

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

“GAs have been shown to be capable of describing extremely complex bahaviour in a range of application domains, including those of molecular recognition and design.” (P. Willett)1
Current Concepts in Computer-based Molecular Design
In the previous Chapters we have addressed some issues related to adaptive optimization methods and fitness calculation in the context of drug design tasks, in particular evolutionary algorithms and artificial neural networks. To close the design cycle depicted in Figure 1.4, we still have to define the molecule generator. This will be the main focus of this Chapter. We will highlight only selected approaches, which we have chosen either because they illustrate a general principle, or we have particular experience with these methods. Again the focus will be on evolutionary techniques.
Generally, current computer-based molecular design approaches may be regarded as being guided by two major strategies:
- Structure-based design relying on a 3D receptor model of the ligand-binding pocket
- Ligand-based design starting from the knowledge of one or several known actives without taking the 3D receptor structure into account.
The majority of the current structure-based design tools are based on a computer model of a binding site and require a scoring function that computes an estimate of the binding affinity of a molecule—e.g., a potential inhibitor—in a given conformation (also called a pose) within the binding pocket. In contrast, ligand-based tools usually build on a scoring function that implements some sort of similarity principle rather than estimating binding affinity in a receptor-ligand docking experiment. Of course, both approaches complement each other and can be combined-depending on how much biostructure information is available at the beginning or becomes available during a project. New structures can for example be docked into or grown within a binding pocket (provided a receptor structure is available) or compared to a known active reference molecule.
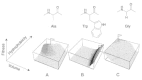
De novo design attempts to generate novel molecules matching a given binding pattern (pharmacophore), i.e., the spatial arrangement of relevant receptor-ligand interaction points. Among the most prominent software solutions for structure-based de novo design are the packages LUDI,2 BUILDER 3 and CAVEAT.4 These algorithms identify potential ligand-receptor interaction or attachment points in the receptor binding pocket and construct novel molecular entities by combinatorial or sequential assembly of atoms and molecular fragments (Fig. 1).
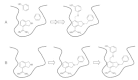
As mentioned above the compatibility (quality, fitness) of novel structures or an individual molecular fragment in a given position is often estimated by empirical scoring functions.5Although fast combinatorial docking procedures clearly proved their applicability to de novo design,6one of the major problems still to be solved is the accurate prediction of binding energies.7,8 This problem has been approached in many different ways, e.g., by force-field based methods,9-17techniques based on the Poisson-Boltzmann equation,18–21 potentials of mean force,22-27 free energy perturbation,28 and linear response approximations.29,30 In this context it is common to differentiate between empirical and knowledge-based scoring functions. The term “empirical scoring function” stresses that these quality functions approximate the free energy of binding, δGbinding, as a sum of weighted interactions that are described by simple geometrical functions, fi, of the ligand and receptor co-ordinates r (Eq.1).8 Most empirical scoring functions are calibrated with a set of experimental binding affinities obtained from protein-ligand complexes, i.e., the weights (coefficients) δGi are determined by regression techniques in a supervised fashion. Such functions usually consider individual contributions from hydrogen bonds, ionic interactions, hydrophobic interactions, and binding entropy. As with many empirical approaches the difficulty with empirical scoring arises from inconsistent calibration data.
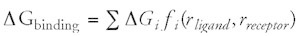
Knowledge-based scoring functions have their foundation in the inverse formulation of the Boltzmann law computing an energy function that is also referred to as a “potential of mean force” (PMF). The inverse Boltzmann technique can be applied to derive sets of atom-pair potentials (energy functions) favoring preferred contacts and penalizing repulsive interactions. The various approaches differ in the sets of protein-ligand complexes used to obtain these potentials, the form of the energy function, the definition of protein and ligand atom types, the definition of reference states, distance cutoffs, and several additional parameters. Scoring functions provide a very active and rapidly changing research field. Thorough treatments of historical and actual concepts and achievements can be found in the literature.7,31–33
A complementary approach to starting from a receptor structure is to build upon a pharmacophore hypothesis that was derived from a known bioactive molecule or ligand.34 based on a pharmacophore model, alternative molecular architectures can be virtually assembled mimicking the pharmacophore pattern present in the original template structure (for review, see Chapters 2 and 3). This methodology and related tactics represent workable approaches to ligand de novo design when a high-resolution receptor structure is not available, which is especially the case for many membrane-bound neuroreceptors in central nervous system research, including the large group of various G-protein-coupled receptors.
Irrespective of the availability of a receptor model and the choice of the fitness function, there are two alternative approaches of how to assemble new molecular structures, namely atom-based methods and fragment-based methods. Furthermore, the assembly process itself may be used to categorize the different molecular design concepts. One may differentiate between incremental-growth and construct-and-score techniques. The first method starts from a small molecular fragment and sequentially adds and modifies parts (atoms, fragments) to obtain the final design (Fig. 1). At each step the intermediate solution is scored and evaluated. The alternative is to first build a complete molecule and then perform a single scoring step for the virtual product. Table 1 contains a compilation of selected software tools, which implement different procedures and are commonly used in molecular design studies. Several additional algorithms have been proposed during the last decade, most of them are not named but only mentioned in the context of the respective publication. Textbooks and several recent review articles provide in-depth treatments of this field of computational chemistry, with a historical focus on structure-based approaches.33,35–40
Table 1
Selected examples of de novo design algorithms.
While atom-based techniques build up a molecule atom by atom, fragment-based design methods use sets of pre-defined molecular building blocks that are connected by a virtual synthesis scheme. This approach can have several advantages, particularly in combination with evolutionary algorithms:
- Fragments can be regarded as molecular “modules”. both whole molecules and fragments are easily encoded in a “genotype” representation, e.g., a molecular “chromosome”;
- the definition of a fragment is variable; it may mean large molecular building blocks (e.g., synthons of combinatorial or parallel chemistry) as well as small fragments like functional groups or even single atoms. In this view, atom-based design encompasses a special case of fragment definition;
- the chance of designing a synthetically feasible and “drug-like” structure will be high if physically accessible reaction educts (synthons) or retro-synthetically obtained sets of fragments are used in virtual synthesis; and
- the size of the search space is greatly reduced by the use of the fragment-concept compared to purely atom-based techniques.
Despite their appeal and ease of implementation fragment-based techniques have some limitations. Most important, they are often restricted to relatively coarse-grained designs, because fine-tuning of structures can be hampered due to a restricted fragment set—especially during the final optimization cycles. A chemically meaningful selection of fragments for the design process is crucial for success. Very often the usefulness of a particular fragment set depends on the design task. It can be very beneficial to use different sets for the design of GPCR modulators or kinase inhibitors, for example. Furthermore, during the first virtual design cycle—while a coarse-grained search is performed—the fragment sets can differ from the sets used during the later stages. In the ideal case the fragment sets should adapt in a similar way to the output structures. Only recently this idea of adaptive fragment definitions has been incorporated into evolutionary de novo design algorithms (G. Schneider, K. Bleicher, J. Zuegge et al, unpublished).
Evolutionary Structure Generators
The idea of using evolutionary algorithms for de novo design is not new, and several extensive reviews and compilations of articles treating this topic are available.1,41-44 The first examples of EA-based design programs were published around 1990 (Table 2). Initially, these approaches focused on biopolymers like peptides and small RNA molecules using 2D similarity scores as fitness function, but most of the more recent developments concentrate on the design of small molecules as potential drug candidates. The aim of de novo design within a receptor binding pocket (in situ) is to generate molecules that satisfy as many of the potential or known interaction sites as possible without violating the steric constraints of the binding pocket. Characteristic programs for this task are PRO-LIGAND,45,46 LeapFrog,47 ChemicalGenesis,48 and the early work of Blaney and coworkers.49 Typical evolutionary techniques for the de novo design of molecules to fit 2D constraints include the algorithms developed by Venkatasubramanian et al,50–52 Nachbar,53 Globus et al,54 Devillers et al,55 Weber et al,56,57 Douget et al,58 and Schneider and coworkers.59,60 Usually such algorithms treat molecules as a linear string, a tree-structure, or as a molecular graph. The SMILES representation of molecules is frequently applied.61 In 3D design the molecule is usually manipulated directly.
Table 2
Selected evolutionary de novo design methods (adapted from Gillet).
