

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Power Laws in Genomic Quantities
A few years ago it was noticed that the distributions of gene-family sizes in fully-sequenced genomes follow power-law distributions.1,2 Since then different authors have shown that there is in fact a large array of genomic features that show power law distributions. Almost all of these concern the distributions of genomic features within a single genome. For instance, it is shown in reference 3 that the number of genomic occurrences of DNA words, protein folds, superfamilies, and families all follow power-law distributions. Power-law distributions are also found in the structure of the ‘protein universe’;4 the number of protein families per fold is power-law distributed, and so is the number of different assigned biological functions per fold.3 Power-laws also appear in the structure of cellular interaction and regulatory networks. For example, the number of genes that a given gene interacts with is power-law distributed. This holds both when one defines ‘interaction’ between genes on the level of the proteins that they encode5,7 or if one defines it at the level of coregulation of the expression of the genes.8 The experimental data on transcription regulatory networks is rather incomplete but they also suggest that the number of genes regulated per transcription factor might have power-law tails.9,10 Finally, power-laws also appear in cellular metabolic networks; the number of substrates that any given substrate interacts with is power-law distributed.11,12
Comparing Genomic Features across Genomes
Note that almost all the power-law distributions just mentioned refer to statistics that are taken over a single genome or cellular network. The statistics of genomic features across genomes has been much less (if at all) investigated. To a large extent this may be because until recently there simply weren't enough fully-sequenced genomes to obtain meaningful statistics across genomes. However, this situation is changing rapidly.
There are currently about 150 fully-sequenced microbial genomes in GenBank and this number appears to grow exponentially as I have shown in reference 13.
Figure 1 shows an updated plot of the number of fully sequenced microbial genomes as a function of time (see Methods section). The current number of available microbial genomes is only large enough to allow for meaningful cross-genome comparisons of the most basic statistics of gene-content and organization and this is what I will focus on in this chapter. However, the exponential fit in Figure 1 predicts that the number of sequenced genomes doubles roughly every 17 months. This implies that by 2010 we may have as many as 3000 fully-sequenced microbial genomes available. It is therefore clear that much more detailed comparative genomic analyses than the ones presented in this chapter will become possible over the next decade.
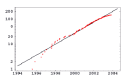
Figure 1
The number of fully-sequenced microbial genomes submitted to the genbank database as a function of time (in years). The vertical axis is shown on a logarithmic scale. The black line is the least-square fit to an exponential. n = 2(t-1993.4)/1.38
In reference 13 I compared the number of genes in high-level functional categories across all sequenced genomes and showed that they follow power-laws as a function of the total number of genes in the genome. In this chapter I will recapitulate these results and augment them in several ways. In particular, I have extended the analysis to all functional categories that are represented by at least 1 gene in each bacterial genome and have recalculated the observed exponents based on the latest genomic data. Second, I will go into more detail regarding the implications of the observed scaling laws for the general organization of gene-content across genomes and discuss an evolutionary model that relates the observed scaling behavior in gene content to fundamental constants of the evolutionary process. Finally, I will discuss theoretical explanations for the category of transcription regulatory genes which shows approximately quadratic scaling with the total number of genes in the genome.
Scaling in Functional Gene-Content Statistics
To count and compare the number of genes in different functional categories for all sequenced genomes one needs to first define a set of functional categories and then annotate all genomes in terms of these functional categories. I used the biological process hierarchy of the Gene Ontology14 to define functional categories, and Interpro annotations of fully-sequenced genomes to associate genes with GO categories. The details of the annotation procedure are described in the Methods section. The result is a count of the number of genes associated with each of the GO categories in the biological process hierarchy for each of the sequenced genomes.
The set of genomes in this study consists of 116 bacteria, 15 archaea, and 10 eukaryota. In this chapter I will focus solely on the bacterial data since this is the only kingdom for which there is sufficient data to obtain meaningful statistics. The reader is referred to reference 13 for a discussion of the observed scaling laws in archaea and eukaryota.
There are 154 GO categories in the biological process hierarchy that have at least 1 associated gene in each of the 116 bacterial genomes. I will refer to these categories as the ‘ubiquitous’ categories. The results for a selection of 6 of these ubiquitous GO categories are shown in Figure 2. The figure shows the dependence of the number of genes in each category on the total number of genes with annotation in the genome.
