

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

MDM2 is an oncogene that is frequently over-expressed in various human cancers, including sarcomas, gliomas, melanomas, and breast cancers.1 The primary function of MDM2 is to inhibit the activity of the p53 tumor suppressor protein. P53 inhibits cell proliferation in response to DNA damage and other stresses by activating the transcription of genes that mediate either cell-cycle arrest or apoptosis. MDM2 binds the transactivation domain of p53 and inhibits the ability of p53 to activate transcription. Importantly, MDM2 binding also promotes the ubiquitin-mediated degradation of p53, as well as the export of p53 from the nucleus to the cytoplasm. The MDM2 gene itself is transcriptionally activated by p53, thus forming an autoregulatory feedback loop in which p53 promotes the expression of its own inhibitory factor. The ability of MDM2 to promote the degradation and nuclear export of p53 depends on an intact RING finger domain located in the MDM2 C terminus. This chapter will discuss the mechanisms by which MDM2 inhibits p53 function, with an emphasis on the requirement of the MDM2 RING finger domain in p53 inhibition.
Introduction
The MDM2 gene was first discovered in 1987 as a gene that was amplified in spontaneously transformed murine 3T3 fibroblasts.2 Subsequent studies revealed that the MDM2 protein interacts strongly with the tumor suppressor protein p53 and inhibits p53 activity.3,4 It is now well-established that the primary function of MDM2 is to inhibit p53.5,6 The importance of p53 inhibition by MDM2 is best illustrated through the attempted generation of mice that lack MDM2. These MDM2 “knockout” mice are not viable, dying at an early stage of embryonic development.7,8 Importantly, however, the concomitant knockout of p53 allows survival of the mice. These results demonstrate that MDM2 normally inhibits p53 function, and that this inhibition is critically important during embryonic development.
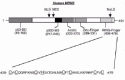
Human MDM2 is a 491 amino acid protein that localizes mostly in the nucleus, though it can also localize in the cytoplasm.6 The MDM2 gene has been mapped to chromosome 12q13-14 and includes several exons that encode the MDM2 protein. Alternative splicing of the primary mRNA transcript can result in the generation of different MDM2 isoforms, some of which inhibit the activity of the wild-type protein in a dominant-negative fashion.9,10 Analysis of the MDM2 coding sequence has revealed several structural and functional domains (fig. 1). These include the p53-binding domain located in the N terminus between residues 19-102, the p300-binding domain between residues 217-246, an acidic domain located between residues 222-272, a zinc finger domain located between residues 289-331, and a RING finger domain located in the MDM2 C terminus between residues 438-478. RING fingers are zinc binding domains with a defined octet of cysteine (C) and histidine (H) residues spaced at varying intervals in the coding sequence.11-14 In the RING H2 subtype, the cysteine and histidines are arranged in the order C3H2C3, while in the RING HC subtype the order is C3HC4. The MDM2 RING finger is of the C3HC4 subtype and, like other RING fingers, is predicted to bind two zinc atoms. RING finger domains are believed to function in the assembly and architecture of large protein complexes, including enzyme complexes of the ubiquitin system of protein degradation.15,16 As we will see below, the MDM2 protein can function as an ubiquitin-system E3 ligase that promotes the specific degradation of p53, and this activity requires the MDM2 RING finger domain.
MDM2 Promotes p53 Degradation
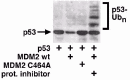
In 1997, research groups led by Moshe Oren in Israel and Karen Vousden in Frederick, Maryland were investigating the binding interactions between MDM2 and p53.17,18 These studies were complicated by the fact that p53 levels were consistently decreased with coexpression of MDM2 (see fig. 2) as an example), which made a quantitative analysis of the p53:MDM2 interaction difficult. The researchers decided to investigate the molecular basis for the decreased levels of p53. Small-molecule inhibitors of the proteasome prevented the decrease in p53 levels observed with MDM2 expression, suggesting that MDM2 was somehow promoting p53 degradation by the proteasome. Moreover, a high molecular weight ladder of p53 species was generated when p53 was coexpressed with MDM2 (see fig. 2) as an example). The size of these p53 species was consistent with the addition of multiple ubiquitin-moieties to p53, and analysis with anti-ubiquitin antibodies identified them as p53:ubiquitin conjugates. These seminal studies provided the first evidence that MDM2 can promote the degradation of p53 through the ubiquitin-proteasome pathway.17,18
MDM2 Is an E3 Ubiquitin-Protein Ligase
The ubiquitin-proteasome system is the major pathway in the cell for selective protein degradation.19,20 The covalent attachment of multiple ubiquitin molecules to lysine residues of a target protein serves to signal its recognition and rapid degradation by the 26S proteasome. Ubiquitination of a protein substrate requires the concerted action of three classes of enzymes designated E1, E2, and E3. The ubiquitin activating enzyme E1 initially activates ubiquitin in an ATP-dependent reaction through the formation of a thiol-ester bond between the carboxyl terminus of ubiquitin and the thiol group of a specific cysteine residue of E1. Ubiquitin is then transferred to a specific cysteine residue on one of several ubiquitin-conjugating enzymes (Ubcs or E2s). E2 enzymes in turn may transfer the ubiquitin either directly to a substrate or to the final class of enzymes known as ubiquitin protein ligases (or E3s). The E3 enzymes catalyze the formation of an isopeptide bond between the carboxyl terminus of ubiquitin and the ε-amino group of lysine residues on the target protein. A substrate may then be multiply ubiquitinated through the attachment of additional ubiquitin molecules to specific lysine residues of ubiquitin itself. In many cases the E1, E2, and E3 enzymes involved form large, multi-protein complexes. This increases the efficiency of the process by allowing the rapid thiol-ester transfer of ubiquitin molecules between proteins.
