

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Interruption of blood flow to the liver is an unavoidable consequence of liver transplantation and resectional surgery. A growing body of experimental evidence suggests that reperfusion of the ischemic liver initiates hepatocellular injury and inflammation culminating in severe liver injury and organ dysfunction that may necessitate retransplantation. A large number of studies have implicated reactive oxygen species as potential mediators of this post-ischemic tissue injury. Recent developments in genetic engineering as well as chemical modeling have allowed for the production of novel superoxide dismutase (SOD) isoforms and low molecular weight SOD mimics with extended circulating half-lives or significant membrane permeabilities, respectively. Application of these newly developed free radical scavengers have shown promising results in animal models of liver I/R and may become powerful tools in the treatment of post-ischemic liver injury.
Introduction
Interruption of blood flow to the liver is an unavoidable consequence of liver transplantation and resectional surgery as well as hemorrhagic shock and thermal or burn injury.1 There is a growing body of experimental and clinical data to suggest that the ischemia and reperfusion (I/R) induced by these surgical procedures or pathophysiological events injures the liver and may ultimately lead to tissue dysfunction and possibly liver failure.2 Recent evidence suggests that I/R-induced liver injury occurs in two distinct phases consisting of an acute phase occurring within the first 6 hours of reperfusion followed by a later, sub-acute phase occurring from 6 to 24 hours post-ischemia.3 The acute phase is characterized by the polymorphonuclear leukocyte (PMN)-independent activation of resident Kupffer cells resulting in enhanced production of reactive oxygen species (ROS) in association with alterations in the redox state of the tissue in favor of a more oxidative environment.4-6 Historically, enhanced ROS production has been thought to injure tissue by virtue of its ability to degrade membrane lipids and/or proteins. However, more recent studies suggest that oxidative degradation of biomolecules is not produced at a time or in sufficient amounts to account for I/R-induced liver injury.7 In fact, it is becoming increasingly appreciated that ROS may mediate the early I/R-induced liver injury via their ability to enhance the expression of certain redox-sensitive genes known to be important in promoting hepatocellular injury. For example, several lines of evidence implicate I/R-induced ROS production in mediating hepatocellular injury by activating the NF-κB and AP-1-dependent expression of certain cytokines known to be involved in the pathophysiology of I/R-induced injury.8,9 Indeed, Kupffer cell-derived expression of tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and interleukin 12 (IL-12) have been implicated as important mediators of reperfusion injury in the liver.10-12
In contrast, the late, sub-acute phase of I/R injury has been shown to be a PMN-dependent process in which I/R-induced ROS generation is associated with cytokine and chemokine expression.13,14 There is good evidence to suggest that these inflammatory mediators promote the invasion of PMNs into the interstitium via the up-regulation of adhesion molecules and formation of chemotactic gradients.14,15 Adherent PMNs become metabolically activated and transmigrate through the sinusoidal and microvascular endothelial cells to the underlying hepatocytes where they generate additional reactive oxygen metabolites in conjunction with the release of extracellular matrix degrading enzymes such as collagenase and matrix metalloproteases.6,16,17 The net result is an amplification of the acute responses resulting in extensive inflammatory tissue injury.
Reactive Oxygen Species in the Post-Ischemic Liver
It is becoming increasingly apparent that ROS are crucial for the initiation and propagation of this post-ischemic liver injury. Several studies have demonstrated the protective effect of various antioxidants including N-acetyl cysteine (NAC) and glutathione (GSH), as well as others.18-20 While these studies have demonstrated a role for ROS in the generation of hepatic I/R injury, they have not identified the sources of these oxidants and free radicals, the specific species produced, or the mechanisms by which these reactive metabolites mediate their effects.