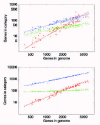
The upper panel shows the categories “signal transduction” (red), “carbohydrate metabolism” (blue), and “DNA repair” (green), while the lower panel shows the categories “transcription regulation” (red), “biological process” (blue), and “protein biosynthesis” (green). Each dot represents the counts in a single bacterial genome and both axes in Figure 2 are shown on a logarithmic scale. Note that in reference 13 a similar plot was shown but with the horizontal axis representing the total number of genes rather than the total number of annotated genes. Since the total number of annotated genes is to a very good approximation proportional the total number of genes, the results are virtually identical whether one uses the total number of genes or the total number of annotated genes. I decided to use the total number of annotated genes on the horizontal axis in Figure 2 partly to illustrate this fact. In addition, the genome size in bacteria is also to a very good approximation proportional to the total number of genes in the genome. Thus, if we had used the genome size instead of the number of annotated genes on the horizontal axis Figure 2 would again have looked virtually identical.
The dots of each color in Figure 2 fall approximately on a straight line. Thus, the logarithms of the number of genes nc in a category c and the total number of genes g (or the number of annotated genes or the genome size) are approximately linearly related:
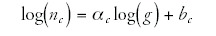
In other words, the number of genes nc in a category increases as a power-law in the total number of genes g:
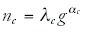
For the 6 functional categories shown, the exponents of the best power-law fits are indicated in the figure caption. The fits were obtained using the procedure described in the Methods section. The exponents range from α = 0.16 for protein biosynthesis to α = 1.95 for signal transduction.
To further show the range and variation of the observed exponents Figure 3 shows the inferred exponents and their 99% posterior probability intervals for a selection of 20 functional categories.
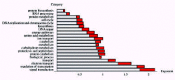
The exponents range from close to zero to roughly 2. Note that, for a category c with exponent αc the relative proportion pc of genes in the genome scales as pc = λcgα-1. That is, when αc <1the proportion of genes in the category will decrease with genome size, while for αc>1 the proportion of genes in the category will increase with genome size. Thus, for a categoryc with exponent close to 2, the proportion pc will increase almost linearly with genome size. The behavior of different categories thus ranges from categories where the number of genes is almost constant with genome size ( αc ≈ 0), to categories where the proportion of genes in the genome increases linearly with genome size ( αc ≈ 2).
The general picture that emerges from Figure 3 is that the proportion of genes in essential low-level functional categories such as protein biosynthesis and DNA replication decreases with genome size, whereas the proportion of genes that play regulatory roles such as genes involved in signal transduction and transcription regulation increases approximately linearly with genome size. In between these extremes is a number of categories, including different metabolic functions, for which the exponent is roughly 1, indicating that the genomic percentage of genes in these categories is roughly independent of genome size.
Upper Bound on Genome Size
The observed quadratic scaling of the number of regulatory genes with the total number of genes in the genome obviously cannot extend to arbitrarily large genome sizes. If we extend the red curves, corresponding to “transcription regulation” and “signal transduction”, in Figure 2 to the right, we will eventually reach a point where the number of signal transducers and transcription regulators would be larger than the total number of genes in the genome and this is obviously impossible. Thus, if all bacterial genomes obey the relations indicated in our figure, there must be an upper bound on bacterial genome size. A naive upper bound is obtained by demanding that the number of genes in any category is less than the total number of genes in the genome, i.e., nc = λcg ≤ g. If one substitutes the values of the fits for transcription regulation into this equation one obtains an upper bound of approximately g ≤ 70000 genes. A tighter upper bound is obtained when one demands that nc cannot increase by more than 1 gene when g is increased by 1 gene, i.e., λc(g+1) ≤ λcg + 1.* One then obtains an upper bound of g ≤ 34000 when the values for transcription regulation are substituted. In reference 15 an upper bound is derived by assuming that the number of genes involved in transcription regulation has to be less than half of the genome size, i.e.nc ≤ g/2.With our fit this leads to an upper bound of about g ≤ 30000.
It is clear that all these upper bounds substantially overestimate the approximately 10000 genes of the largest observed bacterial genomes and that a more realistic theory is needed to plausibly explain the apparent size constraint on bacterial genomes. In this regard it is also interesting to note that in all the upper bounds just proposed, the proportion of genes that are transcription regulators is at least 50% at the maximal genome size, whereas the percentage is at most 11% in the currently sequenced bacterial genomes.