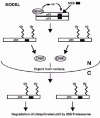
In vitro studies with purified E1 enzyme, E2 enzyme, MDM2, and p53 showed that MDM2 could form a direct thiol-ester linkage with ubiquitin at Cys464 in the MDM2 RING finger domain, and could transfer the ubiquitin to p53.21 Removal of either the E1 or E2 enzymes prevented formation of the thiol-ester bond between ubiquitin and MDM2, and also prevented the transfer of ubiquitin to p53. Moreover, mutations that converted Cys464 to Ala464 (C464A) also inhibited the transfer of ubiquitin to p53. Taken together, these data support a model in which MDM2 functions as an E3 ubiquitin ligase that can accept activated ubiquitin from an E2 enzyme, and then transfer the ubiquitin to p53 (fig. 3).
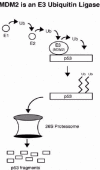
Figure 3
MDM2 is an E3 ubiquitin-ligase for p53. Current models suggest that MDM2 accepts activated ubiquitin from an E2 enzyme and transfers the ubiquitin to multiple lysine residues in the p53 C terminus.
The nature of p53 ubiquitination by MDM2 has not been fully clarified. MDM2 can stimulate the formation of a “ladder” of p53:ubiquitin conjugates in cells transiently expressing both proteins (see fig. 2 as an example). This ladder could result from the addition of single ubiquitin molecules to multiple lysine residues in p53, or from the formation of poly-ubiquitin chains of varying lengths on one or only a few lysines. Mutational analysis of p53, as well as in vitro studies using ubiquitin-aldehyde, support the notion that MDM2 mono-ubiquitinates p53, transferring single ubiquitin moieties to multiple lysine residues located in the extreme C terminus of p53.22,23 However, the fact that formation of poly-ubiquitin chains is necessary to target a protein to the proteasome24 indicates that a so called “E4” enzyme must exist that can promote p53 poly-ubiquitination. Recent studies suggest p300 may be such a factor. P300 is a large (approx. 300 kilodalton) protein that can bind the central region of MDM2 (fig. 1), as well as two separate regions of p53. While purified MDM2 can promote p53 mono-ubiquitination in an in vitro system, addition of p300 stimulates the poly-ubiquitination of p53,25 probably through the formation of a p53:MDM2:p300 trimeric complex. These and other studies indicate that p300 can participate with MDM2 to promote p53 degradation by promoting p53 poly-ubiquitination25-27 (fig. 4 Left). However, it is important to note that p300 can also function as an activator of p53 by promoting p53 acetylation at one or more lysine residues in the p53 C terminus (Fig 4 Right). This acetylation increases the DNA binding activity of p53, thus leading to an activation of p53-responsive genes, and will be discussed in more detail below. Regulatory interactions between p53, MDM2, and p300 remain both interesting and complex.
P53 Is Stabilized and Its Levels Increase in Response to Stress
P53 levels increase in response to multiple genotoxic and nongenotoxic stresses, including DNA damage, hypoxia, ribonucleotide depletion, and the abnormal activation of oncogene signaling pathways, among others.28-30 The effect of increasing p53 is to inhibit cell proliferation through either cell cycle arrest or apoptosis (fig. 5). These effects are mediated by p53-responsive genes, such as p21 and bax. The importance of signaling either a cell cycle arrest or apoptosis in response to these stresses is abundantly clear. DNA damaging stresses have the potential to damage chromosomes, alter genomic integrity, and cause mutations that may activate oncogenes and/or inhibit tumor suppressors. By triggering a cell cycle arrest or apoptosis in response to DNA damaging stress, p53 prevents the propagation of cells that may have an altered genome or acquired potentially cancer-promoting mutations. Similarly, the mutational activation of oncogenes is an early step in the development of most or all human cancers. By triggering an arrest or apoptosis in response to the mutational activation of oncogenes, p53 prevents the growth of cells that would otherwise be prone to becoming cancerous. It is worth noting that many chemotherapeutic drugs used in cancer treatment in some way modify DNA, resulting in the generation of single or double-stranded breaks. In cancers that retain wild-type p53, these DNA breaks trigger an increase in p53 levels, and a resulting activation of p53-induced cell death pathways.31,32 In contrast, the same thing does not occur in cancers that express mutant p53 or in which the wild-type p53 gene has been deleted.31,32 This explains the increased sensitivity to chemotherapeutics observed in cancers with wild-type p53 compared with cancers that lack wild-type p53 expression.
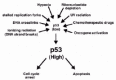
Figure 5
p53 levels increase in response to diverse stresses, resulting in cell cycle arrest or apoptosis.
