory, Methods and Applications Leiden: ESCOM Science Publishers B.V.,1993. [PMC free article: PMC158788]
- 171.
- Norinder U. Experimental design based 3-D QSAR analysis of steroid-protein interactions: application to human CBG complexes. J Comput-Aided Mol Des. 1990;4:381–389. [PubMed: 2092083]
- 172.
- de Gregorio C, Kier LB, Hall LH. QSAR modeling with the electrotopological state indices: Corticosteroids. J Comput-Aided Mol Des. 1998;12:557–561. [PubMed: 9879503]
- 173.
- Robert D, Amat L, Carbo-Dorca R. Three-dimensional quantitative structure-activity relationships from tuned molecular quantum similarity measures: Prediction of the corticosteroid-binding globulin binding affinity for a steroid family. J Chem Inf Comput Sci. 1999;39:333–344. [PubMed: 10192946]
- 174.
- Polanski J. The non-grid technique for modeling 3D QSAR using self-organizing neural network (SOM) and PLS analysis: Application to steroids and colchicinoids. SAR QSAR Environ Res. 2000;11:245–261. [PubMed: 10969874]
- 175.
- Polanski J, Walczak B. The comparative molecular surface analysis (CoMSA): A novel tool for molecular design. Comput Chem. 2000;24:615–625. [PubMed: 10890372]
- 176.
- Liu SS, Yin CS, Li ZI. et al. QSAR study of steroid benchmark and dipeptides based on MEDV-13. J Chem Inf Comput Sci. 2001;41:321–329. [PubMed: 11277718]
- 177.
- Carbó R, Leyda L, Arnau M. An electron density measure of the similarity between two compounds. Int J Quantum Chem. 1980;17:1185–1189.
- 178.
- Hodgkin EE, Richards WG. Molecular similarity based on electrostatic potential and electric field. Int J Quantum Chem, Quantum Biol Symp. 1987;14:105–110.
- 179.
- Good AC. The calculation of molecular similarity: Alternative formulas, data manipulation and graphical display. J Mol Graph. 1992;10:144–151. [PubMed: 1361360]
- 180.
- Kauffman GW, Jurs PC. Prediction of inhibition of the sodium ion-proton antiporter by bezoylguanidine derivatives from molecular structure. J Chem Inf Comput Sci. 2000;40:753–761. [PubMed: 10850779]
- 181.
- Rogers DR, Hopfinger AJ. Application of genetic function approximation to quantitative structure-activity relationships and quantitative structure-property relationships. J Chem Inf Comput Sci. 1994;34:854–866.
- 182.
- Jouan-Rimbaud D, Massart DL, de Noord OE. Random correlation in variable selection for multivariate calibration with a genetic algorithm. Chemo Intell Lab Sys. 1996;35:213–220.
- 183.
- Tetko IV, Livingstone DJ, Luik AI. Neural network studies. 1. Comparison of overfitting and overtraining. J Chem Inf Comput Sci. 1995;35:826–833.
- 184.
- Selassie CD, Li RL, Poe M. et al. On the optimization of hydrophobic and hydrophilic substituent interactions of 2,4-diamino-5-(substituted-benzyl)pyrimidines with dihydrofolate reductase. J Med Chem. 1991;34:46–54. [PubMed: 1899453]
- 185.
- Lucic B, Trinajstic N. Multivariate regression outperforms several robust architectures of neural networks in QSAR modeling. J Chem Inf Comput Sci. 1999;39:121–132.
- 186.
- Lucic B, Amic D, Trinajstic N. Nonlinear multivariate regression outperforms several concisely designed neural networks on three QSPR data sets. J Chem Inf Comput Sci. 2000;40:403–413. [PubMed: 10761147]
- 187.
- Schneider G, Wrede P. Optimizing amino acid sequences by simulated molecular evolution In: Jesshope C, Jossifov V, Wilhelmi W, eds. Parallel Computing and Cellular Automata Berlin: Akademie-Verlag,1993335–346.
- 188.
- Hansch C, Leo A. A substituent constants for correlation analysis in chemistry and biology New York: John Wiley & Sons, Inc., 1979. [PMC free article: PMC281483]
- PubMedLinks to PubMed
- Modeling Structure-Activity Relationships - Madame Curie Bioscience DatabaseModeling Structure-Activity Relationships - Madame Curie Bioscience Database
- Regional Differences and Variability in Left Ventricular Wall Motion - Madame Cu...Regional Differences and Variability in Left Ventricular Wall Motion - Madame Curie Bioscience Database
- UI-H-BW1-anf-g-03-0-UI.s1 NCI_CGAP_Sub7 Homo sapiens cDNA clone IMAGE:3082180 3'...UI-H-BW1-anf-g-03-0-UI.s1 NCI_CGAP_Sub7 Homo sapiens cDNA clone IMAGE:3082180 3', mRNA sequencegi|11599688|gnl|dbEST|7041874|gb|BF 9.1|Nucleotide
Your browsing activity is empty.
Activity recording is turned off.
See more...