Consequences for the Topology of the Transcription Regulatory Network
The approximately quadratic scaling of the number of transcription regulatory genes also has some interesting consequences for the structure of the transcription regulatory network as a function of genome size. The class of transcription regulatory genes consists for the most part of DNA-binding transcription factors that regulate transcription through the binding to specific regulatory motifs in intergenic regions. We can imagine the transcription regulatory network by a set of arrows pointing from each of the regulators to each of the genes that they regulate. The total number of arrows in this network for a genome of size g is the product of the number of genes g times the average number of incoming arrows per gene <r(g)>, i.e. <r(g)> represents the average number of different regulators regulating each gene in a genome of size g. Note that we can also write the total number of arrows as the number of transcription factors ntr (g)in a genome of size g times the average number of genes <n(g)> that each regulator regulates. We thus have
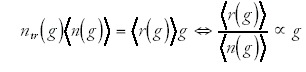
There is currently very little data to decide if real regulatory networks are closer to the limit where <r(g)> increases linearly, or closer to the limit where <n(g)>∝1/g. One piece of indirect evidence is the dependence of the number of operons and the amount of intergenic region on genome size. If the average number of transcription factors <r(g)> regulating each gene were to increase with genome size, then one might expect that, as genome size increases, the average operon size should decrease and that the amount of intergenic region per gene should increase. It is of course a nontrivial task to identify the number of operons from genome sequence alone. However, as a proxy we may consider runs of consecutive genes that are located on the same strand of the DNA (see ref. 16 for a method of estimating operon number using this statistic). Since all genes in an operon necessarily have to be transcribed in the same direction, a decrease in operon number would likely be reflected by a decrease in the average length of runs of consecutive genes on the same strand. Figure 4 (upper panel) shows this average length of iso-strand genes as a function of genome size for all bacterial genomes that are currently in the NCBI database of fully-sequenced genomes.
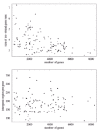
The figure suggests a slight decrease of the length of these runs, consistent with what was reported in reference 16, but there is a large amount of variation and the trend is far from convincing, i.e,r2=0.23 under simple regression. Note also that the drop in operon size is at most a factor of two between the largest and smallest genomes, while total gene number increases almost a factor of 20. The lower panel shows the average number of intergenic bases per gene as a function of the total number of genes in the genome. In this case a correlation between genome size and the amount of intergenic region is completely absent, i.e, r2 = 0.0005.Thus these two statistics provide little evidence that <r(g)> increases substantially with genome size. However, one cannot exclude the possibility that in small genomes a large proportion of genes is not transcriptionally regulated at all, and that as genome size increases this proportion drops dramatically. This would still lead to a substantial increase of <r(g)> with genome size. It does seem plausible, however, that larger genomes may have a larger number of ‘specialized’ regulons that typically regulate a smaller number of genes compared to the more general regulators that one expects to find in all organisms, and that <n(g)> thus decreases with genome size.
Quality of the Fits
The power-laws observed in Figure 2 are observed for the large majority of the 154 ubiquitous functional categories. I assessed the quality of the power-law fits by a measure F that measures the fraction of the variance in the data that is explained by the power-law fit (see Methods). Figure 5 shows the cumulative distribution of F for all 154 ubiquitous functional categories.
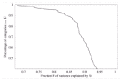
As can be seen from Figure 5 about two thirds of the categories have more than 90% of the variance explained by the fit. More than 95% of the categories have more than 80% of the variance explained by the fit. We thus see that most ubiquitous functional categories follow scaling laws like the ones shown in Figure 2.
Principle Component Analysis
Instead of considering the scaling behavior of one functional category at a time, we can consider all functional categories at once. We can consider a 154-dimensional ‘function space’ in which each axis represents an ubiquitous functional category, and represent the ‘functional gene content’ of a genome by a point in this 154-dimensional space. That is, if we number the categories 1 through 154, and nc is the number of genes in category c, then ñ= (n1,n2,...,n154) represents the functional gene content of the genome as a point in the function space. The set of all sequenced genomes thus forms a scatter in this function space.
We can now ask what the shape is of this cloud in function space. To do this, it is convenient to again consider all axes on logarithmic scales. That is, we consider the scatter of points →x with xc = log(nc).The results in the previous section showed that, to a good approximation, almost all functional categories obey the linear equations
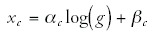
where g is the total number of genes in the genome. If this equation were to hold exactly for all categories, then the numbers xc and xc', for any two categories c and c', would also be linearly related:
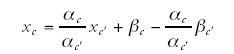
Thus, the statement that equation (4) holds for all categories is equivalent to the statement that the scatter of points in function space falls on a straight line. Of course, since equation (4) holds only approximately for each category, the scatter of points falls only approximately on a line. To illustrate this Figure 6 shows three projections of the scatter of points onto three-dimensional subspaces each representing three functional categories.
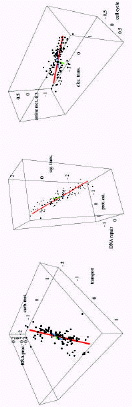
That is, each of these three figures shows the scatter of points with respect to only three of the 154 axes. The functional categories that were used for the projections are indicated in each of the plots and in the caption. As can be seen, the scatter of points indeed approximately falls on a straight line in each of these projections.