The increase in p53 levels following stress results from a stabilization of the p53 protein.33,34 This is illustrated in (fig. 6.) RKO cells (human colon cancer cells that express wild-type p53) were untreated, or exposed to UV or ionizing (IR) radiation. Six hours after radiation exposure, the cells were treated with cyclohexamide (CHX) to inhibit de novo protein synthesis, and p53 steady state levels were monitored at various time points after CHX addition. The rate at which p53 levels decrease under these conditions is a measure of protein stability. In untreated cells, p53 levels decrease rapidly following CHX treatment, with a half-life (T1/2) of approximately 30 min. In contrast, p53 levels are elevated in the UV and IR-treated cells and remain elevated for up to 2 hours following CHX treatment. The half-life (T1/2) of p53 is increased following UV or IR-treatment to about 100 minutes or greater than 2 hours, respectively. These results demonstrate that the stress-induced increase in p53 levels is due, at least in part, to an increase in p53 protein stability.

Stress Inhibits MDM2-dependent p53 Degradation
Given that MDM2 promotes p53 ubiquitination and degradation, it is perhaps not surprising that the stress-induced stabilization of p53 results from an inhibition of these MDM2 activities.34 Agents that damage or modify DNA, such as IR and UV radiation, DNA cross-linkers, and chemotherapeutic drugs, activate a signal transduction cascade that culminates in the phosphorylation of p53 at multiple sites, including sites within or near the N-terminal MDM2-binding domain (fig.7).28-30,35 Phosphorylation of Ser15 and Ser20 in p53 is critical for the stabilization and activation of p53 in response to DNA damage,36-38 and kinases that can phosphorylate p53 at these sites have been identified. One of these kinases, referred to as the ataxia telangiectasia mutated (ATM) kinase, is activated in response to DNA strand breaks, such as those that might occur in response to IR, and can directly phosphorylate p53 at Ser15 and other residues 28-30,35 (fig. 7). A second kinase that is related to ATM and referred to as the ATM and Rad3-related (ATR) kinase, is activated in response to stalled replication forks, such as might occur in response to UV radiation. ATR can also phosphorylate p53 at Ser15. P53 phosphorylation at Ser20 requires ATM or ATR, but is not phosphorylated by either of these kinases directly. Instead, ATM and ATR activate other kinases referred to as Chk1 (activated by ATR) and Chk2 (activated by ATM) that can directly phosphorylate p53 at Ser20.39 Most current models propose that Ser20 phosphorylation stabilizes p53 by blocking the interaction between p53 and MDM2, whereas the phosphorylated Ser15 residue is believed to activate p53 transcriptional activity by recruiting p300 or CBP.40,41 As mentioned earlier, p300 activates p53 promoting its acetylation at C-terminal lysine residues. This acetylation is believed to increase the DNA binding affinity of p53, thus leading to an activation of p53 responsive genes. It is important to note that ATM can also phosphorylate MDM2 (Fig. 7), and this phosphorylation has been reported to diminish MDM2-mediated degradation and nuclear export of p53.42,43
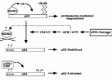
The stabilization of p53 in response aberrant oncogene signaling occurs through a pathway different from that which stabilizes p53 following DNA damage.44,45 In this case, activated oncogenes signal an increase in the transcription of a tumor suppressor protein referred to as either p14 or Arf (referred to as p19/Arf in the mouse). P14 forms a direct complex with MDM2 and inhibits the MDM2-mediated ubiquitination of p53 (Fig. 8). Most current models suggest that p14 inactivates MDM2 by sequestering it in the nucleolus,44-47 though there was some suggestion that p14 binding with MDM2 in the nucleoplasm is enough to inhibit MDM2 activity. Regardless, the effect of inhibiting MDM2 is to stabilize p53 and thus increase p53 levels. The tumor suppressor PML has also been implicated in the oncogene-mediated activation of p53. PML is the major component in multi-protein sub-nuclear complexes termed PML-nuclear bodies (PML-NBs).48-49 PML can interact with several different proteins and recruit them to PML-NBs, including p5350 and p300/CBP.51 Further, PML can stimulate p53 transcriptional activity in transiently transfected cells, and this requires recruitment of p53 to PML-NBs.50 Studies from the laboratories of Pier Pandolfi at Memorial Sloan Kettering in New York and Scott Lowe at the Cold Spring Harbor Laboratories in New York have provided compelling evidence that the oncogene-mediated activation of p53 requires PML. In these studies, p53 activity was measured in PML+/+ and PML-/- mouse embryo fibroblasts (MEFs) infected with retroviruses expressing an activated ras oncogene (Ras V12). Ras V12 expression in PML+/+ cells resulted in the activation of p53, recruitment of p53 into PML-NBs, and the induction of premature senescence. In contrast, p53 was neither activated nor recruited into PML-NBs in PML-/- cells, and the cells were resistant to ras-induced senescence.52,53 From these studies emerge a model in which p14/Arf stabilizes p53 in response to aberrant oncogene signaling by inhibiting the activity of MDM2, whereas the activation of p53 involves recruitment of p53 to PML-NBs, and its subsequent acetylation by p300/CBP52-54 (fig. 8).