Sources of ROS in the Post-Ischemic Liver
There are several potential sources of ROS in the post-ischemic liver. Jaeschke and others have demonstrated a role for Kupffer cells in the increased oxidative stress to the tissue.6,17,21 Situated within the lumen of the sinusoids, the Kupffer cells are in direct contact with the endothelial surface (fig. 1). From this position, they are capable of releasing a variety of factors including ROS, pro-inflammatory cytokines, and oxidants into the circulation as well as directing them to the endothelial layer and the underlying hepatocytes. Using isolated Kupffer cells from post-ischemic rat livers, they were able to demonstrate an enhanced basal and stimulated production of superoxide (O2-), the one electron reduction product of oxygen (O2), from these cells when compared to sham operated controls. More recent studies have examined the effects of these cells in vivo. Using gadolinium chloride (GdCl3), a potent inhibitor of Kupffer cell function, several investigators have been able to demonstrate substantial protection against liver I/R injury during the neutrophil-independent phase of reperfusion.22,23 It therefore appears that Kupffer cell-derived oxidant production accounts for much of the observed post-ischemic liver injury. It should be noted, however, that Kupffer cells also represent a significant source of other inflammatory mediators including cytokines and prostanoids which are themselves capable of mediating some of the neutrophil-independent post-ischemic liver injury and may account for at least part of the observed protection.1
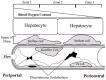
In addition to early Kupffer cell-derived oxidant production, neutrophils present in the late phase of injury have also been shown to contribute.21,24 Activated by factors produced during the acute phase, neutrophils can be seen adhering to the post-ischemic sinusoidal endothelium. 25 Once extravasated into the interstitium (i.e., the space of Disse), these cells release oxidants including O2- and hydrogen peroxide (H2O2) as well as produce hypochlorous acid (HOCl), a strong oxidant produced by myeloperoxidase (MPO).21,26 A number of studies have confirmed this hypothesis demonstrating the protective effect of inhibition of neutrophil infiltration on hepatocellular injury following I/R.14
While identification of the potential cellular sources of ROS are essential to our understanding of oxidant-induced liver injury, characterization of the specific enzymatic or nonenzymatic sources of these oxidants may prove more useful for potential therapeutic interventions. Early reports demonstrated the possible role of the xanthine dehydrogenase (XDH) to xanthine oxidase (XO) conversion in the synthesis of post-ischemic oxygen radicals.27,28 Xanthine oxidase is the cleavage (or oxidized) product of the molybdenum iron-sulfur containing flavin hydroxylase which normally catalyzes the conversion of hypoxanthine and xanthine to uric acid utilizing NAD+ as the final electron acceptor.29 During a variety of cell stresses including I/R, XDH can be converted either via irreversible proteolytic cleavage or reversible oxidation of specific sulfhydryl groups to its oxidase form now utilizing O2 as the final electron acceptor.30 Several recent reports would suggest that this system is indeed active in the post-ischemic liver. Using a model of total hepatic ischemia in rats, Muller and colleagues demonstrated significant increases in the circulating XO activity during the reperfusion period. In addition, these investigators showed significant protection using the XO inhibitor allopurinol following 24 hours of reperfusion.31 Additional studies have further identified XO as an important mediator of early midzonal and pericentral cellular injury in an isolated, perfused model of liver I/R injury.32 Even with these studies, the extent to which XO-derived oxidants contribute to post-ischemic liver injury remains controversial. This relates to the time-course of XDH to XO conversion and the ability of XO inhibitors to act as antioxidants themselves.33,34 Recent studies indicate that the conversion of XDH to XO occurs well after the initial cellular injury and that the absolute amount of XO produced following liver ischemia is minimal, leading some to question its potential role in the mediation of liver injury following I/R.33 Some of this conflicting data may, however, be explained by the cellular distribution of XO. Examination of isolated liver cells revealed a substantial increase in XDH to XO conversion, not in hepatocytes or endothelial cells, but in Kupffer cells.35 Many of the studies described above analyzed XO formation from total liver homogenates. Given that 95% of the liver volume is hepatocytes, assessment of total homogenates would reveal primarily the changes associated with hepatocytes. Kupffer cell XO may therefore represent a small, but important source of XO-derived O2- and H2O2.
In addition to XO, the hepatocyte and/or Kupffer cell mitochondria have been implicated in the generation of post-ischemic ROS. Following even brief periods of ischemia, mitochondrial electron transport malfunction has been observed.36,37 Normally, 4 electrons are passed down the electron transport chain to their terminal acceptor, O2, resulting in the formation of water. However, disruption in this transit has been reported following ischemia and is thought to contribute at least partially to the observed oxidant production and tissue injury.38 In addition to the direct effects of I/R on the mitochondrium, release of certain pro-inflammatory mediators like TNF-α and IL-1b have been associated with the stimulation of ROS formation from this source.39,40 Given the success of immunoneutralization of TNF-α and IL-1β, ROS generation from this site may be a principle mediator of early reperfusion injury.
A third potential source of O2- in the post-ischemic liver is NADPH oxidase. Present in phagocytic leukocytes (monocytes, macrophages, neutrophils) and possibly endothelial cells, this system is capable of generating large quantities of O2- via the reduction of O2 utilizing NADPH as the electron donor.41 This enzyme is composed of several protein sub-units including the membrane spanning gp91 and p22 sub-units, the cytosolic p47 and p67 sub-units as well as the gp91-associated rac-1 GTPase protein.41 Upon stimulation by a variety of factors including bacterial infection, NADPH oxidase is assembled at the cell membrane allowing for the direct release of O2- to the extracellular space.42 Either enzyme catalyzed or spontaneous dismutation of this O2- forms H2O2 which is capable of mediating a variety of effects including induction of an oxidant stress to the surrounding tissue.43,44 However, little data exists demonstrating a role for this complex in the mediation of post-ischemic liver injury. This has been due, in part, to the lack of specific inhibitors for in vivo assessment of its function. Recently, mice were rendered genetically deficient in a functional gp91 sub-unit (gp91-/-) and therefore the capacity to produce O2- via this system.45 Initial studies verified the lack of a functional respiratory burst in neutrophils isolated from these animals proving that the disruption was effective and that this system represents the primary means by which these cells generate ROS.45 Ozaki and others have recently examined this complex in a mouse model of liver ischemia for 45 minutes.46 In their studies, gp91-/- mice were not protected from the damaging effects of ischemia at either 1, 3, or 8 hours of reperfusion as assessed by liver enzyme release and histopathology when compared to wild type controls. Subjection of these same mice to hepatic ischemia for 90 minutes followed by reperfusion for 6 hours has, however, demonstrated substantial and significant protection against the post-ischemic liver injury when compared to their wild type controls.47 These data would indicate possible differences in the mechanisms of post-ischemic liver injury following brief versus extended periods of ischemia. Nevertheless, it appears that O2- or one of its products from this source is an important mediator of I/R-induced liver injury and this may therefore represent an important therapeutic target.