The extent to which the scatter of points falls on a line can be quantified most directly using principle component analysis.17 In principle component analysis one aims to represent a scatter of points in a high-dimensional space by a scatter in a much lower dimensional space. To this end it finds an ordered set of orthogonal coordinate axes in the n-dimensional space that have the property that, for any m ≤ n, the sum of the squared distances of the data points to their projections on the first m axes of the coordinate system is minimized (i.e., no set of m axes has a lower sum of squared distances). That is, the first principle axis is chosen such that the average squared-distance of the data points to this axis is minimized. The second principle axis is chosen such that the average squared-distance of the data points to the surface spanned by the first and second principle axis is minimized. And so on for the further principle components.
The ability of this coordinate system to represent the scatter of points is again measured by the fraction of the variance in the data that is captured by consecutive axes. For the scatter of 116 bacterial genomes in the function space of 154 ubiquitous categories, the first principle axis captures 22% of all the variance in the data. The second principle axis captures 6% of the variance, the third 5% of the variance, etcetera. Thus, the amount of variance explained drops sharply between the first and second principle axis. After that, the amount of variance explained by the data decreases smoothly, dropping between 5 and 10 percent between consecutive axes. As many as 55 axes are needed to cover 95% of the variance in the data. These statistics suggest that only the first principle axis captures an essential characteristic of gene-content in bacterial genomes. Once this largest component of the variance is taken into account, one needs almost as many dimensions as there are genomes to explain the remaining variance in gene-content.
Table 1 summarizes the statistics of the first three principle axes. Each of the axes is a vector ã whose direction in function space is reflected by the relative sizes of the components ac. In the leftmost column of Table 1 I have listed, for each axis, the 4 categories with the highest components ac and the 4 categories with the lowest components ac (redundant categories were omitted). The values of these components ac are shown in the second column. The projection of all genomes onto one of the principle axes gives another vector →v where vg is the component of genome g in the direction of the principle axis. The third column in Table 1 shows, for each principle axis, the 4 genomes with the highest components and the 4 genomes with the lowest components vg. The components themselves are shown in column 4. These first three principle axes are also indicated as the red, blue, and green lines in Figure 6.
Table 1
Summary of the first three principle axes.
As can be easily derived from equation (4), the components ac of the first principle axis correspond precisely to the inferred exponents of Figure 3. Thus, the entire collection of scaling laws is summarized by this single vector. In summary, a large fraction of the variation in functional gene-content among all bacterial genomes can be summarized by a single vector ã which encodes how the numbers of genes in different functional categories increase and decrease as the total size of the genome varies. That is, as the genome size increases or decreases the numbers of genes in each functional category c increase or decrease as g .The vector ã thus reflects a basic functional architecture of gene-content that holds across all bacterial genomes.
The meaning of the second and third principle axis is less clear, and given that they only capture a relatively small amount of the variance it is not clear that they are very meaningful at all. For both these axes the genomes at the extremes tend to be small parasitic organisms. This suggests that these axes may reflect different types of parasitic lifestyles.
Evolutionary Interpretation
What is the origin of the scaling laws discussed in the previous sections? In this section I will show that the observed scaling laws in fact suggest that there are fundamental constants in the evolutionary dynamics of genomes.
Consider a particular genome with numbers of genes nc in different functional categories c. Consider next the evolutionary history of this genome. With this I mean that the current genome can be followed back in time through the life of the cell the genome was taken from, through the cell division that produced it, through the life of its ancestral cell, and its ancestors, and so on. In this way the history of the genome can be traced back all the way to an ancestral prokaryotic cell from which all currently existing bacteria stem. During this evolutionary history the numbers nc have of coursed increased and decreased in ways that are unknown to us. That is, there are (unknown) functions of time nc(t) that describe the evolution of the numbers of genes in each category c in the genome under study. Similarly, there will be a function g(t) that describes the evolutionary history of the total number of genes in the genome.
One can now ask what constraints the functions g(t) and nc(t) should obey such that the observed scaling laws hold. To this end it is convenient to write the dynamics of nc(t) and g(t) in terms of effective duplication and deletion rates. That is, we write
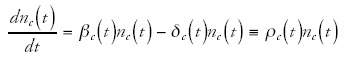
and
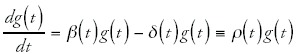
In these equations, βc(t) is the (time-dependent) average duplication rate of genes in category c, β(t) is the average duplication rate of all genes, δc(t) is the average deletion rate of genes in category c, and δ(t) is the average deletion rate of all genes. I have also defined the differences of duplication rates and deletion rates as ρc(t) and ρ(t). Notice that, since in the above equations ρc(t) and ρ(t) can be arbitrary functions of time, any time-dependent function can still be obtained as the solution of the above equations. Formally solving these equations we obtain
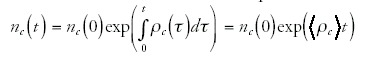
and
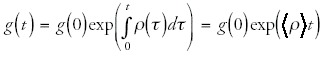
where I have written the integrals as the time averages of ρc and ρ times the total time t. Note that these averages are a function of the evolutionary history of the genome under study. If we now express nc(t) in terms of g(t) we obtain
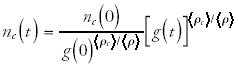
Note that, although this equation may appear to already imply the general scaling relations that were found, in fact it is completely general and holds for any set of evolutionary histories nc(t) and g(t). The equation is nothing more than a way of rewriting the relation between nc(t) and g(t) in terms of the averages <ρc> and <ρ. of this genome's history.