It is also possible to use fragment-based structure generators like TOPAS59 (vide infra for a detailed description of this algorithm) in combination with heuristic 3D conformer builders like CORINA, CONCORD, or CONFORT,62-65 and feed the designs into fast docking programs-;e.g.,FlexX,66,67 DOCK,68,69 or GLIDE70—for fitness determination. The program CONJURE was developed by researchers at Vertex Pharmaceuticals and represents an early version following this approach.71
One such construct-and-score technique was explored at Roche for the design of novel serine protease inhibitors (F. Hoffmann-La Roche Ltd.; K. Bleicher, G. Schneider, M. Stahl; unpublished). Structures were assembled by TOPAS and each new molecule was docked into the binding pocket of the target. The FlexX docking score was used as a measure of fitness.66 In Fig. 2a, a set of 50 docked structures are shown, forming the offspring during one of the last generations of an evolutionary optimization run. In this example, the serine protease Factor VIIa served as the target enzyme. Many of the known residue structures (mainly benzamidine derivatives) for hydrogen-bonding to the aspartic acid Asp189 at the bottom of the S1 pocket have evolved, and the designed molecules reveal a preference for lipophilic moieties potentially binding to an adjacent lipophilic pocket. It must be noted that the docking step for fitness estimation comes at a price, i.e., significantly longer computing time than simple similarity searching. In the particular study we used a population size of 50 individuals with non-parallel program execution, as a consequence, one generation took 45 minutes of computation time. Despite this drawback, evolutionary 3D design methods can be of considerable value if a high-resolution structure or model of the receptor pocket is available. The success of the approach largely depends on the accuracy of the scoring function involved. Sometimes molecules are grown which receive a high docking score (high fitness), but turn out to be inactive. One example is shown in Fig. 2b. This urea derivative possesses poor aqueous solubility, and—as a consequence—its predicted tight binding could not be confirmed in an enzyme inhibition test. This example reminds us not to restrict fitness measures to a single quality (e.g., the predicted binding energy) but to consider several drug-like properties in parallel. One of the most critical physicochemical properties is the aqueous solubility (see Chapter 4). Drug design is a multi-dimensional optimization task.

In the remaining part of this Chapter we will restrict the discussion of evolutionary structure generators to evolution strategy-based systems (see Chapter 1 for details about evolution strategies). First, the special case of peptide design will be presented; and finally the generalization of this reduced approach to fragment-based small molecule design will be made, taking the TOPAS approach as an example. It must be emphasized that there are many ways in which an EA can be implemented,1and the approaches discussed in this Chapter are intended to provide only an entry point only to demonstrate some general principles.
Peptide Design by “Simulated Molecular Evolution”
Cell biology, Genomics, and Proteomics provide three important pillars for rational drug design, and computer algorithms combined with a sophisticated combinatorial chemistry provide a useful toolbox for lead identification. Progress in cell biology has led to deep insights into important cellular processes like differentiation, division, and adaptation. Details of the underlying regulation and control mechanisms have been elucidated, and many signal molecules—in particular peptides which function as endogenous ligands to membrane receptors—have been identified. Protein-protein and peptide-protein interactions also provide a molecular basis for information transfer between cellular compartments. Many diseases are caused by aberrant protein-protein interactions. A possible therapeutic strategy is to design peptides specifically blocking these undesired interactions. PepMaker was one of the first fully automated computer algorithms for designing such blocking peptides.72 It was successfully applied to the design of several bioactive peptides.73,74 The appeal of the PepMaker method described here lies in the design of novel peptides starting just from a single known active peptide, the “seed peptide”. The PepMaker system allows exploration of the neighboring area of the seed peptide in amino acid sequence space. It is expected that several functional peptides can be found in the vicinity of the seed peptide which may have similar or even an improved activity.73,74 This idea will now be explained in more detail because it provides an insight into more advanced “simulated molecular evolution” techniques.
Compared to small molecule design, peptide design is a simple task because of the inherent modular architecture of amino acid sequences, their easy synthetic accessibility, and the restricted size of sequence space. Peptide design can be viewed as a special combinatorial chemistry approach with a restricted set of building blocks (e.g., the 20 standard amino acid residues) and amide bond formation as the only coupling reaction. A typical task is to identify a linear sequence of amino acid residues exhibiting a desired biological activity, e.g., binding affinity to a target molecule. In the simplest case there are only two parameters subject to optimization i) the number of sequence positions (peptide length), and ii) the type of residue present at each sequence position The typical experimental approach is to perform large-scale random screening which has become feasible due to recent advances in peptide synthesis and activity detection techniques.75–77 blind screening is essential if no information about function determining residue patterns is available. This is particularly true when conventional structure-based molecular modeling cannot be performed due to the lack of high-resolution receptor structures. If however a template peptide (“seed”) or other information is already known that can be used to limit the search space, it is worthwhile following some kind of rational design.74,78-80
The PepMaker algorithm generates variant peptides stemming from sequence space regions around the seed structure, thereby approximating a unimodal bell-shaped distribution. It is assumed that molecules with an improved function can be identified among the peptides located close to the seed peptide in sequence space. This assumption is supported by a number of observations:81-88
- In natural evolutionary processes, large alterations of a protein may occur within a generation, but these extremely different mutants rarely survive.
- Most observed mutations leading to a slightly improved function are single-site substitutions keeping the vast majority of the sequence unchanged.
- Conservative replacements tend to prefer substitutions of amino acids which are similar in their intrinsic physicochemical properties.
The algorithm generates a bell-shaped distribution of variants for construction of a biased peptide library, which is thought to approximately reflect some of these aspects of natural protein evolution. In addition to incremental optimization by small steps, large sequence alterations can also lead to improved function. This might be the case if, for example several optima exist in sequence space.82,89,90This idea consequently follows the evolution strategy approach discussed in Chapter 1.
The width of the distribution, σ, may be altered to generate more or less focused libraries. As a result the “diversity” of the libraries can be expected to increase from low to large s values. s functions as a strategy parameter in the evolutionary design principle (see Chapter 1). To quantify “diversity”, the Shannon uncertainty measure can be applied (Eq. 2).91,92 A similar entropy measure has been shown to be useful for assessment of the molecular diversity of large collections of small organic molecules and descriptor analysis.93-95 Details on the concept of entropy and information theory can be found in the literature.96-98 Here we will give a brief introduction to the general concept.
The Shannon entropy of a sequence alignment block has been proposed as a measure of the randomness of the residue distribution at each aligned position.92,99,100 Various interpretations of the meaning of “entropy” or “information content” are possible, including treatment as a chemical diversity measure or the degree of feature conservation. If Pi(xk) gives the frequency of the symbol xk from the alphabet x1, x2, x3,..., xA at the alignment position i, the Shannon entropy Hi at this position is defined by
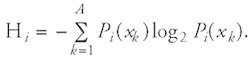
In this definition Pi(xk)log2 Pi(xk) is taken to be zero if Pi(xk) = 0. The unit of the Shannon entropy is “bit” because the base of the logarithm in the formula is two. The Shannon information Ri at the position i in the sequence alignment block is defined as the difference of two entropies:
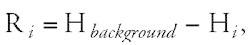
where Hbackground corresponds to the average sequence entropy. Hbackground is maximal when the symbols of the alphabet are evenly distributed:
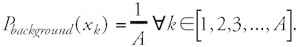
In this case the formula for Hbackground simplifies to
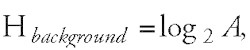
with all calculated values for Ri equal to or larger than zero. Therefore, an even background distribution of all residues is usually assumed when information theory is applied to sequence alignment blocks. This means that the information content at a given position is high, when the distribution of the symbols is far from random. As a result, calculating the information content for each position in a sequence alignment can be used to spot conserved symbols in the block.
However, the naïve assumption of evenly distributed symbols in the background might lead to false interpretations, if the background distribution is highly biased towards certain symbols. Unfortunately, when Hbackground is calculated using the true background distribution, other problems in the interpretation of Ri might occur. It could be the case that Ri is calculated to be zero, although the frequency of each symbol differs in Hbackground and Hi, simply because both distributions are equally far from a totally random distribution. This is because Ri only tells us about the whole distribution of symbols, without comparing the frequency of each specific symbol directly. As a possible solution to this problem one can calculate the relative entropy Hi (Pi ∥ Pbackground), also known as Kullback-Leibler “distance”, defined by
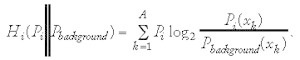
The relative entropy equals the Shannon information for an even background distribution, but differs otherwise. Hi (Pi ∥ Pbackground) is always equal to or greater than zero. It vanishes only, if every single symbol has the same frequency in both the background distribution and within a sequence block position. Contrary to the Shannon entropy, relative entropy is not a “state function”, and although it is often useful to think of relative entropy as a distance between two probability distributions, it is not symmetric and is not a correct mathematical distance measure.92 For potential applications of Equation 4, see e.g., the textbooks of Durbin and coworkers and baldi and brunak.98,100 The H-bloX software provides an easy-to-use web interface to entropy calculation and diversity assessment and may be downloaded as HTML/JavaScript, or accessed online at the URL: http://www.modlab.de.95 The H-bloX analysis is not restricted to protein, DNA or RNA sequences. It may be applied to arbitrary chemical libraries, provided a sensible alignment of structures can be accomplished. As long as the molecular structure can be described by a limited set of building blocks—which is often straightforward for combinatorial libraries—a corresponding sequence-representation may be used as the input data for H-bloX.