Yet other possible sources of ROS are the sub-cellular organelles known as microsomes. The liver plays a primary role in the detoxification of a number of endogenously produced as well as ingested and absorbed compounds. To accomplish these tasks, it is heavily fortified with a number of oxidases including the family of cytochrome p450 enzymes. Held primarily within the microsome, these enzymes consume nearly 30% of the hepatocytes O2 and, in the process, produce ROS. While their role in hepatic I/R has not been fully examined, they remain as a potential high output source of ROS within the post-ischemic liver and may therefore contribute extensively to the observed injury. From this discussion, it is apparent that the liver is capable of generating large amounts of ROS from a number of different sources.
Mechanisms of ROS-Induced Liver Injury
It has been appreciated for some time that oxidants and free radicals may damage cellular components leading to cell death. From these initial studies, it was determined that oxidants/free radicals were capable of modifying a number of cellular constituents including membrane proteins and lipids as well as nucleic acids.48 While biochemical studies have demonstrated the relatively poor oxidizing capabilities of certain species such as O2- or H2O2, interaction of these molecules with certain transition metals or metal containing proteins results in the formation of extremely strong, although short lived, oxidant species. For example, interaction of O2- with ferric iron (Fe3+) in the presence of H2O2 can lead to the formation of the potent oxidant, hydroxyl radical (OH) through the Fenton reaction.49 Direct oxidation of cellular constituents by this pathway was held as the primary means of post-ischemic liver injury for some time, however, recent experimental data would indicate otherwise. Jaeschke and associates examined the oxidation of membrane lipids as a marker of direct post-ischemic cellular oxidation7 Their data revealed low levels of cell membrane oxidation following liver I/R. This is not surprising given the relatively low levels of free iron in the liver or the circulation. Without this catalytic component, strong oxidants (i.e., OH.) may not be produced thereby inhibiting the oxidative capacity of reactive species such as O2- and H2O2. To further characterize the ability of lipid peroxidation to injure the liver, this same group infused tert-butyl hydroperoxide (tBH), a stable peroxide derivative capable of oxidizing membrane lipids while being resistant to degradation by catalase. In this system, tBH was able to induce tissue levels of lipid peroxidation much greater than those found in post-ischemic liver, in the absence of significant hepatocellular damage. Given these data, it is apparent that ROS may act to damage the post-ischemic liver through other indirect mechanisms.
Even though widespread oxidation of cellular lipids may not be a principal mechanism of ROS-induced liver injury, interaction of these species, and in particular O2-, with other cellular constituents and metabolites still remains a possibility. O2- may interact with other intracellular as well as extracellular radical species including nitric oxide (NO). NO is a short-lived radical species generated by the conversion of L-arginine to L-citrulline in the presence of NADPH by a family of enzymes known as nitric oxide synthases. NO was originally identified as the endothelium-derived relaxing factor, a molecule capable of interacting with guanylyl cyclase leading to the relaxation of vascular smooth muscle.50 Since its discovery as a mediator of vascular reactivity, this molecule has been shown to interact with and inhibit the action of a number of cellular constituents including members of the caspase family and certain transcription factors as well as mediate direct cellular damage depending on the levels produced51. One possible mechanism for NO-mediated cellular damage is through its interaction with O2-. In this reaction, NO reacts with O2- forming peroxynitrite (ONOO-) and its conjugate peroxynitrous acid (ONOOH): rapid and spontaneous decomposition of ONOOH to yield nitrate (NO3-) and nitrite (NO2-) produces one or more potent oxidants as intermediates capable of oxidizing and nitrating a number of biological molecules. If O2- mediates its apparent damaging effects through production of ONOO-, then one would expect protection from inhibition or genetic deletion of NOS. This is not the case however. Mice rendered genetically deficient in endothelial and inducible NOS demonstrate substantial increases in post-ischemic liver injury.52-54 Further, infusion of authentic ONOO- failed to increase liver injury, but instead actually reduced the levels of liver injury observed following ischemia although these data are difficult to interpret given the relative instability of ONOO- at physiological pH.55 However, interaction and degradation of NO by O2- within the post-ischemic liver may serve to remove an important hepatoprotective molecule. For example, reductions in the availability of NO in the liver are known to increase microvascular pressure through the liver via decreases in vessel diameter.56 This occurs because, under normal circumstances, NO counteracts other vasoconstrictor substances including endothelin and phenylephrine.57,58 Inhibition or genetic disruption of the production of NO by eNOS results in an imbalance in the vasorelaxer/vasoconstrictor ratio leading to contraction of the extrasinusoidal hepatic stellate (or Ito) cells (fig. 1).58 These cells, in addition to being important storage cells for certain vitamins and lipids and sources of certain growth factors and collagen synthesis, are capable of contracting thereby reducing the diameter of the sinusoid. Support for this concept is found in studies using endothelin receptor antagonists as well as endothelin receptor agonists prior to liver I/R which demonstrate the importance of sinusoidal perfusion in the post-ischemic liver.59,60 The action of NO on stellate cells may therefore represent an important perfusion-related function of NO and interaction of this molecule with O2- would effectively remove it from the system.