The observed scaling relations only follow from (10) if the same equation holds for all genomes. That is, if the variables nc(0), g(0) and <ρc>/<ρ> are the same for all evolutionary histories. This requirement is illustrated in Figure 7.
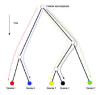
The figure shows the evolutionary histories of a set of genomes in a phylogenetic tree. Each leaf of the tree represents one of the bacterial genomes, and at the root is the common ancestor genome of all the genomes at the leafs. We thus have a separate equation (10) for each of the genomes at the leafs. The initial numbers nc(0)and g(0) in each of these equations are just the numbers of genes in category c and in the whole genome of the common ancestor, and it is therefore clear that the variables nc(0) and g(0) are indeed the same for all the equations.*
Much more interesting is the requirement that the ratios < ρc>/< ρ> are the same for all evolutionary histories. These different evolutionary histories are indicated as colored lines in Figure 7. The requirement that <ρc>/<ρ> be equal for all evolutionary histories thus demands that if one takes the average of the duplication minus deletion rate of genes in category c over the entire evolutionary history of a genome, and divides this by the average duplication minus deletion rate of all genes in the genome, then this ratio always comes out the same, independent of what evolutionary history is averaged over. That is, the ratios <ρc>/<ρ> are universal evolutionary constants and correspond precisely to the exponents αc = <ρc>/<ρ> of the observed scaling laws.
So what determines <ρc> and <ρ> ? It seems reasonable to assume that the rate at which genes are duplicated and deleted mostly reflects the effects of selection. That is, as a first approximation we may assume that the rate at which duplications and deletions are introduced during evolution are approximately the same for all genes but that different genes have different probabilities of being selectively beneficial when duplicated or deleted. The rate ρc(t) is then given by some overall rate γ at which duplications and deletions are introduced* times the difference between the fraction of genes ƒ+c(t) in the category that would benefit the organism when duplicated and the fraction ƒ-c(t) of genes in the category that would benefit the organism when deleted:
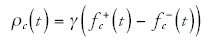
We then have
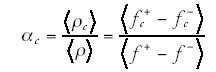
The fractions ƒ+c(t) and ƒ-c(t) of course depend on the selective pressures of the environment at time t. Thus, as one follows the genome through its evolutionary history, the demand for genes of a certain function fluctuates up and down, and this will be reflected in the fluctuations of ƒ+c(t) - ƒ-c(t). It seems intuitively clear that the size of the fluctuations is going to depend strongly on the functional class. That is, one would expect that the demand for genes that provide an essential and basic function fluctuates relatively little. For instance, one would expect that even as the selective environment changes it is rare that existing protein biosynthesis genes become dispensable or that duplicates of these genes become desirable. On the other hand, the desirability of transcription regulatory genes and signal transducers is going to depend crucially on the selective environment in which the organism finds itself. It is thus not implausible that, if one averages over sufficiently long evolutionary times that the ratio <ƒ+c - ƒ-c>/<ƒ+ - ƒ-> always reaches the same limit, and that this limit is large for highly environment-dependent categories such as transcription regulation, and small for environment-insensitive categories such as protein biosynthesis and replication. The main open question that remains is the origin of the precise numerical values of the ratios <ƒ+c - ƒ-c>/<ƒ+ - ƒ-> for different functional categories.
The Exponent for Transcription Regulatory Genes
The exponent αc for the category of genes involved in transcription regulation is close to 2, with the 99% posterior probability interval running from 1.72 to 1.95. Thus, even though the current data suggests that the exponent is slightly less than 2, it is tempting to think that the scaling might be simply quadratic. This is especially tempting given that this allows one to speculate more easily about the origin of this exponent. That is, it is easier to theorize about a quadratic scaling law then about a scaling law with exponent 1.83. Some theoretical explanations for the quadratic scaling of transcription regulators have recently been put forward.15 In this section I want to discuss these proposals and contrast them with my own suggestions in this regard in reference 13.
