

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Introduction to Cell Death
Death is the inevitable fate for all living things including individual cells. However, untimely death results in dysfunction and disease. Understanding the normal regulation of cell death and cell survival is critical for the development of successful therapies for preventing and treating diseases.
Cell death can occur by two different, well-characterized mechanisms, necrosis (accidental cell death)1 and apoptosis (programmed cell death).2 Necrosis is a rapid cell death process in response to extreme insults. Loss of cellular homeostasis upon injury leads to influx of extracellular ions and water that causes swelling of both the cell and intracellular organelles. Subsequent plasma membrane breakdown and random fracture of chromosomal DNA during necrotic cell death engenders inflammation in the adjacent tissue in vivo. (See chapter XX, “PARP-1 and the shape of cell death”). Apoptosis or programmed cell death (PCD) is a structurally distinctive, physiological cell death responsible for cell loss in embryogenesis, normal cell turnover, tissue atrophy induced by endocrine or other stimuli, negative selection in the immune system, and development of the nervous system. Apoptosis may also occur in pathophysiological conditions. Apoptotic features include cytoplasmic shrinkage, accompanied by phosphatidylserine exposure, blebbing from the cell surface, and the formation of apoptotic bodies. Although cytoplasmic compartments display intact structure, the nucleus suffers chromatin condensation, initiated at sublamellar foci and frequently broadening to generate densely heterochromatic regions. Changes in several cell surface molecules substantiate that, in tissues, apoptotic cells are immediately recognized and phagocytosed by their neighbors within a short time.3 Necrosis and apoptosis are distinct mechanisms of cell death with defined morphological and biochemical features. However, there are specialized death programs observed in many chronic neurodegenerative diseases, heart failure, exposure to cytotoxic compounds, hypoxia or viral infection that express features common to both necrosis and apoptosis. In recent years it has become clear that cell death programs are more complex and diverse than can be explained simply by the two pathways, necrosis and apoptosis. There are death programs that are unique to cell type and lethal stimulus. The current challenge is to define the biochemical events that occur in diverse cell death programs.
Release of apoptosis-inducing factor (AIF) mediated by poly(ADP-ribose) polymerase-1 (PARP-1), also called poly(ADP-ribose) synthetase (PARS) or poly(ADP-ribose) transferase (pADPRT), is an important novel cell death program. Pharmacologic inhibition of PARP-1 or genetic knockout of PARP-1 results in profound protection against excitotoxic cell death and ischemic injury in the nervous system.4-6 How PARP-1 activation kills cells has been a mystery. Identification of AIF as the cell death mediator following PARP-1 activation was surprising. In this chapter, we will focus on PARP-1 over activation and mitochondrial AIF release in cell death signaling.
PARP-1 and Nuclear Death Signals
Oxidative DNA Damage
In contrast to the highly structured signal cascades identified for the mitochondria-mediated cell death, our knowledge of nuclear events that participate in the cell death process is limited. The morphologic observation of nuclear condensation and fractionation into apoptotic bodies is the fundamental criterion for defining apoptosis. Changes in nuclear structure are often considered as final, irreversible events in cell death. PARP-1-mediated cell death initiates from the damage of chromosomal DNA and activation of a signal cascade that terminates with large scale DNA cleavage and pyknotic nuclei.
DNA damage is a common cell death-inducing signal but the death program that is activated varies by cell type. DNases and reactive oxygen species (ROS) can damage DNA. The mitochondrion is a significant source of ROS that are associated with the pathogenesis of many diseases and with aging. The oxygen species that are typically linked to oxidative stress include superoxide anion, hydroxyl radical (OH), hydrogen peroxide (H2O2), nitric oxide (NO) and peroxynitrite (ONOO-). Although generation of these species from molecular oxygen is a normal feature of mammalian respiration, ROS can cause severe damage to cellular proteins, lipids and DNA when produced at high levels by disease processes such as ischemia, atherosclerosis, diabetes, pulmonary fibrosis, neurodegenerative disorders and arthritis.7 Under conditions of elevated calcium and energy failure, xanthine dehydrogenase is converted to xanthine oxidase, resulting in additional superoxide anion formation.8 The biochemical reactions involving NO are still being characterized, however, NO is likely to produce peroxynitrite in the presence of superoxide. In vitro the rate of this reaction is three times faster than the reaction rate of the enzyme, superoxide dismutase (SOD) catalyzing the dismutation of superoxide anion.9 Therefore, when present at appropriate concentrations, NO can effectively compete with SOD for superoxide anion. Peroxynitrite has the activity of hydroxyl radical and nitrogen dioxide radical, although it does not readily decompose into these entities. Peroxynitrite can directly nitrate and hydroxylate aromatic rings of amino acid residues. It is also a potent oxidant that reacts readily with sulfhydryls, with zinc-thiolate, lipids, proteins and DNA.9 Peroxynitrite induces the breakage of DNA strand and the activation of PARP-1 that results in the depletion of the cellular nicotinamide adenine dinucleotide (NAD+) and ATP pools. Peroxynitrite also suppresses mitochondrial function, thereby inhibiting the ability of the cell to restore NAD+ and ATP. Therefore, preventing the generation of oxyradicals or peroxynitrite by using superoxide-neutralizing strategies or selective inhibition of nitric oxide synthase (NOS) protects against the development of tissue injury. The mitochondrial antioxidant defense systems including superoxide dismutase, glutathione peroxidase, and regulation of the redox state of mitochondrial NAD(P)H, are essential cytoprotective factors.
PARP-1 Activation
The primary cellular response to DNA injury is to repair the damage, but the failure to repair the damage leads to loss of genomic integrity, carcinogenesis or cell death. To restore the damaged DNA, human cells have five major processes that are homologous recombinational repair (HRR), nonhomologous end joining (NHEJ), nucleotide excision repair (NER), base excision repair (BER) and mismatch r7pair 2(MMR).10 Many of the proteins involved in these DNA repair systems can also be recruited to contribute to cell death, including p53, PARPs, BRCA1, ATM, ATR, WRN, BLM, Tip60, catalytic subunit of DNA-PK, XPB, XPD, p33ING1b, Ref-1/Ape, MSH2, MSH6, MLH1 and PMS2 (Table 1).11
Table 1
Factor involved in nuclear morphologic changes during cell death programs.