This short digression to entropy and library diversity was intended to provide a better understanding of the two main problems in evolutionary structure generation and library design. These issues are critical because the diversity of molecular building blocks determines the degree of library focusing (Fig. 1.2):
- An appropriate metric in chemical space (e.g., sequence space in the case of biopolymer design) must be defined. Both the step-size of an evolutionary walk in chemical space and the degree of library diversity will be measured on the basis of this metric.
- A procedure must be available that allows the calculation of mutation rates for pair-wise exchange and modification of molecular fragments or building blocks (e.g., amino acid monomers for peptide design). Similarity between building blocks will be defined based on the metric in chemical space.
To illustrate the idea of a mutation operator, an example of a mutation event is shown in Fig. 3. For reasons of clarity, the mutation of a single amino acid residue is illustrated. However, a similar scheme is appropriate for arbitrarily defined combinatorial building blocks or whole compounds. In the example shown, the amino acid tyrosine (Y) represents the parent structure, and the remaining 19 genetically encoded amino acids provide the stock of structures. With decreasing probability the parent is substituted by more distant residues. In the example, distance between two residues is defined by their Euclidean distance, di,j, in a primitive two-dimensional space spanned by a hydrophobicity and a volume axis (Fig. 2.5a). This amino acid similarity measure proved to be useful in several design exercises.74,101 As indicated by the dashed lines in Fig. 3, based on this model a mutation of tyrosine to phenylalanine (YØF) or leucine (YØL) is very likely to occur, whereas the tyrosine-arginine transition (YØR) is extremely rare. Another set of transition probabilities would result from a different s value or a changed ordering of the residues. In the PepMaker model the residue transition probability P(iØj) is given by Equation 7.72,102
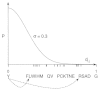
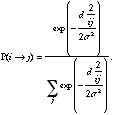
A great variety of substitution matrices have been suggested to measure distance between pairs of amino acid sequences, and it is not a trivial task to select the most appropriate for similarity searching or sequence design (for details, see the literature).103-105 Fig. 4 provides an example of the effect caused by an appropriate mutation matrix. In the example given the mutation matrix given in Table 3 was used. Ten small peptide libraries were designed, synthesized and tested (A. Kramer, G. Schneider, J. Schneider-Mergener, P. Wrede; unpublished). All libraries were generated from the same seed peptide, but a different σ-value was used for each library. The total entropy (diversity) of a library was computed by Equation 8, where N = 11. Low σ-values led to low total entropy, and large σ-values produced many different variant peptides which is reflected by large total library entropy. In this particular case the experimentally determined distribution of active molecules reflects the theoretically expected distribution (Fig. 4). For practical applications of the PepMaker algorithm the width of the variant distribution should be set to σ < 0.2 to obtain focusing libraries containing a sufficient number of active peptides for subsequent SAR modeling. However, this value can only be a rough estimate. It must be kept in mind that an optimal setting depends on the shape of the underlying fitness landscape, the particular amino acid exchange matrix chosen, and other parameters.
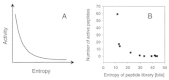
Table 3
Amino acid distance matrix based on physicochemical properties.
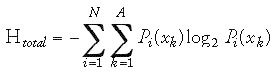
To demonstrate how to derive a seed peptide and apply the PepMaker system, we will now provide a prospective example taking HRV-14 infection as a model system for protein-protein interactions.
The causative agent for the common cold disease is human rhinovirus 14 (HRV-14) which in its first step of infection interacts with intercellular adhesion molecule 1 (ICAM-1), a cell surface receptor. HRV consists of a viral (+)-strand RNA which is packed into an icosahedral protein shell (capsid). The capsid (300 Å diameter) is composed of 60 identical protein complexes (protomers) consisting of four distinct viral proteins each (VP1-VP4). A three-dimensional structure of HRV-14 was determined by X-ray crystallographic analysis to a resolution of 3.0 Å106 based on this structure and electron microscopic investigations, Rossmann and coworkers formulated the canyon hypothesis.107,108 The 25 Å deep canyon is a depression at the surface of each protomer circulating around each of the 12 vertices. The accessible surface of the canyon consists of parts of the capsid proteins VP1 and VP3. It is postulated that the canyon provides the recognition site for the virus-specific receptor ICAM-1. This hypothesis is supported by the effect of point mutations located in the viral canyon on virus adsorption on the host cell.109 Further support stems from experiments with antibodies directed against ICAM-1 and their corresponding anti-idiotypic antibodies:110-112 Anti-idiotypic antibodies do not inhibit HRV-14 adsorption, as antibodies are too big to bind to the canyon.
The receptor ICAM-1 is expressed at the surface of human endothelial and some epithelial cells, monocytes and lymphocyte sub-populations, and is involved in immunological and inflammatory processes.113 The natural ligand of ICAM-1 is the lymphocyte-function-associated antigen-1 (LFA-1) which functions in cell-to-cell adhesion.114 The protein consists of five immunoglobulin-like extracellular domains and belongs to the immunoglobulin superfamily. ICAM-1 is the specific receptor of human rhinoviruses of the major group which comprises 90 of 120 HRV serotypes.115-117 Investigations of point mutations in ICAM-1 and binding studies with HRV-14 revealed that mainly domain 1 of ICAM-1 is involved in HRV-14 binding.118-120 The first hypothetical domain structure was based on sequence alignment, prediction of the secondary structure, and comparison with the tertiary structure of IgG.118 Some years ago, an X-ray structure of domains 1 and 2 at 2.1 Å resolution became available (Fig. 5).120 The sequence of an ICAM-1-derived seed peptide was selected on basis of the X-ray model of domain 1 in combination with binding studies employing ICAM-1 mutants and HRV-14.118-120 This ICAM-1-derived peptide represents a continuous sequence of 9 amino acid residues from a loop in positions 43 to 51 of domain 1 (Fig. 5). This surface-exposed stretch of residues serves as a candidate for constructing an ICAM-1-derived peptide. The peptide might be used for blocking the HRV-14 canyon for recognition of ICAM-1. This could be tested by inhibition of virus adsorption at ICAM-1. The following nonapeptide, therefore, may serve as the seed-peptide for the PepMaker approach: LLPGNNRKV. Table 4 shows two small peptide libraries generated by PepMaker. Library 1 was generated with σ = 0.05, library 2 was generated with s = 0.5. Depending on the biological activity of the seed peptide—i.e., activity in cell-protection against HRV-14 infection (which has not been investigated until now)—we assume that there are several peptides in the vicinity of the seed peptide revealing comparable or possibly even higher cell protective activity. Especially in cases where a large amount of peptide is required for a biological test system this approach might be helpful to select a few promising peptide candidates for activity testing. However, at least one peptide with the desired biological function must already be known.
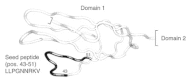
Figure 5
Backbone structure of domain 1 of human ICAM-1 (PDb code: 1iam). The location of the seed peptide is shown in dark color.
Table 4
Peptide libraries generated with PepMaker; The seed peptide was LLPGNNRKV. d: distance to the seed peptide.