Intracellular O2- generation may also interact with and modify a number of cellular enzymes. For example, O2- has been shown to interact with aconitase, an enzymatic component of the citric acid cycle responsible for the conversion of citrate to isocitrate.61 Inactivation of this enzyme can result in significant reductions in cellular ATP production. If inactivation of this and other cellular enzymes occurs to a significant level, as it may during I/R, then hepatocellular dysfunction, injury, and death may occur.
Additional experimental data suggest that ROS may act to modulate the expression of certain pro-inflammatory mediators.62 Activated shortly after reperfusion, nuclear factor kappa B (NF-κB) is capable of activating the transcription of numerous genes including cytokines, adhesion molecules, and certain free radical producing enzymes (e.g., iNOS).26,63-66 Several studies have identified this and other transcription factors as oxidant sensitive.67 Oxidative stress is thought to activate a class of kinases known as inhibitory kappa B kinases (IKK) which phosphorylate the endogenous inhibitor of NF-κB, inhibitory kappa B (IκB).67,68 Once phosphorylated, IκB is targeted for polyubiquination and subsequent degradation by the 26S proteasome. Through this mechanism, ROS and perhaps O2- are capable of indirectly modulating the inflammatory process, actions independent of widespread cellular protein and lipid oxidation. In addition, recent experimental data would indicate that NO may act to inhibit the activation of NF-κB through S-nitrosylation the p50 sub-unit of NF-κB.69 Inactivation of NO by O2- would therefore lead to enhanced activation of this factor leading to increased expression of potentially damaging pro-inflammatory mediators. Taken together, it appears that ROS are capable, through a variety of mechanisms, of causing directly or indirectly hepatocellular injury and organ dysfunction.
Therapeutic Strategies against Post-Ischemic O2- Formation
From the above discussion, it is apparent that ROS and in particular O2- may be capable of directly or indirectly mediating a substantial portion of post-ischemic tissue injury. Early studies utilized nonspecific radical scavengers to implicate these oxidants in the initiation and propagation of tissue injury. Direct measurement of ROS formation by resident and infiltrating immune cells further implicated these species in liver I/R injury. Therefore, if ROS and in particular O2- is indeed important to the initiation and propagation of post-ischemic tissue injury then scavenging of this species should result in substantial reductions in liver injury. In support of this are recent studies demonstrating the profound protective effect of adenoviral transfection of manganese superoxide dismutase (Mn-SOD) in the post-ischemic liver. In this study, selective scavenging of mitochondrial generated O2- resulted in significant reductions in serum enzyme release and NF-κB activation.9
While the use of adenoviral transfection of various SOD enzymes provides profound mechanistic information regarding the source and role of O2- in the post-ischemic liver, their clinical and therapeutic utility are limited due to the known hepatocellular inflammation and injury induced by these adenoviral vectors.70 Exogenous administration of antioxidant enzymes may therefore provide the best therapeutic avenue at this time. Unfortunately, native proteins such as Cu/Zn-SOD or Mn-SOD have extremely short half-lives (<7 minutes) in vivo rendering their use less than effective.71 However, chemical modifications of these proteins such as the attachment of side chains like polyethylene glycol have proved useful in extending their life span in the circulation.18,19 Utilizing glycosylated forms of SOD thought to be taken up by hepatocytes and/or Kupffer cells, Fujita and colleagues were indeed able to significantly attenuate post-ischemic liver injury in the mouse.72 More recent studies by Yabe and associates have confirmed these findings. SOD conjugated to either galactose or mannose resulted in a 50% reduction in serum ALT levels following 30 minutes of ischemia and 1 hour of reperfusion.18 Further, these studies identified the mannosylated form of SOD to be taken up by nonparenchymal cells (i.e., endothelial cells, Kupffer cells, stellate cells, pit cells) resulting in substantial hepatocellular protection. These data would indicate the ability of ROS generated within Kupffer cells or endothelial cells to directly or indirectly mediate liver I/R injury.
Other attempts have been made to increase the half-life of antioxidant enzymes as well. For example, Gao and colleagues developed a modified, polycationic form of human Mn-SOD (pcMn-SOD) containing a 26 amino acid which is capable of binding to the heparan sulfate moieties on the microvascular endothelium as well as the interstitial matrix.73 Similar in structure to the endogenously produced extracellular SOD, this enzyme has been shown to have a greatly extended half-life (>30 hours).73 In contrast to those modified forms of SOD described above, this enzyme remains on the exterior of endothelial cells and hepatocytes allowing for the study of the effects of extracellularly generated O2-. Pretreatment of mice with this pcMn-SOD resulted in substantial protection against the post-ischemic liver injury incurred following 45 or 90 minutes of ischemia and 6 hours of reperfusion.47 The protection observed with this enzyme also correlated well with a dramatic and significant reduction in serum TNF-α, a known mediator of liver cell injury during I/R.