Base excision repair (BER) is a major DNA repair pathway protecting mammalian cells from single-base DNA damage caused by DNA alkylating and oxidizing agents and other genotoxins. The DNA-repairing and protein-modifying enzyme PARP-1 is an abundant nuclear protein that is involved in DNA BER system. In response to DNA damage, PARP-1 activity is rapidly increased up to 500 fold upon binding to DNA strand nicks and breaks. PARP-1 transfers 50-200 residues of poly(ADP-ribose) (PAR) to itself and to the acceptor proteins such as histones, DNA and RNA polymerases, topoisomerases, high mobility group proteins, and transcription factors. Because of its prompt response, the intensity of PARP-1 activation could be a key factor regulating cell death or cell survival induced by DNA damage. As a nick sensor, PARP-1 activation could comprise a signal to regulate cellular DNA repair mechanism, transcription and replication, cytoskeletal organization, protein degradation, and other cellular activities through polymer-binding proteins.12-15 Excessive DNA damage, however, generates subsequent immense poly(ADP-ribosyl)ation by PARP-1 that can activate death programs. PARP-1 overactivation has been reported to mediate cell death in ischemia-reperfusion injury after myocardial infarction, cerebral ischemia, inflammation , ROS induced injury, glutamate excitotoxicity and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) injury.16 Other PARP family members might also play a role in cell death. Overexpression of the PARP family member tankyrase-2 leads to cytotoxicity.17 Although the cell death mechanism mediated by PARP-1 hyperactivation are not yet known, a recent study revealed apoptosis-inducing factor (AIF) as one of the downstream signaling constituents in PARP-1 dependent cell death.18
Poly(ADP-Ribose) Glycohydrolase (PARG)
The synthesis and turnover of ADP-ribose polymers is dynamic. Poly(ADP-ribose) glycohydrolase (PARG) plays the major role in the degradation of the polymer and it controls the level and complexity of polymerized PAR in vivo.19 In contrast to an array of different PARP proteins, only a single member of the PARG gene has been identified in mammals thus far. The cDNAs of PARG have been isolated and cloned from bovine,20 rat,21 human and mouse.22 PARG is translated into a catalytically active protein of approximately 111 kD, which can be cleaved by a protease to a 59 kD protein that is also catalytically active. The deduced amino acid sequence of the bovine PARG shares little or no homology with other known proteins. PARG contains a bipartite nuclear location signal, as would be predicted for a nuclear protein.20 Deletion analysis of PARG cDNA shows that the catalytic site of PARG is located in the C-terminal part of the protein. The C-terminal region is highly conserved in Drosophila and Caenorhabditis elegans PARG, suggesting that this region is essential for PARG activity. This domain has a high pI value (isoelectric point) of 8.6 that is necessary for high affinity binding to negatively charged ADP-ribose polymers.19 The expression of PARG and PARP-1 proteins seem to be regulated independently, as suggested by the fact that PARG expression and activity show no significant differences between wild-type and PARP-1 null mice.23
In vivo, PARG activity is much higher than PARP activity.19 Kinetic analyses of NAD+ consumption and polymer accumulation following DNA damage indicate that the two enzymes PARP-1 and PARG are involved in PAR polymer synthesis. These studies also show that significant PAR polymer is accumulated only when greater than 90% of PARG activity is inhibited. The role played by PARG in the molecular mechanisms of cell death is unclear. Because PARP-1 inhibits itself by automodification it was thought that PARG might be instrumental in cell death by preventing PARP-1 inactivation. Therefore, it has been hypothesized that PARG inhibition would limit and reduce PARP-1-mediated toxicity. Indeed, reports of PARG inhibition by gallotannin suggest that inhibition of PARG is protective.24 Unfortunately, gallotannin also inhibits PARP-1 (M.F. Poitras et al unpublished) and is a free radical scavenger; thus, interpretation for the role of PARG in PARP-1 dependent cytotoxicity is unclear. Furthermore, different lines of evidence suggest that cellular PARG activity plays a protective role against PARP-induced toxicity. Overexpression of PARP-1 or PARP-2 in the yeast Saccharomyces cerevisiae (devoid of PARP and PARG activity) leads to growth inhibition. Coexpression of either PARP-1 or PARP-2 with PARG restores growth, suggesting that PARG can prevent the toxic effects of PARP activation.25 Inactivation of cellular PARG activity leads to increase in PAR polymer and is correlated with growth arrest in cell lines.26 The Drosophila PARG knockout mutant exhibits larval lethality at the normal development temperature of 25°C.27 If the larvae are incubated at 29°C a quarter of the PARG knockout mutants progress to the adult stage but display progressive neurodegeneration with extensive accumulation of poly(ADP-ribose) the central nervous system, reduced locomotor activity and a short lifespan. Taken together these initial studies suggest that poly(ADP-ribose) metabolism is required for maintenance of normal cellular function. Further studies on the role of PARG in PARP-mediated cell death with more selective and specific inhibitors and RNAi (RNA interference) and knockout approaches are warranted.