One of the first things that of course come to mind when attempting to explain a scaling of the form nc∝g2 is that in a genome with g genes, the number of pairs of genes scales precisely as g2.That is, there are g(g - 1)/2 possible interactions in a genome with g genes. Therefore, if one could find and argument that suggests that the number of transcription regulators should be proportional to the number of pairs of genes, this would provide an possible explanation. In a recent paper15 Croft and coworkers put forward two different models for the observed quadratic scaling of the number of transcription regulatory genes that both in essence argue that the number of regulators should be proportional to the number of potential interactions between genes, i.e., the number of pairs of genes in the genome.
Although it is attractive to seek such a simple combinatorial explanation I believe that a simple survey of what is known about the regulatory role of transcription factors in bacteria shows that such models are in fact highly implausible. If each regulator were to somehow ‘correspond’ to one or more pairwise interactions of genes then one would expect that the role of most regulatory genes would be to ensure that certain pairs of genes are expressed together or to ensure that certain pairs of genes are not expressed together.
This is, however, not what is observed. To mention just a few known E. coli regulons: the factor crp responds to the concentration of cAMP in the cell, and its main role is to make the activation of many catabolic pathways conditional on the presence or absence of cAMP. The factor lexA responds to single stranded DNA and activates a number of genes that are involved in the repair of DNA damage. PurR responds to the concentrations of hypoxanthine and guanine and represses a set of genes involved in de novo purine biosynthesis. FadR senses the presence of long chain fatty acyl-coA compounds and in response regulates genes that transport and synthesize fatty acids. Finally, tyrR responds to the levels of phenylalanine, tyrosine, and tryptophan and regulates genes that synthesize and transport aromatic amino-acids.
In all these cases, the role of the regulator is to sense a particular signal and to respond to this signal by activating or repressing a set of genes that implement a specific biological function which is related to the signal. In none of these example does it appear that the role of the regulator is to regulate the interactions between pairs of genes. It thus seems that the number of different transcription regulators that the cell has is much more a reflection of the number of different cellular responses to different environments that the cell is capable of.
In reference 13 I provided a qualitative argument for the approximately quadratic scaling of the number of transcription regulatory genes. According to equation (12) the average difference <ƒ+c - ƒ-c> between the fractions of transcription regulatory genes that would increase fitness when duplicated and those that would increase fitness when deleted is almost twice as large as the average difference <ƒ+ - ƒ-> between the fractions of all genes that would increase fitness when duplicated and those that would increase fitness when deleted. This requirement will of course be satisfied if both <ƒ+c> ≈ 2<ƒ+> and <ƒ-c> ≈ 2<ƒ->. That is, transcription regulators are twice as likely to lead to fitness increase when duplicated as average genes, and transcription regulators are also twice as likely to lead to fitness increase when deleted.
I want to suggest that the origin of the factor 2 in these rates is the switch-like function of transcription factors. Imagine a gene that has just emerged through a gene duplication. Originally the duplicate will be the same as its parent. At that point, the main change caused by this duplication that may affect fitness is a change in the dosage of the gene, i.e., from one to two copies. One would expect that the probability for this dosage change to have strong deleterious effects is approximately equal for transcription regulators as for genes in general. If the duplicated gene is to get fixed in the genome on a longer time scale, then a process of mutation and selection should modify the duplicated gene into a gene that increases the fitness of the organism. I suggest that the probability for this process to be successful is twice as high in transcription regulators as in nonregulatory genes. In nonregulatory genes the only avenue for beneficial change is a change in the molecular function of the gene, e.g., a metabolic gene may evolve to catalyze a new chemical reaction. Transcription regulators, however, may evolve to respond to a signal which the cell was previously insensitive to. They may evolve to affect the state of the cell both when this signal is present and when the signal is absent. Since this gives twice as many opportunities for a beneficial change, one may expect that there is twice as much probability of success.
A similar argument holds for the rate of deletion. When deleting a nonregulatory gene, the cell simply changes to a state without the gene. When a transcription regulator is removed, one effectively removes a ‘switch’ from the genome: the cell becomes insensitive to the signal that the regulator responded to. But there are again two ‘ways’ of implementing this insensitivity. The genes regulated in response to the signal may be turned constitutively on, or constitutively off. Thus there are two independent ways of removing a transcription factor, and one would thus expect the effective rate of transcription factor deletion to be twice the rate of deletion of general genes.
These arguments are of course highly speculative and may well turn out to be incorrect. However, they at least seem consistent with what we know about transcription regulation in bacteria and the process of evolution through gene duplication and deletion. Note also that very similar arguments as the ones just presented for transcription regulatory genes can be put forward for the category of signal-transducing genes. These are indeed also observed to scale approximately quadratically with genome size. Thus, the argument just presented explains the scaling exponent for both the transcription regulation category and the signal transduction category. However, I suspect that it will be impossible to come to any solid conclusions regarding the cause of the approximately quadratic scaling for transcription regulators and signal transducers until we have better data on the genome-wide topology of the transcription regulatory and signal transduction networks in bacteria of differing sizes.