Taking together, these data suggest that intracellular and extracellular O2- generation contributes extensively to the observed tissue injury. Recently, a novel class of nonenzymatic, low molecular weight, membrane permeable, manganese containing SOD mimetics have been created to circumvent some of the problems associated with native or modified enzyme administration. As discussed earlier, administration of native antioxidant enzymes is not therapeutically effective due to their extremely short half-life. Further, administration of large amounts of protein, native or modified, in humans has been associated with immunological complications resulting in their withdrawal from patient use. These manganese-containing protoporphyrin compounds, now in clinical trials, are devoid of these classical side effects. In experimental models of intestinal I/R, these compounds have proven extremely effective. For example, Salvemini et al were able to significantly attenuate post-ischemic neutrophil infiltration in the rat small intestine following 45 minutes of superior mesenteric artery occlusion.62,74 Further, these mimics have been associated with the reduction of TNF-α expression following splanchnic I/R injury. The use of these compounds in hepatic ischemia has only recently been examined. Treatment of mice with M40401 (Metaphore Pharmaceuticals), a prototypical SOD mimetic, at a dose of 3 mg/kg, 15 minutes prior to the initiation of 90 minutes of ischemia resulted in significant attenuation of post-ischemic liver injury at 6 hours of reperfusion as measured by serum ALT release when compared to control (9836.34 ± 998.34 vs. 5628.13 ± 1617.28 for vehicle vs. M40401; p<0.05).47 Additional studies have been performed utilizing another manganese/ porphyrin SOD mimic and have shown dramatic protective effects. For example, treatment of mice with 3 mg/kg of AEOL 10150 (Aeolus Pharmaceuticals, (fig. 2) significantly reduced the post-ischemic rise in serum ALT following both 45 and 90 minutes of ischemia and 6 hours of reperfusion (90 minutes of ischemia: 9836.34 ± 998.34 vs. 5940.48 ± 1342.38 for vehicle vs. AEOL 10150; p<0.05; 45 minutes of ischemia: 974.301 ± 186.47 vs. 359.49 ± 63.03 for vehicle v. AEOL 10150; p<0.05).47 These data, when compared to those obtained with the pcMn-SOD, suggest that both intracellular and extracellular O2- generation are important in the pathogenesis of liver I/R injury and that use of low molecular weight SOD mimics may be as effective in treating I/R-induced liver injury.
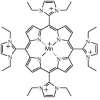
Figure 2
Structure of AEOL-10150 SOD mimetic.
Conclusions
It is now well appreciated that ROS and in particular O2- directly or indirectly mediate a substantial portion of post-ischemic liver injury. Whether through interactions with and decomposition of protective factors like NO or by direct inactivation of cellular enzymes like aconitase, O2- is capable of initiating and propagating post-ischemic hepatocellular injury and tissue inflammation. Novel SOD isoforms and mimics have been developed which overcome the inherent problems associated with native antioxidant enzymes and scavengers. These new compounds may prove useful in the treatment of post-ischemic liver injury.
References
- 1.
- Fan C, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver. J Mol Med. 1999;77(8):577–592. [PubMed: 10543390]
- 2.
- Lemasters JJ, Thurman RG. The many facets of reperfusion injury. Gastroenterology. 1995;108(4):1317–1320. [PubMed: 7698602]
- 3.
- Lichtman SN, Lemasters JJ. Role of cytokines and cytokine-producing cells in reperfusion injury to the liver. Semin Liver Dis. 1999;19(2):171–187. [PubMed: 10422199]
- 4.
- Wink DA, Feelisch M, Fukuto J. et al. The cytotoxicity of nitroxyl: Possible implications for the pathophysiological role of no. Arch Biochem Biophys. 1998;351(1):66–74. [PubMed: 9501920]
- 5.
- Grisham MB. Interaction between nitric oxide and superoxide: Role in modulating leukocyte adhesion in the postischemic microvasculature. Transplant Proc. 1995;27(5):2842–2843. [PubMed: 7482938]
- 6.
- Jaeschke H, Farhood A. Neutrophil and kupffer cell-induced oxidant stress and ischemia- reperfusion injury in rat liver. Am J Physiol. 1991;260(3 Pt 1):G355–G362. [PubMed: 2003603]
- 7.
- Mathews WR, Guido DM, Fisher MA. et al. Lipid peroxidation as molecular mechanism of liver cell injury during reperfusion after ischemia. Free Radic Biol Med. 1994;16(6):763–770. [PubMed: 8070679]
- 8.
- Zhou W, Zhang Y, Hosch MS. et al. Subcellular site of superoxide dismutase expression differentially controls AP-1 activity and injury in mouse liver following ischemia/reperfusion. Hepatology. 2001;33(4):902–914. [PubMed: 11283855]
- 9.
- Zwacka RM, Zhou W, Zhang Y. et al. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-kappaB activation. Nat Med. 1998;4(6):698–704. [PubMed: 9623979]
- 10.
- Colletti LM, Kunkel SL, Walz A. et al. The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 1996;23(3):506–514. [PubMed: 8617430]
- 11.