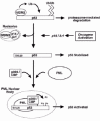
MDM2 Promotes p53 Nuclear Export
In addition to promoting p53 degradation, MDM2 can also promote the export of p53 from the nucleus to the cytoplasm, and this is expected to further inhibit the transcriptional activity of p53. p53 contains two leucine-rich nuclear export signals (NESs), one located in the N-terminal MDM2-binding domain of p53, and the second located in the C terminus within the tetramerization domain.55,56 Both of these NESs can support nuclear export when fused to a heterologous, nuclear protein. Disruption of the C-terminal NES causes p53 to display a more pronounced nuclear localization;55 however, a similar finding has not been demonstrated for the N-terminal NES. Despite the identification of these NESs, the molecular mechanisms that control p53 nuclear export and the role of MDM2 in this process have remained a matter of debate. Initial studies found that endogenous wild-type p53 accumulated in the nucleus and was stabilized in cells exposed to leptomycin B (LMB), a specific inhibitor of nuclear export.55,57 This suggested that nuclear export is required for p53 degradation. Further studies reported that MDM2 contains an NES within its sequence, and that blocking nuclear export could inhibit the ability of MDM2 to promote p53 degradation.57-59 Based on these findings a model was proposed in which MDM2 transports p53 from the nucleus to the cytoplasm for p53 to be degraded, and this transport depends on a functional NES within MDM2. In conflict with this model were reports that p53 contains its own NES, and that this NES is sufficient to promote p53 nuclear export even in cells that lack MDM2 expression.55 The NES identified in these studies was the C-terminal NES located within the p53 tetramerization domain, and structural analyses indicated that this NES would be inaccessible to the export machinery when p53 is a tetramer, but would be exposed and accessible when p53 is in either a dimeric or monomeric form. Taken together, these latter findings supported a second model in which p53 is exported from the nucleus via its own NES and independent of MDM2, and that this export may be regulated by changes in p53 tetramerization.
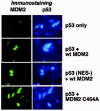
In an attempt to clarify the role of MDM2 in p53 nuclear export, cultured cells were transiently transfected with DNAs encoding p53 and MDM2, and localization of p53 and MDM2 was then monitored by immunofluorescence staining.60,61 As shown in (Figs. 9 and 10), p53 displayed a predominantly nuclear localization when expressed alone, but was relocalized to the cytoplasm in cells coexpressing MDM2. These results are consistent with the hypothesis that MDM2 can promote p53 nuclear export. Importantly, MDM2 remained in the nucleus despite the fact that p53 was relocalized to the cytoplasm, suggesting that nuclear MDM2 can promote p53 nuclear export. Mutants of p53 and MDM2 deficient in their nuclear export signals (NES mutants) were also tested in these experiments. The MDM2 NES mutant promoted the nuclear export of p53 to an extent similar to wild-type MDM2 (fig.10), indicating that the MDM2 NES was not required for this effect. In contrast, the p53 NES- mutant was nuclear when expressed alone, and remained nuclear when coexpressed with wild-type MDM2 (figs. 9 and 10). These results indicate that the MDM2-mediated nuclear export of p53 requires the NES of p53, but not the NES of MDM2. Finally, to assess whether the RING-finger domain of MDM2 has a function in p53 nuclear export, p53 localization was monitored in cells cotransfected with wild-type p53 and the MDM2 C464A mutant. As mentioned earlier, the C464A mutation in the MDM2 RING-finger domain inhibits the ability of MDM2 to promote p53 ubiquitination. As shown in (Figs. 9 and 10), MDM2 C464A was unable to promote the nuclear export of p53, indicating that an intact MDM2 RING finger domain is required for p53 nuclear export. It is important to note that the C464A mutation did not affect the ability of MDM2 to bind p53.60,61 Taken together, these results support the current model for p53 nuclear export (Fig. 11). According to this model, MDM2 can bind p53 and promote its ubiqutination in the nucleus, and this leads to the export of p53 from the nucleus to the cytoplasm. This export requires the p53 NES located in p53s C terminus. Based on the structural studies mentioned earlier, it is proposed that the addition of ubiquitin exposes the C-terminal NES, probably by causing dissolution of p53 tetramers into p53 monomers or dimers. This allows recognition of the C-terminal NES by the export factor CRM1. This model was further supported by reports demonstrating that mutations in specific ubiquitination sites in p53's C terminus can inhibit the nuclear export of p53 that is mediated by MDM2.62,63
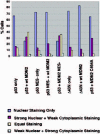
Several studies have supported the notion that nuclear export is an important component of p53 regulation. For example, various tumor types have been described in which p53 is functionally inactivated due to its abnormal sequestration in the cytoplasm, including inflammatory breast carcinoma, undifferentiated neuroblastoma, colorectal carcinoma, and retinoblastoma.64-68 Sequestration of p53 in the cytoplasm is believed to contribute to the generation of these cancers by inhibiting p53 activity. In some cases, treatments that either block nuclear export (exposure to the nuclear export inhibitor leptomycin B), or inhibit endogenous MDM2 expression, result in the redistribution of p53 into the nucleus.55,69 These findings suggest that the cytoplasmic localization of p53 in these cancers results, at least in part, from excessive nuclear export that is mediated by MDM2.
Interactions Between MDM2 and Other Proteins
Notwithstanding its effects on p53, there is abundant evidence that MDM2 has p53-independent activities that may contribute to its oncogenic properties. This evidence includes the following: First, rare cancers bearing both p53 mutations and MDM2 gene amplifications have been described and are more aggressive than those with alterations of either gene alone.70 This suggests that MDM2 over-expression may accelerate tumor growth even in the absence of functional p53. Second, MDM2 over-expression has been reported to transform cells in culture in the absence of a functioning p53.71 Finally, targeted MDM2 over-expression in the mammary glands of mice leads to abnormal cell proliferation and mammary hypertrophy to a similar extent in both a p53 +/+ and p53 -/- background.72 Accordingly, considerable effort has been aimed at identifying cell-cycle regulatory proteins other than p53 with which MDM2 can interact. A number of such factors have been identified, including the p53-related protein p73, the MDM2-related protein MDMX, the tumor suppressor proteins pRB and PML, the ribosomal protein L5, the transcription factor E2F1, and the neuronal differentiation factor Numb, among others73 (fig. 12). In most cases, the effect of these interactions on cell growth has not been completed clarified. In the paragraphs below, we briefly discuss the interactions of MDM2 with these other proteins.