Peptide de novo design was the first successful case of evolutionary design employing a neural network for the fitness function.102,121,122 In Fig. 6, fitness landscapes are shown, which were generated by a neural network system that was trained on the prediction of eubacterial signal peptidase I cleavage sites in amino acid sequences.121 A set of known peptidase substrates served as the training data for feature extraction by ANN.121,123 It turned out that a chemical space spanned by the amino acid properties “hydrophobicity” and “volume” was suited for this particular application. The fitness functions for the three selected sequence positions shown are smooth and separate the set of natural amino acid residues into “low-fitness” and “high-fitness” candidates. In a series of in machina design experiments the alanine residue was selected as best-suited in position -3 (numbered relative to the signal peptidase cleavage site) (Fig. 6a), tryptophan in position -2 (Fig. 6b), and glycine in position -1 (Fig. 6c). Due to the continuous nature of the fitness landscapes evolutionary search for idealized substrates was straightforward. The design run converged after only 52 optimization steps, changing the initial parent sequence FICLTMGYIC into the functional enzyme substrate FFFFGWYGWA*RE (the asterisk denotes the signal peptidase I cleavage site). Its biological activity—activity as a substrate—is comparable to wild-type sequences, which was proven by an in vivo protein secretion assay and subsequent mass-spectrometric sequence and fragment analysis.73 The X-ray structure of the catalytic domain of signal peptidase I from Escherichia coli was published after these ligand-based design experiments were completed.124,125 It is evident from the structure of the active site that the model peptide excellently compliments the structural and electrostatic features within the enzyme.
In a further application of evolutionary search guided by neural networks, antigen-mimicking peptides were developed “from first principles”.74 This design approach included a round of bench experiments for data generation and subsequent computer-assisted evolutionary optimization. The five-step procedure represents a special version of the design cycle shown in Fig. 1.4:
- Identification of a single compound with some desired activity, e.g., by expert knowledge, data base or random screening, combinatorial libraries, or phage display;
- Generation of a focusing library taking the compound obtained in step 1 as a “seed structure”. A limited set of variants is generated approximately Gaussian-distributed in some physicochemical space around the “seed peptide”;
- Synthesis and testing of the new variants for their bioactivity;
- Training of artificial neural networks providing heuristic (Q)SAR based on the activities measured in step 3;
- Computer-based evolutionary search for highly active compounds taking the network models as the fitness function.
A novel peptide was identified fully preventing the positive chronotropic effect of anti-b1-adrenoceptor auto-antibodies from the serum of patients with idiopathic dilated cardiomyopathy (DCM).74 In an in vitro assay the designed active peptide showed more significant effects compared to the natural epitope. The idea was to test whether it is feasible to derive artificial epitope sequences that might be used as potential immuno-therapeutical agents following the design strategy described above. The model peptide GWFGGADWHA exhibits an activity comparable to its natural counterpart (ARRCYNDPKC) but has a significantly different residue sequence. Selection of such antibody-specific “artificial antigens” may be regarded as complementary to natural clonal B-cell selection leading to the production of specific antibodies. The peptide-antibody interaction investigated can be considered as a model of specific peptide-protein interactions. These results demonstrate that computer-based evolutionary searches can generate novel peptides with substantial biological activity.
TOPAS: Fragment-Based Design of Drug-Like Molecules
The software tool TOPAS (TOPology Assigning System)provides an example of a fragment-based molecular structure generator and optimizer based on an evolution strategy.59 Its basic idea is similar to the GA-based software LEA conceived by Douget and coworkers.58 In both programs SMILES representations of molecules are varied by genetic operators. The SMILES strings are assembled from a compilation of molecular building blocks. In the case of TOPAS, these were generated by retro-synthetic fragmentation of the Derwent World Drug Index (WDI version of 1997; as distributed by Daylight Chemical Information Systems Inc., Irvine, CA, USA), in LEA the fragment libraries contain a diverse collection of selected building blocks. The idea behind the TOPAS fragment set is that re-assembly of such drug-derived building blocks by a limited set of chemical reactions might lead to chemically feasible novel structures, from both the medicinal chemistry and the synthesis planning perspective.
To compile a database of drug-like building blocks for evolutionary de novo design by TOPAS, all 36,000 structures contained in the WDI, which had an entry related to “mechanism” or “activity”, were subjected to retro-synthetic fragmentation. The reactions are listed in Table 5. This approach is identical to the original RECAP procedure developed by Hann and coworkers.126 In total, 24,563 distinctive building blocks were generated (“stock of structures”). Of course, there are many other ways to create fragment sets, and we found it useful to have several such collections available for different design tasks. For example, if the task was to design a potential GPCR modulating agent, then a fragment set generated from known GPCR modulators would be a reasonable choice.
TOPAS is grounded on a (1,λ) evolution strategy (see Chapter 1). Starting from an arbitrary point in search space, a set of λ variants are generated, satisfying a bell-shaped distribution centered in the chemical space co-ordinates of the parent structure. This means that most of the variants are very similar to their parent, and with increasing distance in chemical space the number of offspring decreases. In the original implementation of TOPAS, fitness was defined as pair-wise similarity between the template (reference structure) and the offspring. Two different concepts were realized to measure similarity: i) 2D structural similarity as defined by the Tanimoto index on Daylight's 2D fingerprints (Eq. 2.9), and ii) 2D topological pharmacophore similarity (see Chapter 2). Tanimoto similarity varies between zero and one, where the value of 1 indicates structural identity. Topological pharmacophore similarity values vary between zero (indicating identical pharmacophore distribution in the two molecules) and positive values indicating varying degrees of pharmacophore similarity. Thus, optimal fitness values are 1 for the Tanimoto measure, and 0 for the pharmacophore similarity measure. Additional penalty terms such as a modified “rule of 5” and a topological shape filter were added to the fitness function to avoid undesired structures (Note: one particular advantage of the TOPAS approach is that arbitrary quality and penalty functions can be included to compute fitness).
Variant structures are derived from the parent molecule, SP, in a four-step process, following the algorithm outlined in Chapter 1:
- Exhaustive retro-synthetic fragmentation of Sp;
- Random selection of one of the generated fragments;
- Substitution of this fragment by the one from the stock of building blocks having a pair-wise similarity index that is close to Gaussian-distributed random number;
- Virtual synthesis to assemble the novel chemical structure.
To demonstrate step 1, the thrombin inhibitor NAPAP was subjected to fragmentation by TOPAS. Reaction scheme 1 (amide bond cleavage) was applied twice, and reaction 11 (sulfonamide bond cleavage) occurred once, resulting in four fragments (Fig. 7). Depending on the similarity measure selected and the width the variant distribution, offspring is generated, e.g., by subjecting the benzamidine residue to mutation (Fig. 8). The other three fragments remain unchanged. For fitness calculation, each of the new structures is compared to the template, and the most similar one will become the parent of the next generation.
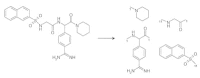
Figure 7
Fragmentation of the thrombin inhibitor NAPAP. Application of the fragmentation scheme given in Table 5 leads to four fragments.
This mutation strategy offers the following advantages:
- An adaptive stochastic search is performed in chemical space;
- The type of molecules that are virtually generated is not restricted to a predefined combinatorial class (e.g., peptides, Ugi-reaction products);
- Novel structures are assembled from drug-derived building blocks using a set of “simple” chemical reactions;
- A large diversity of molecular fragments can be explored.
An example of a TOPAS design experiment aiming at the generation of a NAPAP-like structure is shown in Figure 9. The Tanimo index was used as the fitness measure. Initially, a random structure was generated from the stock of building blocks (“parent” of the first generation). The Tanimoto similarity to NAPAP was 0.31 reflecting a great dissimilarity, as expected. In each of the following generations 100 variants were systematically generated by TOPAS, and the best of each generation was selected as the parent for the subsequent generation. Following this scheme, novel molecules were assembled which exhibited a significantly increased fitness (Fig. 9). After only 12 optimization cycles the process converged at a high fitness level (approximately 0.86), and the standard deviation, σ of the variant distributions around the parent structures decreased. The course of s indicates that first comparably broad distributions were generated (large diversity), after some generations, however, a peak in the fitness landscape was climbed (restricted diversity). The parent structures of each generation are shown in Figure 10. The resulting final design shares a significant set of substructure elements with the NAPAP template. Key features for thrombin binding evolved—the benzamidine group forming hydrogen bonds with Asp-189 at the bottom of the thrombin P1 pocket, a sulfonamide interacting with the backbone carbonyl of Gly-216, and the lipophilic para-tolyl and piperidine rings filling a large lipophilic pocket of the thrombin active site cleft. Automated docking by means of FlexX essentially reproduced the NAPAP binding mode.59 (Note: molecular docking and subsequent scoring of the docked solutions was not used in the selection of the final pool of solutions). This de novo design experiment demonstrated that the algorithm can be used for a fast guided search in a very large chemical space, ending up with rational proposals for novel molecular structures that are similar to a given template.
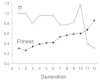
Figure 9
Course of fitness (Tanimoto similarity to NAPAP) and the width (diversity) s of the offspring distribution (“diversity”) during a TOPAS design experiment (cf Fig. 10).