- Lentsch AB, Yoshidome H, Kato A. et al. Requirement for interleukin-12 in the pathogenesis of warm hepatic ischemia/reperfusion injury in mice. Hepatology. 1999;30(6):1448–1453. [PubMed: 10573524]
- 12.
- Shito M, Wakabayashi G, Ueda M. et al. Interleukin 1 receptor blockade reduces tumor necrosis factor production, tissue injury, and mortality after hepatic ischemia- reperfusion in the rat. Transplantation. 1997;63(1):143–148. [PubMed: 9000676]
- 13.
- Jaeschke H. Mechanisms of reperfusion injury after warm ischemia of the liver. J Hepatobiliary Pancreat Surg. 1998;5(4):402–408. [PubMed: 9931389]
- 14.
- Martinez-Mier G, Toledo-Pereyra LH, Ward PA. Adhesion molecules in liver ischemia and reperfusion. J Surg Res. 2000;94(2):185–194. [PubMed: 11104660]
- 15.
- Simpson KJ, Lukacs NW, Colletti L. et al. Cytokines and the liver. J Hepatol. 1997;27(6):1120–1132. [PubMed: 9453442]
- 16.
- Jaeschke H, Smith CW. Cell adhesion and migration. III. Leukocyte adhesion and transmigration in the liver vasculature. Am J Physiol. 1997;273(6 Pt 1):G1169–G1173. [PubMed: 9435541]
- 17.
- Jaeschke H, Bautista AP, Spolarics Z. et al. Superoxide generation by kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic Res Commun. 1991;15(5):277–284. [PubMed: 1666625]
- 18.
- Yabe Y, Nishikawa M, Tamada A. et al. Targeted delivery and improved therapeutic potential of catalase by chemical modification: Combination with superoxide dismutase derivatives. J Pharmacol Exp Ther. 1999;289(2):1176–1184. [PubMed: 10215702]
- 19.
- Yabe Y, Kobayashi N, Nishihashi T. et al. Prevention of neutrophil-mediated hepatic ischemia/ reperfusion injury by superoxide dismutase and catalase derivatives. J Pharmacol Exp Ther. 2001;298(3):894–899. [PubMed: 11504782]
- 20.
- Fukuzawa K, Emre S, Senyuz O. et al. N-acetylcysteine ameliorates reperfusion injury after warm hepatic ischemia. Transplantation. 1995;59(1):6–9. [PubMed: 7839430]
- 21.
- Jaeschke H, Bautista AP, Spolarics Z. et al. Superoxide generation by neutrophils and kupffer cells during in vivo reperfusion after hepatic ischemia in rats. J Leukoc Biol. 1992;52(4):377–382. [PubMed: 1328439]
- 22.
- Mosher B, Dean R, Harkema J. et al. Inhibition of kupffer cells reduced CXC chemokine production and liver injury. J Surg Res. 2001;99(2):201–210. [PubMed: 11469888]
- 23.
- Bremer C, Bradford BU, Hunt KJ. et al. Role of kupffer cells in the pathogenesis of hepatic reperfusion injury. Am J Physiol. 1994;267(4 Pt 1):G630–G636. [PubMed: 7943328]
- 24.
- Farhood A, McGuire GM, Manning AM. et al. Intercellular adhesion molecule 1 (ICAM-1) expression and its role in neutrophil-induced ischemia-reperfusion injury in rat liver. J Leukoc Biol. 1995;57(3):368–374. [PubMed: 7884306]
- 25.
- Yadav SS, Howell DN, Steeber DA. et al. P-selectin mediates reperfusion injury through neutrophil and platelet sequestration in the warm ischemic mouse liver. Hepatology. 1999;29(5):1494–1502. [PubMed: 10216134]
- 26.
- Lentsch AB, Kato A, Yoshidome H. et al. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32(2):169–173. [PubMed: 10915720]
- 27.
- Grisham MB, Hernandez LA, Granger DN. Xanthine oxidase and neutrophil infiltration in intestinal ischemia. Am J Physiol. 1986;251(4 Pt 1):G567–G574. [PubMed: 3020994]
- 28.
- Granger DN, McCord JM, Parks DA. et al. Xanthine oxidase inhibitors attenuate ischemia-induced vascular permeability changes in the cat intestine. Gastroenterology. 1986;90(1):80–84. [PubMed: 3753555]
- 29.
- Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am J Physiol. 1988;255(6 Pt 2):H1269–H1275. [PubMed: 3059826]
- 30.
- McKelvey TG, Hollwarth ME, Granger DN. et al. Mechanisms of conversion of xanthine dehydrogenase to xanthine oxidase in ischemic rat liver and kidney. Am J Physiol. 1988;254(5 Pt 1):G753–G760. [PubMed: 3163235]
- 31.
- Muller MJ, Vollmar B, Friedl HP. et al. Xanthine oxidase and superoxide radicals in portal triad crossclamping- induced microvascular reperfusion injury of the liver. Free Radic Biol Med. 1996;21(2):189–197. [PubMed: 8818634]
- 32.