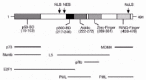
Figure 12
MDM2 interacts with multiple different proteins other than p53. The region of MDM2 involved in binding the indicated protein is shown.
MDM2 Interaction with p73
P73 is a p53-related protein that has a high degree of amino acid sequence and structural similarity to p53.74 P73 can induce apoptosis when over-expressed, and can activate transcription of p53-responsive genes.75 These results indicate that p53 and p73 can carry out redundant functions. However, unlike p53, p73 mutations are not commonly observed in human cancers, and p73-knockout mice are not cancer prone. Instead, p73 knockout mice have neurological, pheromonal, and inflammatory defects.76 MDM2 can bind the transactivation domain of p73 and inhibit its ability to activate transcription and induce apoptosis, but does not induce p73 degradation.77-79 A recent report demonstrated that p73 and p63 (another p53-related protein) are required for p53-dependent apoptosis in response to DNA damage.80 It is unclear at present whether MDM2 binding with p73 in some way inhibits the ability of p73 to cooperate with p53 in this DNA damage response.
MDM2 Interaction with pRb
The retinoblastoma protein pRb is the first characterized tumor suppressor. The Rb protein inhibits growth by sequestering the E2F family of transcription factors, thus inhibiting them from activating the expression of genes that promote G1 to S-phase progression. MDM2 binds to domains of pRb that are required for stable interaction with E2F1, suggesting that MDM2 could inhibit pRb tumor suppressor function by favoring detachment of E2F1 from pRb.81,82 In contrast, pRb was reported to prevent MDM2-dependent degradation of p53 and inhibition of apoptosis in transient transfection studies.83 Taken together, these findings suggest that pRb and MDM2 may antagonize each other's functions through binding one another.
MDM2 Interaction with E2F1
E2F1 is a transcription factor that functions as a dimer with DP1. Together, the E2F1/DP1 dimer can stimulate cell cycle progression by promoting the expression of genes required for the G1 to S-phase transition. In contrast, over-expression of E2F1 stimulates apoptosis, and E2F1 knockout mice are prone to develop certain cancers. These results indicate that E2F1 has both growth-promoting and growth-inhibitory capabilities. In the absence of p53, MDM2 inhibits the pro-apoptotic effect of E2F1/DP1 by favoring degradation of the heterodimer. At the same time, however, MDM2 can stimulate DNA synthesis in cooperation with E2F1/DP1. These seemingly contradictory results were reconciled by the proposal that high levels of E2F1/DP1 were reduced by MDM2 to levels appropriate for the G1-S-phase transition.84
MDM2 Interaction with MDMX
MDMX (also called MDM4) is an MDM2-related protein with a high degree of amino acid sequence and structural similarity with MDM2. MDMX can bind p53 but fails to promote p53 degradation.85 Interestingly, knocking-out MDMX in the mouse resulted in early embryonic death, and this could be rescued by the concomitant knockout of p53,86 similar to the results obtained with MDM2 knockout mice. These results suggest that the function of MDMX is to inhibit p53. MDM2 and MDMX can associate through their RING-finger domains. It is currently believed that MDMX-binding increases the ability of MDM2 to target p53 for degradation, through either stabilizing the MDM2 protein or modulating MDM2s localization.87
MDM2 Interaction with Ribosomal Protein L5
MDM2 forms a complex with ribosomal protein L5 and its associated 5S rRNA.88,89 These interactions have gained interest in recent years in light of the findings that MDM2 contains a nuclear export signal and undergoes active nuclear export. Specifically, it is speculated that MDM2 nuclear export may be linked to the nuclear export of ribosome subunits following their assembly in the nucleolus, and that this may be facilitated by the interaction between MDM2 and L5.
MDM2 Interaction with Numb
Numb is a regulator of neuronal differentiation that can interact with the N terminus of MDM2. Interestingly, Numb levels are decreased in the presence of MDM2, suggesting that MDM2 may target Numb for degradation.90 An effect of MDM2 on neuronal differentiation has not been described.
MDM2 Interaction with PML
As mentioned earlier, PML is a tumor suppressor that can activate p53 by recruiting it to multi-protein nuclear complexes termed PML-NBs.50 MDM2 colocalized with PML in PML-NBs in cells that lacked p53 expression, and purified MDM2 protein was shown to interact with a GST-PML fusion protein generated in bacteria.91 These results indicate that MDM2 can interact with PML in a p53-independent manner. Given that PML can activate p53, it was speculated that the MDM2:PML interaction could indirectly affect the activity of p53. However, experiments to test this possibility have not yet been carried out.
Conclusions
MDM2 is an oncoprotein that is frequently overexpressed in various human cancers. The primary function of MDM2 is to inhibit the tumor suppressor protein p53. MDM2 promotes the ubiquitination of p53 and its degradation by the proteasome, as well as the export of p53 from the nucleus to the cytoplasm. Both of these activities require the RING finger domain located in the MDM2 C terminus. DNA damage and other stresses stabilize p53 through an inhibition of MDM2-mediated degradation. MDM2 interacts with a variety of other proteins in addition to p53, and these interactions may contribute to the oncogenic potential of MDM2.