Mitochondrial Participation in Cell Death Cascades
Recent studies are converging on a central hypothesis that, in many cell death cascades, the mitochondrion and the nucleus play key roles in conducting cell death programs. While mitochondria provide energy production under physiological conditions, under pathophysiologic conditions mitochondria contribute to cell death in many ways. Dysfunction of mitochondrial physiology, such as impaired oxidative phosphorylation, oxygen radical generation, or activation of mitochondrial permeability transition promote the release of mitochondrial death factors, such as cytochrome c, AIF, Smac/DIABLO (second mitochondria-derived activator of caspase/direct IAP binding protein with low pI), EndoG or serine protease Omi/HtrA2 (High temperature requirement protein A2). Among them, cytochrome c, Smac/DIABLO and Omi/ htr2 are involved in caspase-dependent cell death mechanisms, while AIF and EndoG induce caspase-independent cell death (fig. 1). In AIF-interceded cell death, AIF translocates from the mitochondria to the nucleus where it mediates chromatin condensation and large-scale DNA fragmentation possibly through binding to DNA.28,29 Similar to AIF, EndoG also translocates from the mitochondria to the nucleus where it leads to oligonucleosomal DNA fragmentation during apoptosis.30 EndoG can cooperate with exonuclease and DNase I and it catalyzes both high molecular weight DNA cleavage and oligonucleosomal DNA breakdown in a sequential manner in vitro.31 It remains to be explored whether the nuclear relocation of EndoG and subsequent nuclear apoptosis is also involved in PARP-1-dependent cell death.

Apoptosis-Inducing Factor (AIF)
Structure and Function
AIF protein is robustly conserved among mammalian species and it includes three domains; 1) an amino-terminal mitochondrial localization sequence (MLS) of 100 amino acids; 2) a spacer sequence of 27 amino acids; and 3) a carboxyterminal 485 amino acid domain with strong homology to oxidoreductases from other vertebrates (Xenopus laevis), nonvertebrate animals (Caenorhabditis elegans, Drosophila melanogaster), plants, fungi, eubacteria, and archaebacteria (fig. 2).32 The AIF gene is located on the X chromosome (Xq24/225) and it is ubiquitously expressed both in normal tissues and in a variety of cancer cell lines. The mature AIF protein is a flavoprotein with significant homology to plant ascorbate reductases and bacterial NADH oxidases. AIF is normally confined to the mitochondrial intermembrane space,33,34 where it colocalizes with Hsp60.35 Although the mitochondrial function(s) of AIF is yet to be identified, the flavoprotein moiety suggests that AIF may act as an oxidoreductase in mitochondrial electron transport.36 Recently, it was demonstrated that cerebellar granule cells from the Harlequin (Hq) mice, where the AIF expression is markedly reduced, are susceptible to oxidative stress.37 The mitochondrial localization, the sensitivity to oxidative stress in Hq mouse and NADH oxidase activity suggest that AIF might be involved in the process of scavenging ROS in normal cells. Mutation of the oxidoreductase domain does not alter the death promoting activity of AIF activity following translocation to the nucleus, indicating that this function of AIF is not directly involved in cell death. The crystal structure analysis of human AIF revealed there is a strong positive electrostatic potential at the AIF surface implicating this domain in DNA binding.38 DNA-binding defective mutants of AIF fail to induce cell death, which in turn suggests that DNA binding activity of AIF is required for its apoptogenic function in cell death.29

Figure 2
Primary structure of apoptosis-inducing factor (AIF). AIF has an oxidoreductase domain, mitochondrial localization signal (MLS) and nuclear localization signal (NLS).
AIF-Mediated Cell Death
It is apparent that AIF is an essential pro-apoptotic factor in vivo. Genetic studies revealed that AIF-deficient embryonic stem cells are resistant to apoptosis induced by serum deprivation and the suitable removal of cells during embryogenesis is defective in the AIF-deficient mouse embryos.39 The mitochondrial-nuclear translocation of AIF is caspase-independent in some types of cell death18,28,40 and death signal-induced translocation of AIF can be still observed in cells where Apaf-1, caspase-9 or caspase-3 is genetically obliterated.39-41 PARP-1 activators including the DNA-alkylating agent N-methyl-N'-nitro-N-nitrosoguanidine (MNNG), hydrogen peroxide (H2O2) and NMDA induce AIF translocation from the mitochondria to the nucleus, nuclear condensation, annexin-V staining, dissipation of the mitochondrial membrane potential and cell death. Biochemical markers associated with classic apoptosis, including phosphatidylserine exposure, partial chromatin condensation and nuclear condensation are also detected in the absence of caspase activation upon AIF translocation (Table 2).41,42 All these events are caspase independent because broad-spectrum caspase inhibitors do not prevent their expression. These processes, however, are blocked in the presence of PARP-1 inhibitors or in PARP-1 knockouts. It suggests that AIF-mediated cell death is not caspase-dependent and PARP-1 overactivation mediates AIF translocation.
Table 2
Molecular and morphological features in PARP-1/AIF-and caspase-mediated cell death.
While AIF does not require caspase activation to elicit cell death, AIF can participate in caspase-mediated cell death programs. Caspase-8, caspase-2 and the caspase-activated protein t-Bid can trigger the release of AIF from the mitochondria.43-45 Mitochondrial release of AIF and EndoG in HeLa or Jurkat cells treated with staurosporine or actinomycin D, is blocked or delayed by caspase inhibitors.46,47 Heat shock promoter-driven expression of Egl-1 (BH3-only protein) causes the release of mitochondrially localized GFP-WAH-1 (AIF homolog) in all C. elegans wild-type embryos, while mutant cells lacking Ced-3 activity present with more than 80% inhibition in AIF release.48 Transgenic expression of AIF and EndoG in C. elegans kills significantly more wild-type embryo cells than Ced-3-deficient cells. These data suggest that, although AIF and EndoG can function in a caspase-independent mode, AIF and EndoG also act efficiently in a Ced-3-positive background.48 Therefore it is important to determine whether mitochondrial AIF release occurs in a caspase-dependent or caspase-independent manner following different death signals in each cell type in order to develop specific insight into appropriate therapeutic targets for various diseases.