- Marotto ME, Thurman RG, Lemasters JJ. Early midzonal cell death during low-flow hypoxia in the isolated, perfused rat liver: Protection by allopurinol. Hepatology. 1988;8(3):585–590. [PubMed: 3371875]
- 33.
- Frederiks WM, Bosch KS. The proportion of xanthine oxidase activity of total xanthine oxidoreductase activity in situ remains constant in rat liver under various (patho)physiological conditions. Hepatology. 1996;24(5):1179–1184. [PubMed: 8903395]
- 34.
- Das DK, Engelman RM, Clement R. et al. Role of xanthine oxidase inhibitor as free radical scavenger: A novel mechanism of action of allopurinol and oxypurinol in myocardial salvage. Biochem Biophys Res Commun. 1987;148(1):314–319. [PubMed: 2823807]
- 35.
- Wiezorek JS, Brown DH, Kupperman DE. et al. Rapid conversion to high xanthine oxidase activity in viable kupffer cells during hypoxia. J Clin Invest. 1994;94(6):2224–2230. [PMC free article: PMC330048] [PubMed: 7989578]
- 36.
- Lucas DT, Szweda LI. Cardiac reperfusion injury: Aging, lipid peroxidation, and mitochondrial dysfunction. Proc Natl Acad Sci USA. 1998;95(2):510–514. [PMC free article: PMC18450] [PubMed: 9435222]
- 37.
- Schild L, Reinheckel T, Wiswedel I. et al. Short-term impairment of energy production in isolated rat liver mitochondria by hypoxia/reoxygenation: Involvement of oxidative protein modification. Biochem J. 1997;328(Pt 1):205–210. [PMC free article: PMC1218907] [PubMed: 9359854]
- 38.
- Saris NE, Eriksson KO. Mitochondrial dysfunction in ischaemia-reperfusion. Acta Anaesthesiol Scand. 1995;107(Suppl):171–176. [PubMed: 8599272]
- 39.
- Shirasugi N, Wakabayashi G, Shimazu M. et al. Up-regulation of oxygen-derived free radicals by interleukin-1 in hepatic ischemia/reperfusion injury. Transplantation. 1997;64(10):1398–1403. [PubMed: 9392301]
- 40.
- Scales WE, Campbell JrDA, Green ME. et al. Hepatic ischemia/reperfusion injury: Importance of oxidant/tumor necrosis factor interactions. Am J Physiol. 1994;267(6 Pt 1):G1122–G1127. [PubMed: 7810659]
- 41.
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ Res. 2000;86(5):494–501. [PubMed: 10720409]
- 42.
- Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397(2):342–344. [PubMed: 11795892]
- 43.
- Gonzalez-Flecha B, Reides C, Cutrin JC. et al. Oxidative stress produced by suprahepatic occlusion and reperfusion. Hepatology. 1993;18(4):881–889. [PubMed: 8406364]
- 44.
- Gonzalez-Flecha B, Cutrin JC, Boveris A. Time course and mechanism of oxidative stress and tissue damage in rat liver subjected to in vivo ischemia-reperfusion. J Clin Invest. 1993;91(2):456–464. [PMC free article: PMC287955] [PubMed: 8432855]
- 45.
- Pollock JD, Williams DA, Gifford MA. et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9(2):202–209. [PubMed: 7719350]
- 46.
- Ozaki M, Deshpande SS, Angkeow P. et al. Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo. FASEB J. 2000;14(2):418–429. [PubMed: 10657998]
- 47.
- Hines IN, Hoffman JM, Scheerens H. et al. Regulation of postischemic liver injury following different durations of ischemia. Am J Physiol Gastrointest Liver Physiol. 2003;284(3):G536–545. [PubMed: 12444015]
- 48.
- Aust SD, Morehouse LA, Thomas CE. Role of metals in oxygen radical reactions. J Free Radic Biol Med. 1985;1(1):3–25. [PubMed: 3013969]
- 49.
- Grisham MB, Granger DN, Lefer DJ. Modulation of leukocyte-endothelial interactions by reactive metabolites of oxygen and nitrogen: Relevance to ischemic heart disease. Free Radic Biol Med. 1998;25(4-5):404–433. [PubMed: 9741579]
- 50.
- Ignarro LJ, Byrns RE, Buga GM. et al. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res. 1987;61(6):866–879. [PubMed: 2890446]
- 51.
- Grisham MB, Jourd'heuil D, Wink DA. Nitric oxide. I. Physiological chemistry of nitric oxide and its metabolites: Implications in inflammation. Am J Physiol. 1999;276(2 Pt 1):G315–G321. [PubMed: 9950804]
- 52.
- Hines IN, Harada H, Bharwani S. et al. Enhanced post-ischemic liver injury in iNOS-deficient mice: A cautionary note. Biochem Biophys Res Commun. 2001;284(4):972–976. [PubMed: 11409889]
- 53.
- Kawachi S, Hines IN, Laroux FS. et al. Nitric oxide synthase and postischemic liver injury. Biochem Biophys Res Commun. 2000;276(3):851–854. [PubMed: 11027558]
- 54.
- Hines IN, Kawachi S, Harada H. et al. Role of nitric oxide in liver ischemia and reperfusion injury. Mol Cell Biochem. 2002;234-235(1-2):229–237. [PubMed: 12162439]
- 55.