The previous design example was based on similarity searching alone. In this last Section we illustrate a possible interplay of similarity searching, de novo design, and molecular modeling. Here the aim was to design a novel potassium channel (Kv1.5) blocking agent—a so-called “fast follower”—taking a known inhibitor as a starting point.
Inhibitors of voltage-dependent potassium channels induce a decrease in potassium ion movement across the plasma membrane. The biological function of these ion channels is multiple. In cardiac cells a decreased potassium flux leads to the prolongation of the action potential. Increasing myocardial refractoriness by prolonging the action potential can be useful for the treatment of cardiac arrhythmia. Blocking potassium channels and depolarizing the resting membrane potential has been shown to regulate a variety of biological processes, like T-cell activation under immune-reactive conditions. Inherited disorders of voltage-gated ion channels are a recently recognized etiology of epilepsy in the developing and mature central nervous system,127 and quite a number of neurodegenerative diseases are known to be associated or caused by potassium channelopathies.128
The root of this fast-follower design experiment was the structure (a) shown in Fig. 11, which was identified as a potent Kv1.5 blocking agent by Castle and coworkers at Icagen Inc.129 In our electrophysiological studies this compound had an IC50 of 0.1 μM.60 The first hurdle to take was to identify a novel molecular scaffold that may serve as a lead structure candidate (step 1 in Fig. 11). TOPAS was used for this purpose. New structures were generated through assembly or modification of the TOPAS building blocks. A naphthylsulfonamide motif appeared in many of the designs. It was expected that these molecules would be chemically feasible and have some drug-like properties, because the fragments were originally obtained from known bioactive molecules. In fact, the structure (b) shown in Fig. 11 has Kv1.5 blocking potential (IC50= 7 μM).
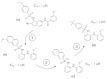
To further optimize structure (b) a pharmacophore matching routine was applied to align it to the original template (a) (step 2 in Fig. 11). The modeling program MOLOC was used for this purpose.130 In this study it was found that removal of a methoxy group present in structure (b) might be beneficial to activity. This hypothesis was proven by electrophysiological studies yielding an IC50 of 1 μM for structure (c).
The final optimization process (Step 3 in Fig. 11) was a CATS similarity search in a virtual combinatorial library. Sulfonylchlorides available from the Roche corporate compound collection were virtually assembled to the free amino functionality of the molecular core of structure (c). A topological pharmacophore similarity to the original template (a)—as implemented in CATS—was determined for each member of the combinatorial library. This procedure led to molecule (d) yielding an IC50 below 1 μM, which is in the same order of magnitude as the template (a). The new structure would now be ready to enter a medicinal chemistry project.
This example shows that evolutionary de novo design algorithms are able to generate novel bioactive classes of compounds. The cyclic interplay between computational design and human reasoning (hypothesis generation), chemical synthesis (structure generation), and biological testing (quality assessment) represents a prototype of Adaptive Drug Design. This and similar strategies will surely provide a basis in the future for drug discovery and lead generation.
Concluding Remarks
Evolutionary algorithms have undoubtedly proven their usefulness for molecular de novo design. Their basic idea is to perform an adaptive stochastic search based on a guided trial-and-error procedure. It must be emphasized that they do not represent the optimal solution to drug design, but may serve as a general optimization strategy, thereby complementing more specific approaches. Their great appeal is the intuitively comprehensible basic algorithm, which perfectly integrates with experimental drug design cycles. Furthermore, they can easily be implemented in molecular design software. EAs excel in situations where the fitness function and the search space are both multidimensional and multimodal. On the other hand, compared to some other design methods EAs can be slow, suffering from premature convergence and leading to sub-optimal solutions. The authors are convinced that with the continuously increasing speed of computers and in combination with specifically tailored chemistry—in particular advanced parallel medicinal chemistry—EAs will increasingly have larger impact on the future drug discovery process and enrich the medicinal chemists' arsenal of structures with novel molecules. The speed of chemical synthesis can be a rate-limiting step in the optimization cycle if conventional routes of de novo synthesis are followed. Parallel medicinal chemistry concepts might provide a solution to this problem, as they represent a smart integration of computational design and combinatorial synthesis.
In the book Hidden Order—How Adaptation Builds Complexity John H. Holland presented a list of seven basic properties and mechanisms that are common to all complex adaptive systems:131 aggregation, non-linearity, flows, diversity, tagging, internal models, and building blocks. Some of these basics have been discussed in the present volume, and simplified computer models have been presented that may serve as a starting point for the implementation of more advanced adaptive systems for drug design. The task of formulating a theory for these systems is difficult, especially because the behaviour of a whole complex adaptive system is more than a simple sum of the behaviours of its parts; complex adaptive systems abound in non-linearities. We must face the fact that the drug design process is inherently non-linear, and the different ways of looking at it lead to different emphases and different models. John H. Holland wrote:131
“Adaptive agents come in startling variety, and their strategies are correspondingly diverse, so we need a language powerful enough to define the feasible strategies for these agents. [..] And we must provide well-defined evolutionary procedures that enable agents to acquire learned anticipations and innovations.”
It is evident that current drug design models are far from perfect; it is also evident that it will be extremely difficult—if not impossible—to formulate a single theory that directly guides the experiment. Selection guided by taste and experience is crucial, and an adaptive drug design process involves a perpetual interplay between theory and experiment.
References
- 1.
- Willett P. Genetic algorithms in molecular recognition and design. Trends Biotechnol. 1995;13:516–521. [PubMed: 8595137]
- 2.
- Böhm HJ. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des. 1992;6:61–78. [PubMed: 1583540]
- 3.
- Roe DC, Kuntz ID. BUILDER v.2: Improving the chemistry of a de novo design strategy. J Comput Aided Mol Des. 1995;9:269–282. [PubMed: 7561978]
- 4.
- Lauri G, Bartlett PA. CAVEAT: A program to facilitate the design of organic molecules. J Comput Aided Mol Des. 1994;8:51–66. [PubMed: 8035213]
- 5.
- Böhm HJ. Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J Comput Aided Mol Des. 1998;12:309–323. [PubMed: 9777490]
- 6.
- Böhm HJ, Banner DW, Weber L. Combinatorial docking and combinatorial chemistry: Design of potent non-peptide thrombin inhibitors. J Comput Aided Mol Des. 1999;13:51–56. [PubMed: 10087499]
- 7.
- Stahl M, Böhm HJ. Development of filter functions for protein-ligand docking. J Mol Graph Model. 1998;16:121–132. [PubMed: 10434251]
- 8.
- Böhm HJ, Stahl M. Rapid empirical scoring functions in virtual screening applications. Med Chem Res. 1999:445–462.
- 9.
- Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins. 1990;8:195–202. [PubMed: 2281083]
- 10.
- Miranker A, Karplus M. An automated method for dynamic ligand design. Proteins. 1995;23:472–490. [PubMed: 8749844]
- 11.
- Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. J Comp Chem. 1992;13:505–524.
- 12.
- Holloway MK, Wai JM, Halgren TA, Fitzgerald PM, Vacca JP, Dorsey BD. et al. A priori prediction of activity for HIV-1 protease inhibitors employing energy minimization in the active site. J Med Chem. 1995;38:305–317. [PubMed: 7830273]
- 13.
- Luty BA, Wassermann ZR, Stouten P F W, Hodge CN, Zacharias M, McCammon JA. Molecular mechanics/grid method for the evaluation of ligand-receptor interactions. J Comput Chem. 1995;16:454–464.
- 14.
- Grootenhuis P D J, van Galen P J M. Correlation of binding affinities with non-bonded interaction energies of thrombin-inhibitor complexes. Acta Cryst. 1995;D51:560–566. [PubMed: 15299844]
- 15.
- Viswanadhan VN, Reddy MR, Wlodawer A, Varney MD, Weinstein JN. An approach to rapid estimation of relative binding affinities of enzyme inhibitors: Application to peptidomimetic inhibitors of the human immunodeficiency virus type 1 protease. J Med Chem. 1996;39:705–712. [PubMed: 8576913]
- 16.
- Vieth M, Hirst JD, Kolinski A, Brooks CL. Assessing energy functions for flexible docking. J Comp Chem. 1998;19:1612–1622.
- 17.
- Shoichet BK, Leach AR, Kuntz ID. Ligand solvation in molecular docking. Proteins. 1999;34:4–16. [PubMed: 10336382]
- 18.
- Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1449. [PubMed: 7761829]
- 19.