AIF may contribute to acute neuronal injury induced by trauma, hypoglycemia or transient ischemia as well as in chronic neurodegenerative diseases (Table 3). The translocation of AIF has been observed in different experimental models of neurotoxicity including death of photoreceptors induced by retinal detachment,49 neuronal cell death induced in vivo by brain trauma50 and cerebral ischemia51 and death of cortical neurons induced in vitro by exposure to heat-inactivated Streptococcus pneumoniae,52 hydrogen peroxide, peroxynitrite,50 the topoisomerase I inhibitor camptothecin, infection with a p53-expressing adenovirus,40 the striatal cell model of Huntington's disease53 or the excitotoxin N-methyl-D-aspartate (NMDA).18 Under excitotoxic conditions, NMDA-induced mitochondrial release of AIF is PARP-dependent and neutralization of AIF by microinjected AIF-specific antibody prevents cell death.40 Blocking NOS, PARP activity or AIF relocation prevents the downstream events in cell death signaling. There are no known specific pharmacologic inhibitors of AIF death-inducing activity. Hsp70 family members and Bcl-2 can prevent AIF translocation and over expression of these proteins provide cytoprotection.54,55 However, these factors also have protective actions independent of their inhibition of AIF translocation. By microinjection or BioPorter, neutralizing antibodies against AIF can be delivered into the cell. Neutralizing antibodies to AIF block NMDA/PARP-1 dependent excitotoxicity in neurons and PARP-1 dependent cytotoxicity in fibroblasts confirming, the key role for AIF in PARP-1 dependent cell death.
Table 3
Molecular and morphological features in PARP-1/AIF-and caspase-mediated cell death.
Mitochondrial Release of AIF
Mitochondria play a key role in cell death by releasing pro-apoptotic proteins from the mitochondrial compartment. Understanding the mechanisms of mitochondrial release following a death signal is an active area of investigation and debate. An array of models has been proposed to describe the mitochondrial release of these factors in the literature.56-59
Bax, Bak or BH3-only proteins can enhance the permeability of the mitochondrial outer mitochondrial membrane and these proteins might promote mitochondrial fission and finally unleash apoptogenic proteins including AIF. Since either Bak or Bax need to couple to BH3-only proteins for the release of pro-apoptotic proteins, BH3-only proteins act upstream of Bax- and Bak-mediated mitochondrial outer membrane permeabilization. It has been proposed that Bax and Bak oligomerization in outer membrane of mitochondria might produce a pore for pro-apoptotic proteins such as cytochrome c (fig. 3). Anti-apoptotic Bcl-2 and Bcl-XL protein and VDAC260 block this event by direct sequestration of pro-apoptotic Bcl-2 family proteins. However, still there is no evidence that AIF can escape through this pore assembled by Bax and Bak. Additionally, Bax and Bak might be involved in mitochondrial fission.61 The oligomerization of Bax and Bak occurs in discrete foci that colocalize with Drp1 and Mfn2 upon apoptosis. This event may suggest Bax and Bak might be involved in mitochondrial fission and fusion and subsequent release of pro-apoptotic proteins.

The activation of mitochondrial membrane transition can cause the mitochondrial release of AIF. Mitochondrial permeability transition (MPT) is triggered by dysregulation of Ca2+, ROS, ganglioside GD3, phosphatases and kinases acting on Bcl-2-like proteins, as well as transcription factors (e.g., p53, and TR3/Nur-77/NGFI-B). MPT, which is a critical event of early apoptosis, results in mitochondrial swelling and leads to outer membrane rupture and thus release of pro-death molecules.57,62 Apoptotic MPT differentially affects the outer and the inner mitochondrial membranes. The inner mitochondrial membrane becomes permeable to solutes up to 1500 Da, yet retains matrix proteins in their normal localization. In contrast, the outer mitochondrial membrane becomes completely permeabilized to proteins, leading to the release of soluble proteins from the mitochondrial intermembrane space to cytoplasm. This model likely suggests a nonspecific release of proapoptotic proteins from the intermembrane space, which occurs in late stage of mitochondrial permeabilization. The role of mitochondrial permeability transition in AIF release is controversial, as microinjection of recombinant AIF results in mitochondrial permeability transition. Therefore does mitochondrial permeability transition release AIF or does release of AIF result in loss of mitochondrial permeability? These events occur in such a short time frame that it is difficult to distinguish the proper sequence of events.
Some cellular signaling molecules can be generated during the cell death event and promote the release of AIF from the mitochondria directly or indirectly. Although the mechanism accounting for AIF release from the mitochondria still remains to be identified, PARP-1-dependent mitochondrial AIF release in PARP-1-mediated cell death could be explained with this hypothesis. Since poly(ADP-ribosyl)ation can provoke divergent signaling events in cell, it will be interesting to observe if signaling pathway through poly(ADP-ribose) formation could lead mitochondrial AIF release in PARP-1-mediated cell death.
Poly(ADP-Ribose) (PAR) Polymer-Mediated Cellular Response
Poly(ADP-ribosyl)ated proteins and/or PAR polymer itself could contribute to the cell death program. Many cytoplasmic and nuclear proteins are substrates of PARPs and thus they are covalently modified with PAR.63 Poly(ADP-ribosyl)ation of acceptor proteins by PARP-1 not only modifies the charge distribution of acceptor proteins, but increases the hindrance of proteins by the addition of a bulky and complex structure. This modification may inhibit the physiological function of acceptor proteins and thus intense poly(ADP-ribosyl)ation of key cellular proteins may lead to cell death. Another possibility is that PAR polymer may participate directly in cell death signaling. Many cellular proteins are recognized to bind to PAR polymer directly and specifically. There is a predicted consensus of PAR polymer binding motif.15 The binding of PAR polymer to this consensus motif, which is composed of hydrophobic and basic amino acid residues, is remarkably specific and in this regard, specific interactions between PAR polymer and another macromolecule could lead to additional signaling routes in many cellular responses. Several proteins are known to interact strongly with PAR, in a noncovalent fashion.63,64 Many of these proteins are related to apoptosis, such as p53, p21, or caspases.64,65 Thus, PAR polymer may be a novel signaling molecule that is produced upon PARP activation. Although the PAR polymer-mediated cell death signaling remains to be further studied, it is possible that PAR polymer may activate cell death mechanisms (fig. 4).
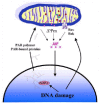
Figure 4
Possible mechanisms of mitochondrial release of apoptosis-inducing factor (AIF).