- Liu P, Xu B, Quilley J. et al. Peroxynitrite attenuates hepatic ischemia-reperfusion injury. Am J Physiol Cell Physiol. 2000;279(6):C1970–C1977. [PubMed: 11078713]
- 56.
- Mittal MK, Gupta TK, Lee FY. et al. Nitric oxide modulates hepatic vascular tone in normal rat liver. Am J Physiol. 1994;267(3 Pt 1):G416–G422. [PubMed: 7943239]
- 57.
- Moy JA, Bates JN, Fisher RA. Effects of nitric oxide on platelet-activating fa. J Biol Chem. 1991;266(13):8092–8096. [PubMed: 1673678]
- 58.
- Rockey DC. Hepatic blood flow regulation by stellate cells in normal and injured liver. Semin Liver Dis. 2001;21(3):337–349. [PubMed: 11586464]
- 59.
- Kitayama Y, Yamanaka N, Kawamura E. et al. Hepatoprotective effect of the endothelin receptor antagonist TAK-044 against ischemia-reperfusion injury in the canine liver. Hepatology. 1997;25(4):938–942. [PubMed: 9096601]
- 60.
- Yokoyama Y, Baveja R, Sonin N. et al. Altered endothelin receptor subtype expression in hepatic injury after ischemia/reperfusion. Shock. 2000;13(1):72–78. [PubMed: 10638673]
- 61.
- Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269(47):29405–29408. [PubMed: 7961919]
- 62.
- Cuzzocrea S, Mazzon E, Dugo L. et al. Protective effects of a new stable, highly active SOD mimetic, M40401 in splanchnic artery occlusion and reperfusion. Br J Pharmacol. 2001;132(1):19–29. [PMC free article: PMC1572533] [PubMed: 11156557]
- 63.
- Janssen-Heininger YM, Poynter ME, Baeuerle PA. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappa B. Free Radic Biol Med. 2000;28(9):1317–1327. [PubMed: 10924851]
- 64.
- Kokura S, Wolf RE, Yoshikawa T. et al. Molecular mechanisms of neutrophil-endothelial cell adhesion induced by redox imbalance. Circ Res. 1999;84(5):516–524. [PubMed: 10082473]
- 65.
- Weber C, Erl W, Pietsch A. et al. Antioxidants inhibit monocyte adhesion by suppressing nuclear factor- kappa B mobilization and induction of vascular cell adhesion molecule-1 in endothelial cells stimulated to generate radicals. Arterioscler Thromb. 1994;14(10):1665–1673. [PubMed: 7522548]
- 66.
- Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem. 1994;269(7):4705–4708. [PubMed: 7508926]
- 67.
- Li N, Karin M. Is NF-kappaB the sensor of oxidative stress? FASEB J. 1999;13(10):1137–1143. [PubMed: 10385605]
- 68.
- Karin M, Ben NeriahY. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. [PubMed: 10837071]
- 69.
- Marshall HE, Stamler JS. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry. 2001;40(6):1688–1693. [PubMed: 11327828]
- 70.
- Muruve DA, Barnes MJ, Stillman IE. et al. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum Gene Ther. 1999;10(6):965–976. [PubMed: 10223730]
- 71.
- Gao B, Flores SC, McCord JM. A site-directed mutant of Cu,Zn-superoxide dismutase modeled after native extracellular superoxide dismutase. Biol Trace Elem Res. 1995;47(1-3):95–100. [PubMed: 7779581]
- 72.
- Fujita T, Nishikawa M, Tamaki C. et al. Targeted delivery of human recombinant superoxide dismutase by chemical modification with mono- and polysaccharide derivatives. J Pharmacol Exp Ther. 1992;263(3):971–978. [PubMed: 1469654]
- 73.
- Gao B, Flores SC, Leff JA. et al. Synthesis and anti-inflammatory activity of a chimeric recombinant superoxide dismutase: SOD2/3. Am J Physiol Lung Cell Mol Physiol. 2003;284(6):L917–925. [PubMed: 12736188]
- 74.
- Salvemini D, Wang ZQ, Zweier JL. et al. A nonpeptidyl mimic of superoxide dismutase with therapeutic activity in rats. Science. 1999;286(5438):304–306. [PubMed: 10514375]
- Role of Superoxide in Post-Ischemic Liver Injury - Madame Curie Bioscience Datab...Role of Superoxide in Post-Ischemic Liver Injury - Madame Curie Bioscience Database
- Structure and Replication of Hepatitis Delta Virus RNA - Madame Curie Bioscience...Structure and Replication of Hepatitis Delta Virus RNA - Madame Curie Bioscience Database
- Neural Crest and the Development of the Enteric Nervous System - Madame Curie Bi...Neural Crest and the Development of the Enteric Nervous System - Madame Curie Bioscience Database
- Calcium Signaling During Phagocytosis - Madame Curie Bioscience DatabaseCalcium Signaling During Phagocytosis - Madame Curie Bioscience Database
- Regulation of Cell Adhesion Responses by Abl Family Kinases - Madame Curie Biosc...Regulation of Cell Adhesion Responses by Abl Family Kinases - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...