Finally, I note that most of the functional categories have exponents that do not appear to equal small integers or even simple rational numbers. It is thus clear that simple arguments such as the ones discussed above will not be capable of explaining these exponents. What determines the average <ƒ+c - ƒ-c>/<ƒ+ - ƒ-> for these categories is a fascinating question that at this point is completely open.
Methods
The Number of Genomes as a Function of Time
The data for Figure 1 were obtained by extracting the submission dates of all the microbial genomes in genbank at ‘ftp://ftp.ncbi.nih.gov/genbank/genomes/Bacteria’ from the ‘gbs’ file for each genome. When there were multiple submission dates the earliest date was taken. The logarithm of the number of genomes as a function of time was then fitted to a straight line using standard least-square regression. This least-square fit gives the number of genomes n(t) as a function of time (in years) as n(t) = 2(t-1993.4)/1.38. The exponential provides a reasonable fit, i.e. r2 = 0.98 even though the data clearly suggest a decrease in the rate at which new genomes appear for the last 1 to 1.5 years.
Gathering Functional Gene-Content Statistics
The numbers of genes in different functional categories for each sequenced genome were obtained in the following way. Interpro annotations18 of fully-sequenced genomes were obtained from the European Bioinformatics Institute.19 Functional categories were taken from the gene ontology biological process hierarchy.14 A mapping from Interpro to GO-categories was also obtained from the gene ontology website. Using this mapping I gathered, for each GO-category, all Interpro entries that map to it or to one of its descendants in the biological process hierarchy. I then counted, for each gene in each fully-sequenced genome and each GO-category, the number of Interpro hits h that the gene has to Interpro categories associated with the GO-category. A gene with many independent Interpro hits that are associated with the same GO-category is of course more likely to be a member of the GO-category than a gene with only a single hit. To quantify this, I chose the probability for a gene to be a member of a GO-category to which it has h hits to be 1 - exp(- λh), with λ = 3. The results presented in this chapter are largely insensitive to changes in λ (see the discussion in ref. 13).
In this way the number of genes associated with each GO-category in each genome was counted. I then selected all GO categories that have a nonzero number of counts in all bacterial genomes. There are 154 such ubiquitous GO-categories. I also counted the total number of genes that have any annotation at all for each genome. These are defined as genes that have at least one Interpro hit. Further discussion of this annotation procedure and its robustness can be found in reference 13.
Power-Law Fitting
In order to fit the data to power-law distributions I used a Bayesian procedure that is described in reference 13. The main advantage of this fitting procedure with respect to simple regression is that the results are explicitly rotationally invariant. That is, the best line that the fitting procedure produces doesn't depend on the orientation of the coordinate axes with respect to the scatter of data points.
The result is that the posterior distribution P(α | D)dα for the slope α of the line, given the data D, is given by
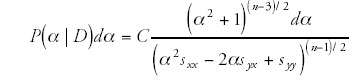
where n is the number of genomes in the data, sxx is the variance in x-values (logarithms of the total gene numbers), syy the variance in y-values (logarithms of the number of genes in the category), syx is the covariance, and C is a normalizing constant. For each of the 154 ubiquitous categories we then calculated the 99% posterior probability interval for the slope from equation.13
Quality of the Power-Law Fits
To calculate the quality of the power-law fits, I first log-transform the data points (gi,ni) to (xi,yi) = (log(gi),log(n,i)). The best power-law fit is a straight line y = αx+β in the (x,y) plane. For each data point (xi,yi) I then find the distance di(l) to this line, and the distance di(c) to the center of the scatter of points. That is, with <x> the average of the x-values, and <y> the average of the y-values, the distance di(c) is given by
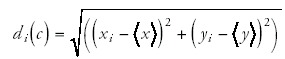
and the distance to the line is given by
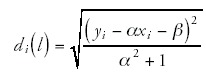
I then define the quality of the fit F as the fraction of the variance that is explained by the fit.
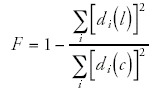
References
- 1.
- Huynen M, van NimwegenE. The frequency distribution of gene family sizes in complete genomes. Mol Biol Evol. 1998;15:583–589. [PubMed: 9580988]
- 2.
- Gerstein M. A structural census of genomes: Comparing bacterial, eukaryotic, and archael genomes in terms of protein structure. J Mol Biol. 1997;274:562–576. [PubMed: 9417935]
- 3.
- Luscombe NM, Qian J, Zhang Z. et al. The dominance of the population by a selected few: Power-law behavior applies to a wide variety of genomic properties. Genome Biology. 2002;3(8) [PMC free article: PMC126234] [PubMed: 12186647]
- 4.