Summary
Mitochondria and the nucleus play critical roles in cell death. The signaling between these two organelles is dynamic. Understanding the signaling process between the nucleus and the mitochondria in cell death programs could lead us to more options in the development of new therapeutic targets. We discussed in this chapter how AIF could be released from the mitochondria and how AIF could act in PARP-dependent cell death mechanism. The molecular mechanisms accounting for PARP-dependent cell death are still being investigated but mitochondrial release of AIF and its translocation to the nucleus appears to be essential in this death process. We speculate that the PARP-mediated depletion of NAD and ATP or novel signals from PARP over activation might trigger AIF translocation, however, the authentic factors and the legitimate mechanism of mitochondrial AIF release remain to be explored.
References
- 1.
- Walker NI, Harmon BV, Gobe GC. et al. Patterns of cell death. Methods Achiev Exp Pathol. 1988;13:18–54. [PubMed: 3045494]
- 2.
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–57. [PMC free article: PMC2008650] [PubMed: 4561027]
- 3.
- Ravichandran KS. “Recruitment signals” from apoptotic cells: Invitation to a quiet meal. Cell. 2003;113(7):817–20. [PubMed: 12837239]
- 4.
- Eliasson MJ, Sampei K, Mandir AS. et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3(10):1089–95. [PubMed: 9334719]
- 5.
- Endres M, Wang ZQ, Namura S. et al. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17(11):1143–51. [PubMed: 9390645]
- 6.
- Zhang J, Dawson VL, Dawson TM. et al. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263(5147):687–9. [PubMed: 8080500]
- 7.
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–47. [PubMed: 11089981]
- 8.
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21(1):2–14. [PubMed: 11149664]
- 9.
- Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: Cause, effect, or association? J Clin Invest. 2003;111(2):163–9. [PMC free article: PMC151889] [PubMed: 12531868]
- 10.
- Wood RD, Mitchell M, Sgouros J. et al. Human DNA repair genes. Science. 2001;291(5507):1284–9. [PubMed: 11181991]
- 11.
- Bernstein C, Bernstein H, Payne CM. et al. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: Fail-safe protection against carcinogenesis. Mutat Res. 2002;511(2):145–78. [PubMed: 12052432]
- 12.
- Bouchard VJ, Rouleau M, Poirier GG. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol. 2003;31(6):446–54. [PubMed: 12829019]
- 13.
- Virag L, Szabo C. The therapeutic potential of poly(ADP-Ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. [PubMed: 12223530]
- 14.
- Chiarugi A. Poly(ADP-ribose) polymerase: Killer or conspirator? The _suicide hypothesis' revisited. Trends Pharmacol Sci. 2002;23(3):122–9. [PubMed: 11879679]
- 15.
- Gagne JP, Hunter JM, Labrecque B. et al. A proteomic approach to the identification of heterogeneous nuclear ribonucleoproteins as a new family of poly(ADP-ribose)-binding proteins. Biochem J. 2003;371(Pt 2):331–40. [PMC free article: PMC1223283] [PubMed: 12517304]
- 16.
- Apoptosis Inducing Factor and PARP mediated injury in the MPTP mouse model of Parkinson's disease: AnnalsIn: Wang H, Shimoji M, Yu SW, Dawson TD, Dawson VD, eds.New York:Acad. Sci 2003 . [PubMed: 12846982]
- 17.
- Kaminker PG, Kim SH, Taylor RD. et al. TANK2, a new TRF1-associated poly(ADP-ribose) polymerase, causes rapid induction of cell death upon overexpression. J Biol Chem. 2001;276(38):35891–9. [PubMed: 11454873]
- 18.
- Yu SW, Wang H, Poitras MF. et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259–63. [PubMed: 12114629]
- 19.
- Davidovic L, Vodenicharov M, Affar EB. et al. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268(1):7–13. [PubMed: 11461113]
- 20.
- Lin W, Ame JC, Aboul-Ela N. et al. Isolation and characterization of the cDNA encoding bovine poly(ADP-ribose) glycohydrolase. J Biol Chem. 1997;272(18):11895–901. [PubMed: 9115250]
- 21.
- Shimokawa T, Masutani M, Nagasawa S. et al. Isolation and cloning of rat poly(ADP-ribose) glycohydrolase: Presence of a potential nuclear export signal conserved in mammalian orthologs. J Biochem (Tokyo). 1999;126(4):748–55. [PubMed: 10502684]
- 22.
- Ame JC, Apiou F, Jacobson EL. et al. Assignment of the poly(ADP-ribose) glycohydrolase gene (PARG) to human chromosome 10q11.23 and mouse chromosome 14B by in situ hybridization. Cytogenet Cell Genet. 1999;85(3-4):269–70. [PubMed: 10449915]
- 23.
- Ame JC, Jacobson EL, Jacobson MK. Molecular heterogeneity and regulation of poly(ADP-ribose) glycohydrolase. Mol Cell Biochem. 1999;193(1-2):75–81. [PubMed: 10331641]
- 24.
- Ying W, Swanson RA. The poly(ADP-ribose) glycohydrolase inhibitor gallotannin blocks oxidative astrocyte death. Neuroreport. 2000;11(7):1385–8. [PubMed: 10841343]
- 25.
- Perkins E, Sun D, Nguyen A. et al. Novel inhibitors of poly(ADP-ribose) polymerase/PARP1 and PARP2 identified using a cell-based screen in yeast. Cancer Res. 2001;61(10):4175–83. [PubMed: 11358842]
- 26.
- Jonsson GG, Jacobson EL, Jacobson MK. Mechanism of alteration of poly(adenosine diphosphate-ribose) metabolism by hyperthermia. Cancer Res. 1988;48(15):4233–9. [PubMed: 3390818]
- 27.
- Hanai S, Kanai M, Ohashi S. et al. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci USA. 2004;101(1):82–6. [PMC free article: PMC314142] [PubMed: 14676324]
- 28.