- Zhang T, Koshland D E Jr. Computational method for relative binding energies of enzyme-substrate complexes. Protein Sci. 1996;5:348–356. [PMC free article: PMC2143351] [PubMed: 8745413]
- 20.
- Schapira M, Trotov M, Abagyan R. Prediction of the binding energy for small molecules, peptides and proteins. J Mol Recognit. 1999;12:177–190. [PubMed: 10398408]
- 21.
- Majeux N, Scarsi M, Apostolakis J, Ehrhardt C, Caflisch A. Exhaustive docking of molecular fragments with electrostatic solvation. Proteins. 1999;37:88–105. [PubMed: 10451553]
- 22.
- Verkhivker G, Appelt K, Freer ST, Villafranca JE. Empirical free energy calculations of ligand-protein crystallographic complexes. I. Knowledge-based ligand-protein interaction potentials applied to the prediction of human immunodeficiency virus 1 protease binding affinity. Protein Eng. 1995;8:677–691. [PubMed: 8577696]
- 23.
- Wallqvist A, Jernigan RL, Covell DG. A preference-based free-energy parameterization of enzyme-inhibitor binding. Applications to HIV-1-protease inhibitor design. Protein Sci. 1995;4:1881–1903. [PMC free article: PMC2143230] [PubMed: 8528086]
- 24.
- DeWitte RS, Shakhnovich EI. SMoG: De novo design method based on simple, fast, and accurate free energy estimates. 1. methodology and supporting evidence. J Am Chem Soc. 1996;118:11733–11744.
- 25.
- Mitchell J B O, Laskowski RA, Alex A, Thornton JM. BLEEP-Potential of mean force describing protein ligand interactions: I Generating potential. J Comput Chem. 1999;20:1165–1177.
- 26.
- Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: a simplified potential approach. J Med Chem. 1999;42:791–804. [PubMed: 10072678]
- 27.
- Muegge I, Martin YC, Hajduk PJ, Fesik SW. Evaluation of PMF scoring in docking weak ligands to the FK506 binding protein. J Med Chem. 1999;42:2498–2503. [PubMed: 10411471]
- 28.
- Kollmann PA. Advances and continuing challenges in achieving realistic and predictive simulations of the properties of organic and biological molecules. Acc Chem Res. 1996;29:461–469.
- 29.
- Aqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. [PubMed: 8177887]
- 30.
- Hansson T, Marelius J, Aqvist J. Ligand binding affinity prediction by linear interaction energy methods. J Comput Aided Mol Des. 1998;12:27–35. [PubMed: 9570087]
- 31.
- Rarey M, Kramer B, Lengauer T. Docking of hydrophobic ligands with interaction-based matching algorithms. Bioinformatics. 1999;15:243–250. [PubMed: 10222412]
- 32.
- Gohlke H, Hendlich M, KleBe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol. 2000;295:337–356. [PubMed: 10623530]
- 33.
- Stahl M. Structure-based liBrary designIn: Böhm HJ, Schneider G, eds. Virtual Screening for Bioactive Molecules Weinheim, New York: Wiley-VCH, 2000229–264.
- 34.
- Good A, Mason JS, Pickett SD. Pharmacophore pattern application in virtual screening, library design and QSARIn: Böhm HJ, Schneider G, eds. Virtual Screening for Bioactive Molecules Weinheim, New York: Wiley-VCH, 2000131–159.
- 35.
- Müller K. Ed. De Novo Design Leiden: Escom 1995.
- 36.
- Böhm HJ. Computational tools for structure-based ligand design. Prog Biophys Mol Biol. 1996;66:197–210. [PubMed: 9284450]
- 37.
- Kirkpatrick DL, Watson S, Ulhaq S. Structure-based drug design: Combinatorial chemistry and molecular modeling. Comb Chem High Throughput Screen. 1999;2:211–221. [PubMed: 10469881]
- 38.
- Gane PJ, Dean PM. Recent advances in structure-based rational drug design. Curr Opin Struct Biol. 2000; 10:401–404. [PubMed: 10981625]
- 39.
- Klebe G. Recent developments in structure-based drug design. J Mol Med. 2000;78:269–281. [PubMed: 10954199]
- 40.
- Böhm HJ, Stahl M. Structure-based library design: molecular modelling merges with combinatorial chemistry. Curr Opin Chem Biol. 2000;4:283–286. [PubMed: 10826972]
- 41.
- Devillers J.Ed. Genetic Algorithms in Molecular Modeling New York: Adacemic Press, 1996. [PMC free article: PMC172907]
- 42.
- Clark DE.Ed. Evolutionary Algorithms in Molecular Design Weinheim: Wiley-VCH, 2000. [PMC free article: PMC149136]
- 43.
- De Julian-Ortiz JV. Virtual Darwinian drug design: QSAR inverse proBlem, virtual combinatorial chemistry, and computational screening. Comb Chem High Throughput Screen. 2001;4:295–310. [PubMed: 11375744]
- 44.
- Gillet VJ. De novo molecular designIn: Clark DE, ed. Evolutionary Algorithms in Molecular Design Weinheim: Wiley-VCH, 200049–69.
- 45.
- Clark DE, Frenkel D, Levy SA, Li J, Murray CW, Robson B. et al. PRO-LIGAND: An approach to de novo molecular design. 1. Application to the design of organic molecules. J Comput Aided Mol Des. 1995;9:13–32. [PubMed: 7751867]
- 46.
- Frenkel D, Clark DE, Li J, Murray CW, Robson B, Waszkowycz B, Westhead DR. PRO_LIGAND: An approach to de novo molecular design. 4. Application to the design of peptides. J Comput Aided Mol Des. 1995;9:213–225. [PubMed: 7561974]
- 47.
- LeapFrog is availaBle from TRIPOS Inc,1699 South Hanley Road, Suite 303, St Louis, MO 63144, USA .
- 48.
- Glen RC, Payne AW. A genetic algorithm for the automated generation of molecules within constraints. J Comput Aided Mol Des. 1995;9:181–202. [PubMed: 7608749]
- 49.
- Blaney JM, Dixon JS, Weininger D. Molecular Graphics Society Meeting on Binding Sites: Characterising and Satifying Steric and Chemical Restraints York, UK,
March 1993. Weininger D, WO095/01606. - 50.
- Venkatasubramanian V, Chan K, Caruthers JM. Computer-aided molecular design using genetic algorithms. Computers Chem Eng. 1995;18:833–844.
- 51.
- Venkatasubramanian V, Sundaram A, Chan K, Caruthers JM. Computer-aided molecular design using neural networks and genetic algorithms In: Devillers J, ed. Genetic Algorithms in Molecular Modeling New York: Adacemic Press, 1996271–302.
- 52.
- Venkatasubramanian V, Chan K, Caruthers JM. Evolutionary design of molecules with desired properties using a genetic algorithm. J Chem Inf Comput. Sci1998;38: 1177–1191.
- 53.
- Nachbar RB. Molecular evolution: A hierarchical representation for chemical topology and its automated manipulationIn: Proceedings of the Third Annual Genetic Programming Conference Madison: University of Wisconsin 22-25
July 1998246–253. - 54.
- Globus A, Lawton J, Wipke T. Automatic molecular design using evolutionary techniques. Nanotechnology. 1999;10:290–299.
- 55.
- Devillers J, Putavy C. Designing biodegradable molecules from the combined use of a backpropagation neural network and a genetic algorithm In: Devillers J, ed. Genetic Algorithms in Molecular Modeling New York: Academic Press, 1996303–314.
- 56.
- Weber L, Wallbaum S, Broger C, Gubernator K. Optimization of the biological activity of combinatorial compound libraries by a genetic algorithm. Angew Chemie Int Ed Engl. 1995;34:2280–2282.
- 57.
- Illgen K, Enderle T, Broger C, Weber L. Simulated molecular evolution in a full combinatorial library. Chem Biol. 2000;7:433–441. [PubMed: 10873838]
- 58.
- Douguet D, Thoreau E, Grassy G. A genetic algorithm for the automated generation of small organic molecules: drug design using an evolutionary algorithm. J Comput Aided Mol Des. 2000;14:449–466. [PubMed: 10896317]
- 59.
- Schneider G, Lee ML, Stahl M, Schneider P. De novo design of molecular architectures by evolutionary assembly of drug-derived building blocks. J Comput Aided Mol Des. 2000;14:487–494. [PubMed: 10896320]
- 60.
- Schneider G, Clement-Chomienne O, Hilfiger L, Schneider P, Kirsch S, Böhm HJ. et al. Virtual Screening for Bioactive Molecules By Evolutionary De Novo Design. Angew Chem Int Ed Engl. 2000;39:4130–4133. [PubMed: 11093229]
- 61.