References
- 1.
- Landers JE, Cassel SL, George DL. Translational enhancement of mdm2 oncogene expression in human tumor cells containing a stabilized wild-type p53 protein. Cancer Res. 1997;57:3562–3568. [PubMed: 9270029]
- 2.
- Cahilly-Snyder L, Yang-Feng T, Francke U. et al. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somat Cell Molec Genet. 1987;13:235–244. [PubMed: 3474784]
- 3.
- Momand J, Zambetti GP, Olson DC. et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. [PubMed: 1535557]
- 4.
- Oliner JD, Pietenpol JA, Thiagalingam S. et al. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. [PubMed: 8479525]
- 5.
- Momand J, Wu H, Dasgupta G. MDM2-master regulator of the p53 tumor suppressor protein. Gene. 2000;242:15–29. [PubMed: 10721693]
- 6.
- Deb SP. Function and dysfunction of the human oncoprotein MDM2. Front Biosci. 2002;7:235–243. [PubMed: 11779693]
- 7.
- Jones SN, Roe AE, Donehower LA. et al. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. [PubMed: 7477327]
- 8.
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. [PubMed: 7477326]
- 9.
- Perry ME, Mendrysa SM, Saucedo LJ. et al. p76(MDM2) inhibits the ability of p90(MDM2) to destabilize p53. J Biol Chem. 2000;275:5733–5738. [PubMed: 10681559]
- 10.
- Evans SC, Viswanathan M, Grier JD. et al. An alternatively spliced HDM2 product increases p53 activity by inhibiting HDM2. Oncogene. 2001;20:4041–4049. [PubMed: 11494132]
- 11.
- Deshaies RJ. SCF and cullin/RING H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–467. [PubMed: 10611969]
- 12.
- Tyers M, Jorgensen P. Proteolysis and the cell cycle: With this RING I do thee destroy. Curr Opin Genet Dev. 2000;10:54–64. [PubMed: 10679394]
- 13.
- Jackson PK, Eldridge AG, Freed E. et al. The lore of the RINGs: Substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 2000;10:429–439. [PubMed: 10998601]
- 14.
- Borden KLB. RING domains: Master builders of molecular scaffolds. J Mol Biol. 2000;295:1103–1112. [PubMed: 10653689]
- 15.
- Lorick KL, Jensen JP, Fang S. et al. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA. 1999;96:11364–11369. [PMC free article: PMC18039] [PubMed: 10500182]
- 16.
- Joazeiro CA, Weissman AM. RING finger proteins: Mediators of ubiquitin ligase activity. Cell. 2000;102:549–552. [PubMed: 11007473]
- 17.
- Haupt Y, Maya R, Kazaz A. et al. MDM2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. [PubMed: 9153395]
- 18.
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by MDM2. Nature. 1997;387:299–303. [PubMed: 9153396]
- 19.
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. [PubMed: 9759494]
- 20.
- Ciechanover A, Schwartz AL. Ubiquitin-mediated degradation of cellular proteins in health and disease. Hepatology. 2002;35:3–6. [PubMed: 11786953]
- 21.
- Honda R, Tanaka H, and YasudaH. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. [PubMed: 9450543]
- 22.
- Rodriguez MS, Desterro JM, Lain S. et al. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol. 2000;20:8458–8467. [PMC free article: PMC102152] [PubMed: 11046142]
- 23.
- Lai Z, Ferry KV, Diamond MA. et al. Human mdm2 mediates multiple mono-ubiquitination of p53 by a mechanism requiring enzyme isomerization. J Biol Chem. 2001;27:357–367. [PubMed: 11397792]
- 24.
- Deveraux Q, Ustrell V, Pickart C. et al. A 26S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994;269:7059–7061. [PubMed: 8125911]
- 25.
- Grossman SR, Deato ME, Brignone C. et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300:342–344. [PubMed: 12690203]
- 26.
- Grossman SR, Perez M, Kung AL. et al. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2:405–415. [PubMed: 9809062]
- 27.
- Grossman SR. p300/CBP/p53 interaction and regulation of the p53 response. Eur J Biochem. 2001;268:2773–2778. [PubMed: 11358491]
- 28.
- Prives C, Hall PA. The p53 pathway. J Pathol. 1999;187:112–126. [PubMed: 10341712]
- 29.
- Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13:332–337. [PubMed: 11343904]
- 30.
- Woods DB, Vousden KH. Regulation of p53 function. Exp Cell Res. 2001;264:56–66. [PubMed: 11237523]
- 31.
- Lowe SW. Cancer therapy and p53. Curr Opin Oncol. 1995;7:547–553. [PubMed: 8547404]
- 32.
- Schmitt CA, Fridman JS, Yang M. et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. [PubMed: 12015983]
- 33.
- Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689–1694. [PMC free article: PMC368974] [PubMed: 6092932]
- 34.
- Maki CG, Howley PM. Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol Cell Biol. 1997;17:355–363. [PMC free article: PMC231760] [PubMed: 8972216]
- 35.
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998;12:2973–2983. [PubMed: 9765199]
- 36.
- Shieh SY, Ikeda M, Taya Y. et al. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. [PubMed: 9363941]
- 37.