- Koonin EV, Wolf YI, Karev GP. The structure of the protein universe and genome evolution. Nature. 2002;420:218–222. [PubMed: 12432406]
- 5.
- Uetz P, Giot L, Cagney G. et al. A comprehensive analysis of protein-protein interactions in saccharomyces cerevisiae. Nature. 2000;403:623–627. [PubMed: 10688190]
- 6.
- Ito T, Chiba T, Ozawa R. et al. A comprehensive two-hybrid analysis to explore the yeast protein interactions. PNAS USA. 2001;98:4569–4574. [PMC free article: PMC31875] [PubMed: 11283351]
- 7.
- Maslov S, Sneppen K. Specificity and stability in topology of protein networks. Science. 2002;296:910–913. [PubMed: 11988575]
- 8.
- Stuart JM, Segal E, Koller D. et al. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. [PubMed: 12934013]
- 9.
- Guelzim N, Bottani S, Bourgine P. et al. Topological and causal structure of the yeast transcriptional regulatory network. Nat Genet. 2002;31:60–63. [PubMed: 11967534]
- 10.
- Milo R, Shen-Orr S, Itzkovitz S. et al. Network motifs: Simple building blocks of complex networks. Science. 2002;298:824–827. [PubMed: 12399590]
- 11.
- Jeong H, Tombor B, Albert R. et al. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. [PubMed: 11034217]
- 12.
- Wagner A, Fell D. The small world inside large metabolic networks. Proc Roy Soc London Series B. 2001;268:1803–1810. [PMC free article: PMC1088812] [PubMed: 11522199]
- 13.
- van NimwegenE. Scaling laws in the functional content of genomes. Trends in Genet. 2003;19:479–484. [PubMed: 12957540]
- 14.
- The gene ontologyconsortium. Gene ontology: Tool for the unification of biology. Nat Genet. 2000;25:25–29. [PMC free article: PMC3037419] [PubMed: 10802651]
- 15.
- Croft LJ, Lercher MJ, Gagen MJ. et al. Is prokaryotic complexity limited by accelerated growth in regulatory overhead. Genome Biology. 2003;5:P2.
- 16.
- Cherry JL. Genome size and operon content. J theor Biol. 2003;221:401–410. [PubMed: 12642115]
- 17.
- Joliffe IT. Principle component analysis. New York: Springer-Verlag. 1986
- 18.
- Apweiler R, Attwood TK, Bairoch A. et al. The interpro database, an integrated documentation resource for protein families, domains and functional sites. Nucl Acids Res. 2001;29:37–40. [PMC free article: PMC29841] [PubMed: 11125043]
- 19.
- Apweiler R, Biswas M, Fleischmann W. et al. Proteome analysis database: Online application of interpro and clustr for the functional classification of proteins in whole genomes. Nucl Acids Res. 2001;29:44–48. [PMC free article: PMC29822] [PubMed: 11125045]
Footnotes
- *
Note that in principle nothing keeps a genome from increasing nc by more than 1 gene when g is increased by 1 gene. That is, some genes in other categories than c may be removed and replaced by genes in category c.
- *
One caveat is that we implicitly assume that nc(t) > 0 for all t. A slightly more complex treatment is needed for categories that were not present in the ancestor, or that disappeared and reappeared during the evolutionary history of some genomes.
- *
In reality the rate at which duplications are introduced is unlikely to equal the rate at which delections are introduced. We ignore this complication for notational simplicity. The theoretical development with this complication would be analogous.
- Scaling Laws in the Functional Content of Genomes: Fundamental Constants of Evol...Scaling Laws in the Functional Content of Genomes: Fundamental Constants of Evolution? - Madame Curie Bioscience Database
- PDGF Pathways and Growth of Basal Cell and Squamous Cell Carcinomas - Madame Cur...PDGF Pathways and Growth of Basal Cell and Squamous Cell Carcinomas - Madame Curie Bioscience Database
- Phylogenetic, Structural and Functional Relationships between WD-and Kelch-Repea...Phylogenetic, Structural and Functional Relationships between WD-and Kelch-Repeat Proteins - Madame Curie Bioscience Database
- PROMOTER-ASSOCIATED LONG NONCODING RNAs REPRESS TRANSCRIPTION THROUGH AN RNA BIN...PROMOTER-ASSOCIATED LONG NONCODING RNAs REPRESS TRANSCRIPTION THROUGH AN RNA BINDING PROTEIN TLS - Madame Curie Bioscience Database
- The Role of Synaptotagmin and Synaptotagmin-Like Protein (Slp) in Regulated Exoc...The Role of Synaptotagmin and Synaptotagmin-Like Protein (Slp) in Regulated Exocytosis - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...