- Weininger DJ. SMILES-A chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28:31–36.
- 62.
- Gasteiger J, Rudolph C, Sadowski J. Automatic generation of 3D-atomic coordinates for organic molecules. Tetrahedron Comp Method. 1990;3:537–547.
- 63.
- Sadowski J, Gasteiger J. From atoms and bonds to three-dimensional atomic coordinates: automatic model builders. Chem Reviews. 1993;93:2567–2581.
- 64.
- Rusinko A I I I, Skell JM, Balducci R, McGarity CM, Pearlman RS. Concord, a program for the rapid generation of high quality approximate 3-dimensional molecular structures The University of Texas at Austin and Tripos Associates, St. Louis, MO USA, 1988.
- 65.
- Pearlman RS. Rapid generation of high quality approximate 3D molecular structures. Chem Des Aut News. 1987;2:1–6.
- 66.
- Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. [PubMed: 8780787]
- 67.
- Kramer B, Rarey M, Lengauer T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins. 1999;37:228–241. [PubMed: 10584068]
- 68.
- Shoichet BK, ID Kuntz. Protein docking and complementarity. J Mol Biol. 1991;221:327–346. [PubMed: 1920412]
- 69.
- Gschwend DA, Good AC, Kuntz ID. Molecular docking towards drug discovery. J Mol Recognit. 1996;9:175–186. [PubMed: 8877811]
- 70.
- Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. [PubMed: 9385547]
- 71.
- Walters WP, Stahl MT, Murcko MA. Virtual screening-An overview. Drug Discovery Today. 1998;3:160–178.
- 72.
- Schneider G, Grunert HP, Schuchhardt J, Wolf KU, Müller G, Habermehl KO. et al. A peptide selection scheme for systematic evolutionary design and construction of synthetic peptide libraries. Min Invas Med. 1995;6:106–115.
- 73.
- Wrede P, Landt O, Klages S, Fatemi A, Hahn U, Schneider G. Peptide design aided by neural networks: biological activity of artificial signal peptidase I cleavage sites. Biochemistry. 1998;37:3588–3593. [PubMed: 9530285]
- 74.
- Schneider G, Schrödl W, Wallukat G, Müller J, Nissen E, Rönspeck W. et al. Peptide design by artificial neural networks and computer-based evolutionary search. Proc Natl Acad Sci USA. 1998;95:12179–12184. [PMC free article: PMC22805] [PubMed: 9770460]
- 75.
- Gausepohl H, Boulin C, Kraft M, Frank RW. Automated multiple peptide synthesis. Pept Res. 1992;5:315–320. [PubMed: 1283542]
- 76.
- Kramer A, Schneider-Mergener J. Synthesis and screening of peptide libraries on cellulose membrane supports. Methods Mol Biol. 1998;87:25–39. [PubMed: 9523256]
- 77.
- Kramer A, Keitel T, Winkler K, Stocklein W, Höhne W, SchneiderMergener J. Molecular basis for the binding promiscuity of an anti-p24 (HIV-1) monoclonal antibody. Cell. 1997;91:799–809. [PubMed: 9413989]
- 78.
- Schneider G, Wrede P. The rational design of amino acid sequences by artificial neural networks and simulated molecular evolution: de novo design of an idealized leader peptidase cleavage site. Biophys J. 1994;66:335–344. [PMC free article: PMC1275700] [PubMed: 8161687]
- 79.
- Huang P, Kim S, Loew G. Development of a common 3D pharmacophore for delta-opioid recognition from peptides and non-peptides using a novel computer program. J Comput Aided Mol Des. 1997;11:21–28. [PubMed: 9139108]
- 80.
- Mee RP, Auton TR, Morgan PJ. Design of active analogues of a 15-residue peptide using D-optimal design, QSAR and a combinatorial search algorithm. J Pept Res. 1997;49:89–102. [PubMed: 9128105]
- 81.
- Dayhoff MO, Eck RV. A model of evolutionary change in proteins In: Dayhoff MO, ed. Atlas of Protein Sequence and Structure Washington DC:National Biomed Res Found,1968345.
- 82.
- Eigen M, Winkler-Oswatitsch R, Dress A. Statistical geometry in sequence space. A method of quantitative comparative sequence analysis. Proc Natl Acad Sci USA. 1988;85:5913–5917. [PMC free article: PMC281875] [PubMed: 3413065]
- 83.
- Eigen M, McCaskill JS, Schuster P. The molecular quasi-species. Adv Chem Phys. 1989;75:149–263.
- 84.
- Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. [PubMed: 4843792]
- 85.
- Kimura M. The Neutral Theory of Molecular Evolution Cambridge: University Press 1983.
- 86.
- Miyata T, Miyazawa S, Yasunaga T. Two types of amino acid substitutions in protein evolution. J Mol Evol. 1979;12:219–236. [PubMed: 439147]
- 87.
- Rao J K M. New scoring matrix for amino acid residue exchange based on residue characteristic physical parameters. Int J Peptide Protein Res. 1987;29:276–279. [PubMed: 3570667]
- 88.
- Schuster P, Stadler PF. Landscapes: Complex optimization problems and biopolymer structures. Comput Chem. 1994;18:295–324. [PubMed: 7524995]
- 89.
- Fontana W, Stadler PF, BornBerg-Bauer EG, Griesmacher T, Hofacker IL, Tacker M. et al. RNA folding and combinatory landscapes. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics. 1993;E 47:2083–2099. [PubMed: 9960229]
- 90.
- Kauffman SA. The Origin of Order-Self-Organization and Selection in Evolution New York/Oxford: Oxford University Press, 1993.
- 91.
- Shannon CE. A mathematical theory of communication. Bell System Tech J. 1948;27:379–623.
- 92.
- Schneider TD. Measuring molecular information. J Theor Biol. 1999; 201:87–92. [PubMed: 10534438]
- 93.
- Schneider G, Gutknecht EM, Kansy M, Böhm HJ. Diversity assessment tools: Proposed strategy for implementation and impact on the lead discovery process Roche Progress Report1998. unpublished.
- 94.
- Godden JW, Bajorath J. Shannon entropy-A novel concept in molecular descriptor and diversity analysis. J Mol Graph Model. 2000;18:73–76. [PubMed: 10935210]
- 95.
- Zuegge J, Ebeling M, Schneider G. H-BloX: Visualizing alignment block entropies. J Mol Graph Model. 2001;19:304–306. [PubMed: 11449568]
- 96.
- Ash RB. Information TheoryDover: Mineola 1965 reprinted 1990.
- 97.
- Ebeling W, Engel A, Feistel R. Physik der Evolutionsprozesse Berlin: Akademie-Verlag, 1990.
- 98.
- Baldi P, Brunak S. Bioinformatics-The Machine Learning Approach Cambridge: MIT Press, 1998.
- 99.
- Schneider TD, Stephens RM. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. [PMC free article: PMC332411] [PubMed: 2172928]
- 100.
- Durbin R, Eddy S, Krogh A, Mitchinson G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids Cambridge: Cambridge University Press, 1998.
- 101.
- Schneider G, Wrede P. Artificial neural networks for computer-aided molecular design. Prog Biophys Mol Biol. 1998;70:175–222. [PubMed: 9830312]
- 102.
- Schneider G, Schuchhardt J, Wrede P. Peptide design in machina: development of artificial mitochondrial protein precursor cleavage-sites by simulated molecular evolution. Biophys J. 1995;68:434–447. [PMC free article: PMC1281708] [PubMed: 7696497]
- 103.
- Altschul SF. Amino acid substitution matrices from an information theoretic perspective. J Mol Biol. 1991;219:555–565. [PMC free article: PMC7130686] [PubMed: 2051488]
- 104.
- Henikoff S, Henikoff JG. Performance evaluation of amino acid substitution matrices. Proteins. 1993;17:49–61. [PubMed: 8234244]
- 105.
- Trinquier G, Sanejouand YH. Which effective property of amino acids is best preserved by the genetic code? Protein Engineering. 1998;11:153–169. [PubMed: 9613840]
- 106.
- Rossmann MG, Vriend G. et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature. 1985;317:145. [PubMed: 2993920]
- 107.
- Rossmann MG, Palmenberg AC. Conservation of the putative receptor attachment site in picornaviruses. Virology. 1988;164:373–382. [PubMed: 2835857]
- 108.