- Chehab NH, Malikzay A, Stavridi ES. et al. Phosphorylation of ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA. 1999;96:13777–13782. [PMC free article: PMC24141] [PubMed: 10570149]
- 38.
- Unger T, Juven-Gershon T, Moallem E. et al. Critical role for ser20 of p53 in the negative regulation of p53 by MDM2. EMBO J. 1999;18:1805–1814. [PMC free article: PMC1171266] [PubMed: 10202144]
- 39.
- Chehab NH, Malikzay A, Appel M. et al. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article: PMC316357] [PubMed: 10673500]
- 40.
- Dumaz N, Meek DW. Serine 15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999;18:7002–7010. [PMC free article: PMC1171763] [PubMed: 10601022]
- 41.
- Ito A, Lai CH, Zhao X. et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001;20:1331–1340. [PMC free article: PMC145533] [PubMed: 11250899]
- 42.
- Khosravi R, Maya R, Gottlieb T. et al. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci USA. 1999;96:14973–14977. [PMC free article: PMC24757] [PubMed: 10611322]
- 43.
- Maya R, Balass M, Kim ST. et al. ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–1077. [PMC free article: PMC312683] [PubMed: 11331603]
- 44.
- Lowe SW. Activation of p53 by oncogenes. Endocr Relat Cancer. 1999;6:45–48. [PubMed: 10732786]
- 45.
- Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: Progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. [PubMed: 12573439]
- 46.
- Weber JD, Taylor LJ, Roussel MF. et al. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. [PubMed: 10559859]
- 47.
- Zhang Y, Xiong Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol Cell. 1999;3:579–591. [PubMed: 10360174]
- 48.
- Borden KL. Pondering the promyelocytic leukemia protein (PML) puzzle: Possible functions for PML nuclear bodies. Mol Cell Biol. 2002;22:5259–5269. [PMC free article: PMC133952] [PubMed: 12101223]
- 49.
- Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–170. [PubMed: 11832207]
- 50.
- Fogal V, Gostissa M, Sandy P. et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19:6185–6195. [PMC free article: PMC305840] [PubMed: 11080164]
- 51.
- Doucas V, Tini M, Egan DA. et al. Modulation of CREB binding protein function by the promyelocytic (PML) oncoprotein suggests a role for nuclear bodies in hormone signaling. Proc Natl Acad Sci USA. 1999;96:2627–2632. [PMC free article: PMC15819] [PubMed: 10077561]
- 52.
- Ferbeyre G, de StanchinaE, Querido E. et al. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14:2015–2027. [PMC free article: PMC316863] [PubMed: 10950866]
- 53.
- Pearson M, Carbone R, Sebastiani C. et al. PML regulates p53 acetylation and premature senescence induced by oncogenic ras. Nature. 2000;406:207–210. [PubMed: 10910364]
- 54.
- Pearson M, Pelicci PG. PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene. 2001;20:7250–7256. [PubMed: 11704853]
- 55.
- Stommel JM, Marchenko ND, Jimenez GS. et al. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999;18:1660–1672. [PMC free article: PMC1171253] [PubMed: 10075936]
- 56.
- Zhang Y, Xiong Y. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science. 2001;292:1910–1915. [PubMed: 11397945]
- 57.
- Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18:7288–7293. [PMC free article: PMC109310] [PubMed: 9819415]
- 58.
- Roth J, Dobbelstein M, Freedman DA. et al. Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 1998;17:554–564. [PMC free article: PMC1170405] [PubMed: 9430646]
- 59.
- Tao W, Levine AJ. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc Natl Acad Sci USA. 1999;96:3077–3080. [PMC free article: PMC15897] [PubMed: 10077639]
- 60.
- Boyd SD, Tsai KY, and JacksT. An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nature Cell Biol. 2000;2:563–568. [PubMed: 10980695]
- 61.
- Geyer RK, Yu ZK, Maki CG. The MDM2 RING-finger domain is required to promote p53 nuclear export. Nature Cell Biol. 2000;2:569–573. [PubMed: 10980696]
- 62.
- Gu J, Nie L, Wiederschain D. et al. Identification of p53 sequence elements that are required for MDM2-mediated nuclear export. Mol Cell Biol. 2001;21:8533–8546. [PMC free article: PMC100016] [PubMed: 11713288]
- 63.
- Lohrum MA, Woods DB, Ludwig RL. et al. C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol. 2001;21:8521–8532. [PMC free article: PMC100015] [PubMed: 11713287]
- 64.
- Moll UM, Riou G, Levine AJ. Two distinct mechanisms alter p53 in breast cancer: Mutation and nuclear exclusion. Proc Natl Acad Sci USA. 1992;89:7262–7266. [PMC free article: PMC49686] [PubMed: 1353891]
- 65.
- Sun XF, Carstensen JM, Zhang H. et al. Prognostic significance of cytoplasmic p53 oncoprotein in colorectal adenocarcinoma. Lancet. 1992;340:1369–1373. [PubMed: 1360088]
- 66.
- Bosari S, Viale G, Roncalli M. et al. p53 gene mutations, p53 protein accumulation and compartmentalization in colorectal adenocarcinoma. Am J Pathol. 1995;147:790–798. [PMC free article: PMC1870957] [PubMed: 7677190]
- 67.
- Ueda H, Ullrich SJ, Gangemi JD. et al. Functional inactivation but not structural mutation of p53 causes liver cancer. Nat Genet. 1995;9:41–47. [PubMed: 7704023]
- 68.