- Susin SA, Lorenzo HK, Zamzami N. et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441–6. [PubMed: 9989411]
- 29.
- Ye H, Cande C, Stephanou NC. et al. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol. 2002;9(9):680–4. [PubMed: 12198487]
- 30.
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412(6842):95–9. [PubMed: 11452314]
- 31.
- Widlak P, Li LY, Wang X. et al. Action of recombinant human apoptotic endonuclease G on naked DNA and chromatin substrates: Cooperation with exonuclease and DNase I. J Biol Chem. 2001;276(51):48404–9. [PubMed: 11606588]
- 32.
- Lorenzo HK, Susin SA, Penninger J. et al. Apoptosis inducing factor (AIF): A phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999;6(6):516–24. [PubMed: 10381654]
- 33.
- Mate MJ, Ortiz-Lombardia M, Boitel B. et al. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat Struct Biol. 2002;9(6):442–6. [PubMed: 11967568]
- 34.
- Susin SA, Lorenzo HK, Zamzami N. et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441–6. [PubMed: 9989411]
- 35.
- Daugas E, Susin SA, Zamzami N. et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14(5):729–39. [PubMed: 10744629]
- 36.
- Lipton SA, Bossy-Wetzel E. Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell. 2002;111(2):147–50. [PubMed: 12408857]
- 37.
- Klein JA, Longo-Guess CM, Rossmann MP. et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419(6905):367–74. [PubMed: 12353028]
- 38.
- Mate MJ, Ortiz-Lombardia M, Boitel B. et al. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat Struct Biol. 2002;9(6):442–6. [PubMed: 11967568]
- 39.
- Joza N, Susin SA, Daugas E. et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410(6828):549–54. [PubMed: 11279485]
- 40.
- Cregan SP, Fortin A, MacLaurin JG. et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158(3):507–17. [PMC free article: PMC2173837] [PubMed: 12147675]
- 41.
- Susin SA, Daugas E, Ravagnan L. et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192(4):571–80. [PMC free article: PMC2193229] [PubMed: 10952727]
- 42.
- Loeffler M, Daugas E, Susin SA. et al. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 2001;15(3):758–67. [PubMed: 11259394]
- 43.
- Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297(5585):1352–4. [PubMed: 12193789]
- 44.
- Robertson JD, Enoksson M, Suomela M. et al. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem. 2002;277(33):29803–9. [PubMed: 12065594]
- 45.
- Zamzami N, El HamelC, Maisse C. et al. Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene. 2000;19(54):6342–50. [PubMed: 11175349]
- 46.
- Arnoult D, Gaume B, Karbowski M. et al. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003;22(17):4385–4399. [PMC free article: PMC202365] [PubMed: 12941691]
- 47.
- Arnoult D, Parone P, Martinou JC. et al. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol. 2002;159(6):923–9. [PMC free article: PMC2173976] [PubMed: 12486111]
- 48.
- Wang X, Yang C, Chai J. et al. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298(5598):1587–92. [PubMed: 12446902]
- 49.
- Hisatomi T, Sakamoto T, Goto Y. et al. Critical role of photoreceptor apoptosis in functional damage after retinal detachment. Curr Eye Res. 2002;24(3):161–72. [PubMed: 12221523]
- 50.
- Zhang X, Chen J, Graham SH. et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82(1):181–91. [PubMed: 12091479]
- 51.
- Zhu C, Qiu L, Wang X. et al. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem. 2003;86(2):306–17. [PubMed: 12871572]
- 52.
- Braun JS, Novak R, Murray PJ. et al. Apoptosis-inducing factor mediates microglial and neuronal apoptosis caused by pneumococcus. J Infect Dis. 2001;184(10):1300–9. [PubMed: 11679919]
- 53.
- Wang X, Zhu S, Drozda M. et al. Minocycline inhibits caspase-independent and -dependent mitochondrial cell death pathways in models of Huntington's disease. Proc Natl Acad Sci USA. 2003 [PMC free article: PMC193587] [PubMed: 12930891]
- 54.
- Ravagnan L, Gurbuxani S, Susin SA. et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3(9):839–43. [PubMed: 11533664]
- 55.
- Ruchalski K, Mao H, Singh SK. et al. HSP72 inhibits apoptosis-inducing factor release in ATP depleted renal epithelial cells. Am J Physiol Cell Physiol. 2003 [PubMed: 12930708]
- 56.
- Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8(2):115–28. [PubMed: 12766472]
- 57.
- Newmeyer DD, Ferguson-Miller S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell. 2003;112(4):481–90. [PubMed: 12600312]
- 58.
- Loeffler M, Kroemer G. The mitochondrion in cell death control: Certainties and incognita. Exp Cell Res. 2000;256(1):19–26. [PubMed: 10739647]
- 59.
- van Gurp M, Festjens N, van Loo G. et al. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun. 2003;304(3):487–97. [PubMed: 12729583]
- 60.
- Cheng EH, Sheiko TV, Fisher JK. et al. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301(5632):513–7. [PubMed: 12881569]
- 61.
- Karbowski M, Lee YJ, Gaume B. et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159(6):931–8. [PMC free article: PMC2173996] [PubMed: 12499352]
- 62.
- Zamzami N, Kroemer G. The mitochondrion in apoptosis: How Pandora's box opens. Nat Rev Mol Cell Biol. 2001;2(1):67–71. [PubMed: 11413468]
- 63.
- D'Amours D, Desnoyers S, D'Silva I. et al. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–68. [PMC free article: PMC1220459] [PubMed: 10455009]
- 64.
- Pleschke JM, Kleczkowska HE, Strohm M. et al. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem. 2000;275(52):40974–80. [PubMed: 11016934]
- 65.
- Vaziri H, West MD, Allsopp RC. et al. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J. 1997;16(19):6018–33. [PMC free article: PMC1170232] [PubMed: 9312059]
- 66.
- Fan Z, Beresford PJ, Oh DY. et al. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112(5):659–72. [PubMed: 12628186]
- 67.