- Olson NH, Kolatkar PR, Oliveira MA, Cheng RH, Greve JM, McClelland A. et al. Structure of a human rhinovirus complexed with its receptor molecule. Proc Natl Acad Sci USA. 1993;90:507–511. [PMC free article: PMC45692] [PubMed: 8093643]
- 109.
- Colonno RJ, Condra JH, Mizutani S, Callahan PL, Davies ME, Murcko MA. Evidence for the direct involvement of the rhinovirus canyon in receptor binding. Proc Natl Acad Sci USA. 1988;85:5449–5453. [PMC free article: PMC281774] [PubMed: 2840661]
- 110.
- Rossmann MG, Rueckert RR. What does the molecular structure of viruses tell us about viral functions. Microbiol Sci. 1987;4:206–214. [PubMed: 3153613]
- 111.
- McClintock PR, Prabhakar BS, Notkins AL. Anti-idiotypic antibodies to monoclonal antibodies that neutralize Coxsackie Virus B4 do not recognize viral receptors. Virology. 1986;150:352–360. [PubMed: 3962185]
- 112.
- Cromwell RL. Cellular receptors in virus infections. Am Soc Microbiol News. 1987;53:422.
- 113.
- Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL-1 and interferon, tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1). J Immunol. 1986;137:245–254. [PubMed: 3086451]
- 114.
- Kishimoto TK, Larson RS, Corbi AL, Dustin ML, Staunton DE, Springer TA. The leukocyte integrins. Adv Immunol. 1989;46:146–182. [PubMed: 2551146]
- 115.
- Colonno RJ, Callahan PL, Long WJ. Isolation of a monoclonal antibody that blocks attachment of the major group of human rhinoviruses. J Virol. 1986;57:7–12. [PMC free article: PMC252692] [PubMed: 3001366]
- 116.
- Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 1989;56:849–853. [PubMed: 2538244]
- 117.
- Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW. et al. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56:839–847. [PubMed: 2538243]
- 118.
- Staunton DE, Dustin ML, Erickson HP, Springer TA. The arrangement of the immunoglobulin-like domains of ICAM-1 and the binding sites for LFA-1 and rhinovirus. Cell. 1990;61:243–254. [PubMed: 1970514]
- 119.
- Register RB, Uncapher CR, Naylor AM, Lineberger DW, Colonno RJ. Human-murine chimeras of ICAM-1 identify amino acid residues critical for rhinovirus and antibody binding. J Virol. 1991;65:6589–6596. [PMC free article: PMC250720] [PubMed: 1719231]
- 120.
- Bella J, Kolatkar PR, Marlor CW, Greve JM, Rossmann M. The structure of the two amino-terminal domains of human ICAM-1 suggests how it functions as a rhinovirus receptor and as an LFA-1 integrin ligand. Proc Natl Acad Sci USA. 1998;95:4140–4145. [PMC free article: PMC22455] [PubMed: 9539703]
- 121.
- Schneider G, Wrede P. The rational design of amino acid sequences by artificial neural networks and simulated molecular evolution: de novo design of an idealized leader peptidase cleavage site. Biophys J. 1994;66:335–344. [PMC free article: PMC1275700] [PubMed: 8161687]
- 122.
- Schneider G, Schuchhardt J, Wrede P. Development of simple fitness landscapes for peptides by artificial neural filter systems. Biol Cybern. 1995;73:245–254. [PubMed: 7548312]
- 123.
- Schneider G, Wrede P. Development of artificial neural filters for pattern recognition in protein sequences. J Mol Evol. 1993;36:586–595. [PubMed: 8350352]
- 124.
- Paetzel M, Dalbey RE, Strynadka NC. Crystal structure of a bacterial signal peptidase in complex with a beta-lactam inhibitor. Nature. 1998;396:186–190. [PubMed: 9823901]
- 125.
- Carlos JL, Paetzel M, Brubaker G, Karla A, Ashwell CM, Lively MO. et al. The role of the membrane-spanning domain of type I signal peptidases in substrate cleavage site selection. J Biol Chem. 2000;275:38813–38822. [PubMed: 10982814]
- 126.
- Lewell XQ, Judd DB, Watson SP, Hann MM. RECAP-Retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J Chem Inf Comput Sci. 1998;38:511–522. [PubMed: 9611787]
- 127.
- Steinlein OK, Noebels JL. Ion channels and epilepsy in man and mouse. Curr Opin Genet Dev. 2000;10:286–291. [PubMed: 10826987]
- 128.
- Grillner S. Bridging the gap-From ion channels to networks and behaviour. Curr Opin Neurobiol. 1999;9:663–669. [PubMed: 10607645]
- 129.
- Castle NA, Hollinshead SP, Hughes PF, Mendoza JS, Wilson JW, Amato G. et al. Icagen Inc, Eli Lilly and Company; Int Pat Appl 1998. WO98/04521 .
- 130.
- Gerber PR, Müller K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J Comput Aided Mol Des. 1995;9:251–268. [PubMed: 7561977]
- 131.
- Holland JH. Hidden Order-How Adaptation builds Complexity. Reading: Perseus Books. 1995
- 132.
- Pearlman DA, Murcko MA. CONCERTS: Dynamic connection of fragments as an approach to de novo ligand design. J Med Chem. 1996;39:1651–1663. [PubMed: 8648605]
- 133.
- Rotstein SH, Murcko MA. GenStar: A method for de novo drug design. J Comput Aided Mol Des. 1993;7:23–43. [PubMed: 8473916]
- 134.
- Rotstein SH, Murcko MA. Groupbuild: A fragment-based method for de novo drug design. J Med Chem. 1993;36:1700–1710. [PubMed: 8510098]
- 135.
- Bohacek RS, McMartin C. Multiple highly diverse structures complementary to enzyme binding sites: Results of extensive application of a de novo design method incorporating combinatorial growth. J Am Chem Soc. 1994;116:5560–5571.
- 136.
- Eisen MB, Wiley DC, Karplus M, Hubbard RE. HOOK: A program for finding novel molecular architectures that satisfy the chemical and steric requirements of a macromolecule binding site. Proteins. 1994;19:199–221. [PubMed: 7937734]
- 137.
- Nishibata Y, Itai A. Automatic creation of drug candidate structures based on receptor structure. Starting point for artificial lead generation. Tetrahedron. 1991;41:8985–8990.
- 138.
- Gehlhaar DK, Moerder KE, Zichi D, Sherman CJ, Ogden RC, Freer ST. De novo design of enzyme inhibitors by Monte Carlo ligand generation. J Med Chem. 1995;38:466–472. [PubMed: 7853340]
- 139.
- Miranker A, Karplus M. Functionality maps of binding sites: A multiple copy simultaneous search method. Proteins. 1991;11:29–34. [PubMed: 1961699]
- 140.
- Clark DE, Firth MA, Murray CW. MOLMAKER: De novo generation of 3D databases for use in drug design. J Chem Inf Comput Sci. 1996;36:137–145. [PubMed: 8576288]
- 141.
- Tschinke V, Cohen NC. The NEWLEAD program: A new method for the design of candidate structures from pharmacophoric hypotheses. J Med Chem. 1993;36:3863–3870. [PubMed: 8254618]
- 142.
- Schneider G, Todt T, Wrede P. De novo design of peptides and proteins: Machine-generated sequences by the PROSA program. Comput Appl Biosci. 1994;10:75–77. [PubMed: 8193960]
- 143.
- Murray CW, Clark DE, Auton TR, Firth MA, Li J, Sykes RA. et al. PRO_SELECT: Combining structure-based drug design and combinatorial chemistry for rapid lead discovery. 1. Technology. J Comput Aided Mol Des. 1997;11:193–207. [PubMed: 9089436]
- Cytokines in the Pathogenesis of Rheumatoid Arthritis and Collagen-Induced Arthr...Cytokines in the Pathogenesis of Rheumatoid Arthritis and Collagen-Induced Arthritis - Madame Curie Bioscience Database
- Evolutionary De Novo Design - Madame Curie Bioscience DatabaseEvolutionary De Novo Design - Madame Curie Bioscience Database
- The Evolution of CDK-Activating Kinases - Madame Curie Bioscience DatabaseThe Evolution of CDK-Activating Kinases - Madame Curie Bioscience Database
- Cell Cycle Reactivation in Skeletal Muscle and Other Terminally Differentiated C...Cell Cycle Reactivation in Skeletal Muscle and Other Terminally Differentiated Cells - Madame Curie Bioscience Database
- A Conceptual Framework - Madame Curie Bioscience DatabaseA Conceptual Framework - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...