- Schlamp CL, Poulsen GL, Nork TM. et al. Nuclear exclusion of wild-type p53 in immortalized human retinoblastoma cells. J Natl Cancer Inst. 1997;89:1530–1536. [PubMed: 9337350]
- 69.
- Rodriguez-Lopez AM, Xenaki D, Eden TOB. et al. MDM2 mediated nuclear exclusion of p53 attenuates etoposide-induced apoptosis in neuroblastoma cells. Mol Pharmocol. 2000;59:135–143. [PubMed: 11125034]
- 70.
- Cordon-Cardo C, Latres E, Drobnjak M. et al. Molecular abnormalities of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res. 1994;54:794–799. [PubMed: 8306343]
- 71.
- Dubs-Poterszman MC, Tocque B, Wasylyk B. MDM2 transformation in the absence of p53 and abrogation of the p107 G1 cell-cycle arrest. Oncogene. 1995;11:2445–2449. [PubMed: 8570197]
- 72.
- Lundgren K, Montes de Oca Luna R, McNeill YB. et al. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. 1997;11:714–725. [PubMed: 9087426]
- 73.
- Daujat S, Neel H, Piette J. MDM2: Life without p53. Trends Genet. 2001;17:459–464. [PubMed: 11485818]
- 74.
- Davis PK, Dowdy SF. p73. Int J Biochem Cell Biol. 2001;33:935–939. [PubMed: 11470228]
- 75.
- Jost CA, Marin MC, Kaelin WGJr. p73 is a simian p53-related protein that can induce apoptosis. Nature. 1997;389:122–123. [PubMed: 9296498]
- 76.
- Yang A, Walker N, Bronson R. et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. [PubMed: 10716451]
- 77.
- Zeng X, Chen L, Jost CA. et al. MDM2 suppresses p73 function without promoting p73 degradation. Mol Cell Biol. 1999;19:3257–3266. [PMC free article: PMC84120] [PubMed: 10207051]
- 78.
- Gu J, Chen D, Rosenblum J. et al. Identification of a sequence element from p53 that signals for Mdm2-targeted degradation. Mol Cell Biol. 2000;20:1243–1253. [PMC free article: PMC85255] [PubMed: 10648610]
- 79.
- Dobbelstein M, Wienzek S, Konig C. et al. Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene. 1999;18:2101–2106. [PubMed: 10321734]
- 80.
- Flores ER, Tsai KY, Crowley D. et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564. [PubMed: 11932750]
- 81.
- Xiao ZX, Chen J, Levine AJ. et al. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature. 1995;375:694–698. [PubMed: 7791904]
- 82.
- Martin K, Trouche D, Hagemeier C. et al. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature. 1995;375:691–694. [PubMed: 7791903]
- 83.
- Hsieh JK, Chan FS, O'Connor DJ. et al. RB regulates the stability and the apoptotic function of p53 via MDM2. Mol Cell. 1999;3:181–193. [PubMed: 10078201]
- 84.
- Loughran O, Thangue NB. Apoptotic and growth-promoting activity of E2F modulated by MDM2. Mol Cell Biol. 2000;20:2186–2197. [PMC free article: PMC110835] [PubMed: 10688665]
- 85.
- Jackson MW, Berberich SJ. MdmX protects p53 from Mdm2-mediated degradation. Mol Cell Biol. 2000;20:1001–1007. [PMC free article: PMC85217] [PubMed: 10629057]
- 86.
- Parant J, Chavez-Reyes A, Little NA. et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. [PubMed: 11528400]
- 87.
- Gu J, Kawai H, Nie L. et al. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277:19251–19254. [PubMed: 11953423]
- 88.
- Marechal V, Ellenbaas B, Piette J. et al. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol. 1994;14:7414–7420. [PMC free article: PMC359276] [PubMed: 7935455]
- 89.
- Elenbaas B, Dobbelstein M, Roth J. et al. The MDM2 oncoprotein binds specifically to RNA through its RING finger domain. Mol Med. 1996;2:439–451. [PMC free article: PMC2230168] [PubMed: 8827714]
- 90.
- Juven-Gershon T, Shifman O, Unger T. et al. The Mdm2 oncoprotein interacts with the cell fate regulator Numb. Mol Cell Biol. 1998;18:3974–3982. [PMC free article: PMC108982] [PubMed: 9632782]
- 91.
- Wei X, Yu ZK, Ramalingam A. et al. Physical and functional interactions between PML and MDM2. J Biol Chem. 2003 [PubMed: 12759344]
- MDM2: RING Finger Protein and Regulator of p53 - Madame Curie Bioscience Databas...MDM2: RING Finger Protein and Regulator of p53 - Madame Curie Bioscience Database
- Cytogenetics of Human Sperm - Madame Curie Bioscience DatabaseCytogenetics of Human Sperm - Madame Curie Bioscience Database
- Patterning the Vertebrate Neural Plate by Wnt Signaling - Madame Curie Bioscienc...Patterning the Vertebrate Neural Plate by Wnt Signaling - Madame Curie Bioscience Database
- Phylogenomics and Molecular Evolution of Polyomaviruses - Madame Curie Bioscienc...Phylogenomics and Molecular Evolution of Polyomaviruses - Madame Curie Bioscience Database
- Rho Family GTPases and Cellular Transformation - Madame Curie Bioscience Databas...Rho Family GTPases and Cellular Transformation - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...