- Mabley JG, Suarez-Pinzon WL, Hasko G. et al. Inhibition of poly (ADP-ribose) synthetase by gene disruption or inhibition with 5-iodo-6-amino-1,2-benzopyrone protects mice from multiple-low-dose-streptozotocin-induced diabetes. Br J Pharmacol. 2001;133(6):909–19. [PMC free article: PMC1572858] [PubMed: 11454665]
- 68.
- Suarez-Pinzon WL, Mabley JG, Power R. et al. Poly (ADP-ribose) polymerase inhibition prevents spontaneous and recurrent autoimmune diabetes in NOD mice by inducing apoptosis of islet-infiltrating leukocytes. Diabetes. 2003;52(7):1683–8. [PubMed: 12829633]
- 69.
- Pacher P, Liaudet L, Soriano FG. et al. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51(2):514–21. [PubMed: 11812763]
- 70.
- Mandir AS, Poitras MF, Berliner AR. et al. NMDA but not nonNMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J Neurosci. 2000;20(21):8005–11. [PMC free article: PMC6772735] [PubMed: 11050121]
- 71.
- Cosi C, Colpaert F, Koek W. et al. Poly(ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res. 1996b;729(2):264–9. [PubMed: 8876997]
- 72.
- Cosi C, Chopin P, Marien M. Benzamide, an inhibitor of poly(ADP-ribose) polymerase, attenuates methamphetamine-induced dopamine neurotoxicity in the C57B1/6N mouse. Brain Res. 1996a;735(2):343–8. [PubMed: 8911677]
- 73.
- Mandir AS, Przedborski S, Jackson-Lewis V. et al. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci USA. 1999;96(10):5774–9. [PMC free article: PMC21936] [PubMed: 10318960]
- 74.
- Wang H, Shimoji M, Yu SW. et al. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Ann N Y Acad Sci. 2003;991:132–9. [PubMed: 12846982]
- 75.
- Cao G, Clark RS, Pei W. et al. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23(10):1137–50. [PubMed: 14526224]
- 76.
- LaPlaca MC, Zhang J, Raghupathi R. et al. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma. 2001;18(4):369–76. [PubMed: 11336438]
- 77.
- Suh SW, Aoyama K, Chen Y. et al. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J Neurosci. 2003;23(33):10681–90. [PMC free article: PMC6740913] [PubMed: 14627653]
- 78.
- Scott GS, Jakeman LB, Stokes BT. et al. Peroxynitrite production and activation of poly (adenosine diphosphate-ribose) synthetase in spinal cord injury. Ann Neurol. 1999;45(1):120–4. [PubMed: 9894885]
- 79.
- Cookson MR, Ince PG, Shaw PJ. Peroxynitrite and hydrogen peroxide induced cell death in the NSC34 neuroblastoma x spinal cord cell line: Role of poly (ADP-ribose) polymerase. J Neurochem. 1998;70(2):501–8. [PubMed: 9453543]
- 80.
- Imai H, Koumura T, Nakajima R. et al. Protection from inactivation of the adenine nucleotide translocator during hypoglycaemia-induced apoptosis by mitochondrial phospholipid hydroperoxide glutathione peroxidase. Biochem J. 2003;371(Pt 3):799–809. [PMC free article: PMC1223322] [PubMed: 12534348]
- 81.
- Chiang SK, Lam TT. Post-treatment at 12 or 18 hours with 3-aminobenzamide ameliorates retinal ischemia-reperfusion damage. Invest Ophthalmol Vis Sci. 2000;41(10):3210–4. [PubMed: 10967085]
- 82.
- Tabuchi K, Ito Z, Tsuji S. et al. Poly(adenosine diphosphate-ribose) synthetase inhibitor 3-aminobenzamide alleviates cochlear dysfunction induced by transient ischemia. Ann Otol Rhinol Laryngol. 2001;110(2):118–21. [PubMed: 11219517]
- 83.
- Jijon HB, Churchill T, Malfair D. et al. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am J Physiol Gastrointest Liver Physiol. 2000;279(3):G641–51. [PubMed: 10960365]
- 84.
- Zingarelli B, O'Connor M, Hake PW. Inhibitors of poly(ADP-ribose) polymerase modulate signal transduction pathways in colitis. Eur J Pharmacol. 2003;469(1-3):183–94. [PubMed: 12782201]
- 85.
- Koedel U, Winkler F, Angele B. et al. Meningitis-associated central nervous system complications are mediated by the activation of poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 2002;22(1):39–49. [PubMed: 11807392]
- 86.
- Braun JS, Sublett JE, Freyer D. et al. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J Clin Invest. 2002;109(1):19–27. [PMC free article: PMC150815] [PubMed: 11781347]
- 87.
- Miesel R, Kurpisz M, Kroger H. Modulation of inflammatory arthritis by inhibition of poly(ADP ribose) polymerase. Inflammation. 1995;19(3):379–87. [PubMed: 7628865]
- 88.
- Rice WG, Schaeffer CA, Harten B. et al. Inhibition of HIV-1 infectivity by zinc-ejecting aromatic C-nitroso compounds. Nature. 1993;361(6411):473–5. [PubMed: 8429889]
- 89.
- Ferri KF, Jacotot E, Blanco J. et al. Apoptosis control in syncytia induced by the HIV type 1-envelope glycoprotein complex: Role of mitochondria and caspases. J Exp Med. 2000;192(8):1081–92. [PMC free article: PMC2195869] [PubMed: 11034598]
- 90.
- Genini D, Sheeter D, Rought S. et al. HIV induces lymphocyte apoptosis by a p53-initiated, mitochondrial-mediated mechanism. FASEB J. 2001;15(1):5–6. [PubMed: 11099484]
- 91.
- Ha HC, Juluri K, Zhou Y. et al. Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proc Natl Acad Sci USA. 2001;98(6):3364–8. [PMC free article: PMC30659] [PubMed: 11248084]
- 92.
- Oliver FJ, Menissier-de MurciaJ, Nacci C. et al. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18(16):4446–54. [PMC free article: PMC1171519] [PubMed: 10449410]
- 93.
- Veres B, Gallyas JrF, Varbiro G. et al. Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced septic shock. Biochem Pharmacol. 2003;65(8):1373–82. [PubMed: 12694878]
- 94.
- Broaddus VC, Yang L, Scavo LM. et al. Asbestos induces apoptosis of human and rabbit pleural mesothelial cells via reactive oxygen species. J Clin Invest. 1996;98(9):2050–9. [PMC free article: PMC507649] [PubMed: 8903324]
- 95.
- Ray SD, Balasubramanian G, Bagchi D. et al. Ca(2+)-calmodulin antagonist chlorpromazine and poly(ADP-ribose) polymerase modulators 4-aminobenzamide and nicotinamide influence hepatic expression of BCL-XL and P53 and protect against acetaminophen-induced programmed and unprogrammed cell death in mice. Free Radic Biol Med. 2001;31(3):277–91. [PubMed: 11461765]
- 96.
- Cuzzocrea S, McDonald MC, Mazzon E. et al. Effects of 5-aminoisoquinolinone, a water-soluble, potent inhibitor of the activity of poly (ADP-ribose) polymerase, in a rodent model of lung injury. Biochem Pharmacol. 2002;63(2):293–304. [PubMed: 11841805]
- 97.
- Fujita T, Maruyama M, Araya J. et al. Hydrogen peroxide induces upregulation of Fas in human airway epithelial cells via the activation of PARP-p53 pathway. Am J Respir Cell Mol Biol. 2002;27(5):542–52. [PubMed: 12397013]
- 98.
- Liaudet L, Szabo E, Timashpolsky L. et al. Suppression of poly (ADP-ribose) polymerase activation by 3-aminobenzamide in a rat model of myocardial infarction: Long-term morphological and functional consequences. Br J Pharmacol. 2001;133(8):1424–30. [PMC free article: PMC1621138] [PubMed: 11498530]
- 99.
- Pieper AA, Walles T, Wei G. et al. Myocardial postischemic injury is reduced by polyADPripose polymerase-1 gene disruption. Mol Med. 2000;6(4):271–82. [PMC free article: PMC1949947] [PubMed: 10949908]
- 100.
- Varbiro G, Toth A, Tapodi A. et al. Protective effect of amiodarone but not N-desethylamiodarone on postischemic hearts through the inhibition of mitochondrial permeability transition. J Pharmacol Exp Ther. 2003;307(2):615–25. [PubMed: 12970391]
- 101.
- Kim GT, Chun YS, Park JW. et al. Role of apoptosis-inducing factor in myocardial cell death by ischemia-reperfusion. Biochem Biophys Res Commun. 2003;309(3):619–24. [PubMed: 12963035]
- 102.
- Martin DR, Lewington AJ, Hammerman MR. et al. Inhibition of poly(ADP-ribose) polymerase attenuates ischemic renal injury in rats. Am J Physiol Regul Integr Comp Physiol. 2000;279(5):R1834–40. [PubMed: 11049868]
- 103.
- Ruchalski K, Mao H, Singh SK. et al. HSP72 inhibits apoptosis-inducing factor release in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol. 2003;285(6):C1483–93. [PubMed: 12930708]
- 104.
- Li F, Mao HP, Ruchalski KL. et al. Heat stress prevents mitochondrial injury in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol. 2002;283(3):C917–26. [PubMed: 12176748]
- 105.
- Rosenthal DS, Simbulan-Rosenthal CM, Liu WF. et al. PARP determines the mode of cell death in skin fibroblasts, but not keratinocytes, exposed to sulfur mustard. J Invest Dermatol. 2001;117(6):1566–73. [PubMed: 11886524]
- 106.
- Pardo J, Perez-Galan P, Gamen S. et al. A role of the mitochondrial apoptosis-inducing factor in granulysin-induced apoptosis. J Immunol. 2001;167(3):1222–9. [PubMed: 11466337]
- 107.
- Yamamoto T, Ueta E, Osaki T. Apoptosis induction by interleukin-2-activated cytotoxic lymphocytes in a squamous cell carcinoma cell line and Daudi cells - involvement of reactive oxygen species-dependent cytochrome c and reactive oxygen species-independent apoptosis-inducing factors. Immunology. 2003;110(2):217–24. [PMC free article: PMC1783038] [PubMed: 14511235]
- Introduction to Cell Death
- PARP-1 and Nuclear Death Signals
- Poly(ADP-Ribose) Glycohydrolase (PARG)
- Mitochondrial Participation in Cell Death Cascades
- Apoptosis-Inducing Factor (AIF)
- AIF-Mediated Cell Death
- Mitochondrial Release of AIF
- Poly(ADP-Ribose) (PAR) Polymer-Mediated Cellular Response
- Summary
- References
- PARP and the Release of Apoptosis-Inducing Factor from Mitochondria - Madame Cur...PARP and the Release of Apoptosis-Inducing Factor from Mitochondria - Madame Curie Bioscience Database
- THE RNA INFRASTRUCTURE: An Introduction to ncRNA Networks - Madame Curie Bioscie...THE RNA INFRASTRUCTURE: An Introduction to ncRNA Networks - Madame Curie Bioscience Database
- The Tn4371 ICE Family of Bacterial Mobile Genetic Elements - Madame Curie Biosci...The Tn4371 ICE Family of Bacterial Mobile Genetic Elements - Madame Curie Bioscience Database
- Idiopathic Parkinson's Disease: Staging an α-Synucleinopathy with a Predictable ...Idiopathic Parkinson's Disease: Staging an α-Synucleinopathy with a Predictable Pathoanatomy - Madame Curie Bioscience Database
- The Role of Heat Shock Proteins during Neurodegeneration in Alzheimer's, Parkins...The Role of Heat Shock Proteins during Neurodegeneration in Alzheimer's, Parkinson's and Huntington's Disease - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...